Two-Phase Equilibrium Conditions in Nanopores

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Theory
2.1. Thermodynamic Relations for Small Systems
2.2. The Integral Pressure of a Representative Volume Element
2.3. A Mechanical Interpretation of the Pressures
3. Simulations
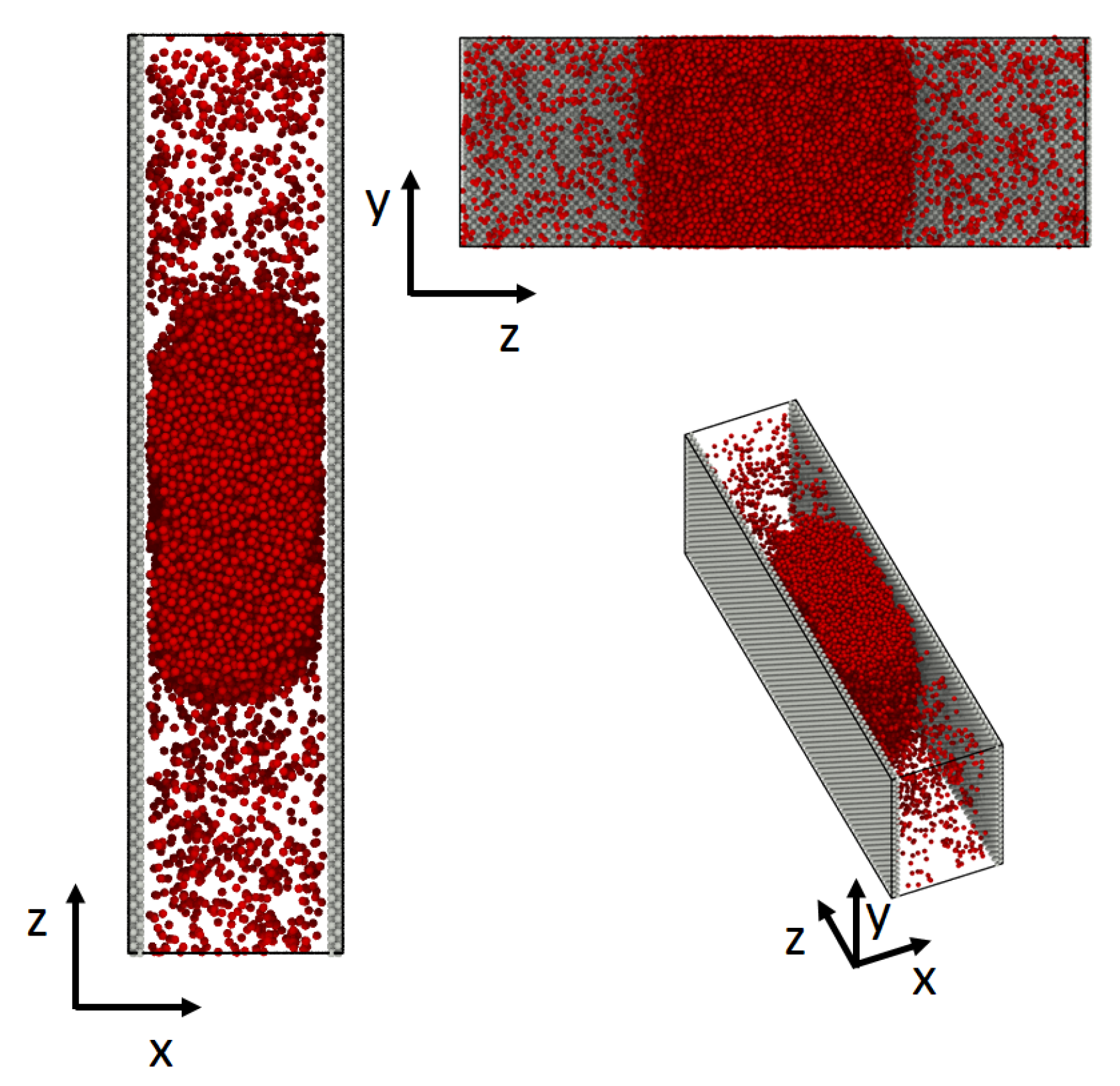
3.1. Molecular Dynamic Simulations
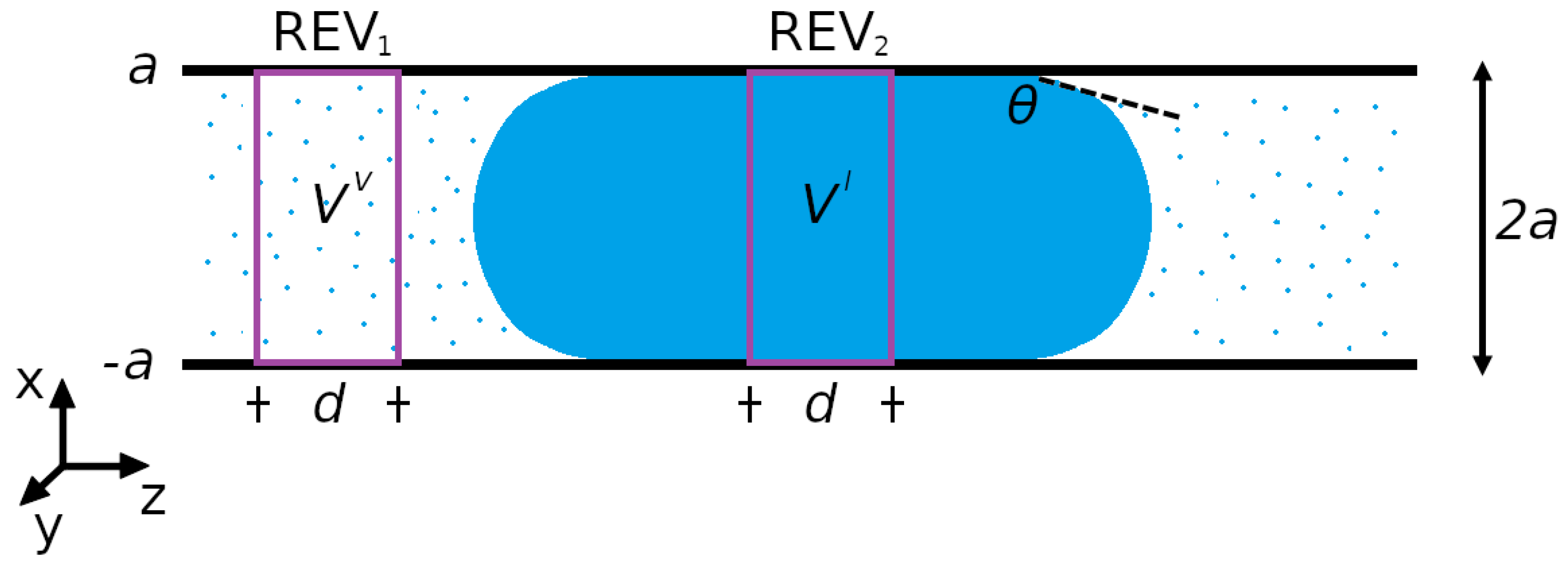
3.1.1. System
3.1.2. Particle Interaction Potential
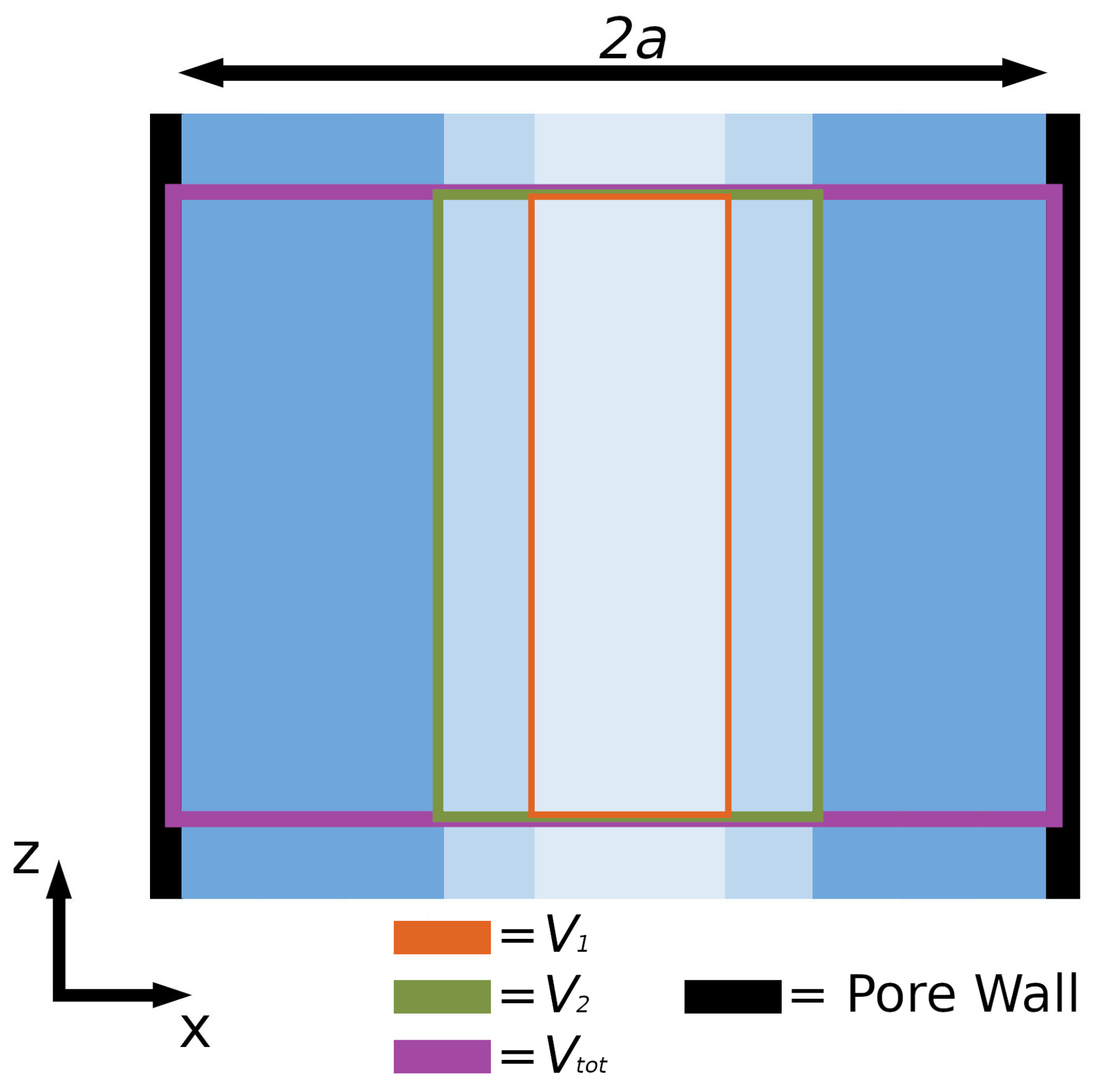
3.1.3. Computation of REV Pressures and Wall-Fluid Surface Tension
3.2. Gibbs Ensemble Monte Carlo
4. Results and Discussion
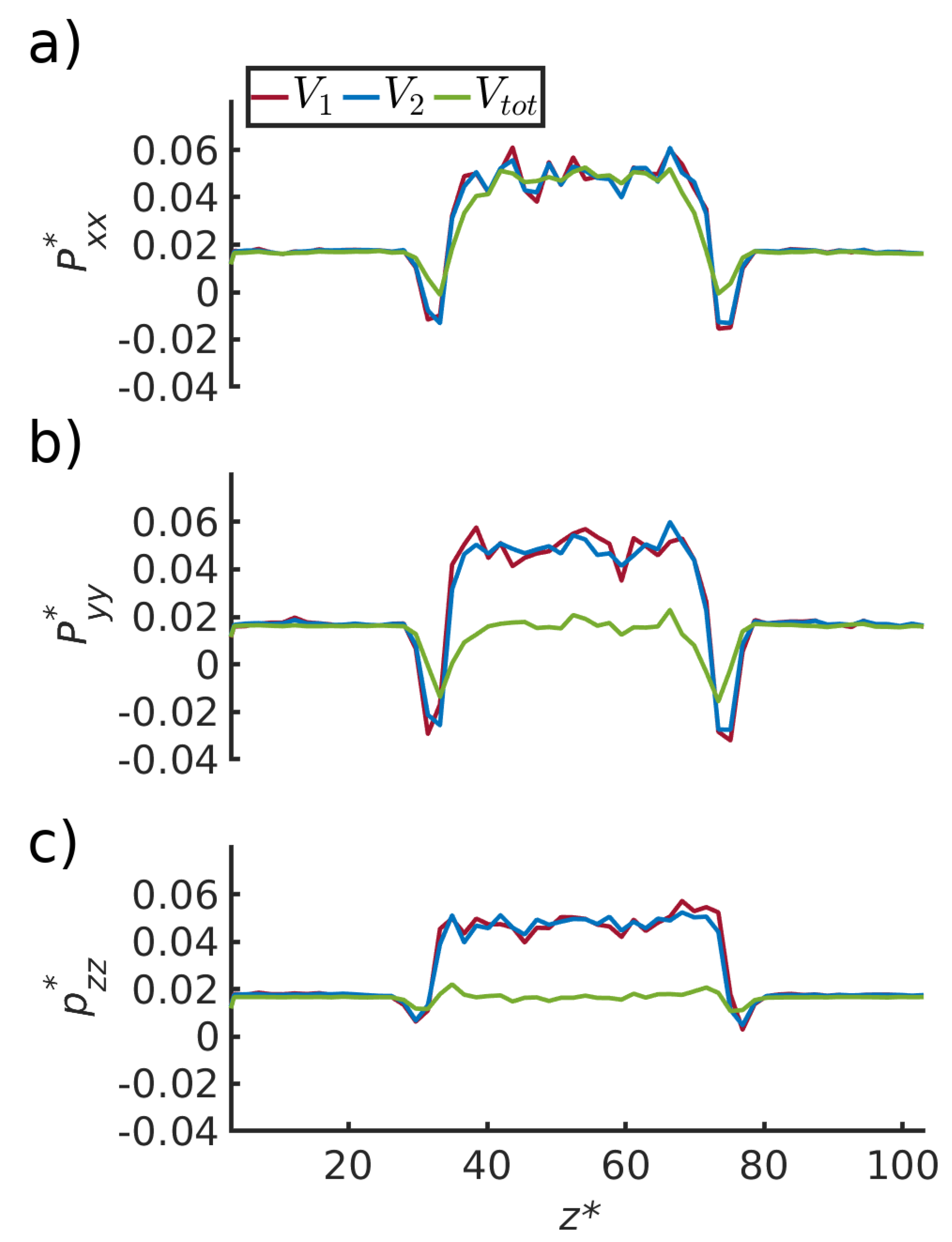
4.1. Pressure Component Normal to the Pore Wall
4.2. Pressure Variation in the Direction Normal to the Pore Wall
4.3. Pressure Variation along the Pore
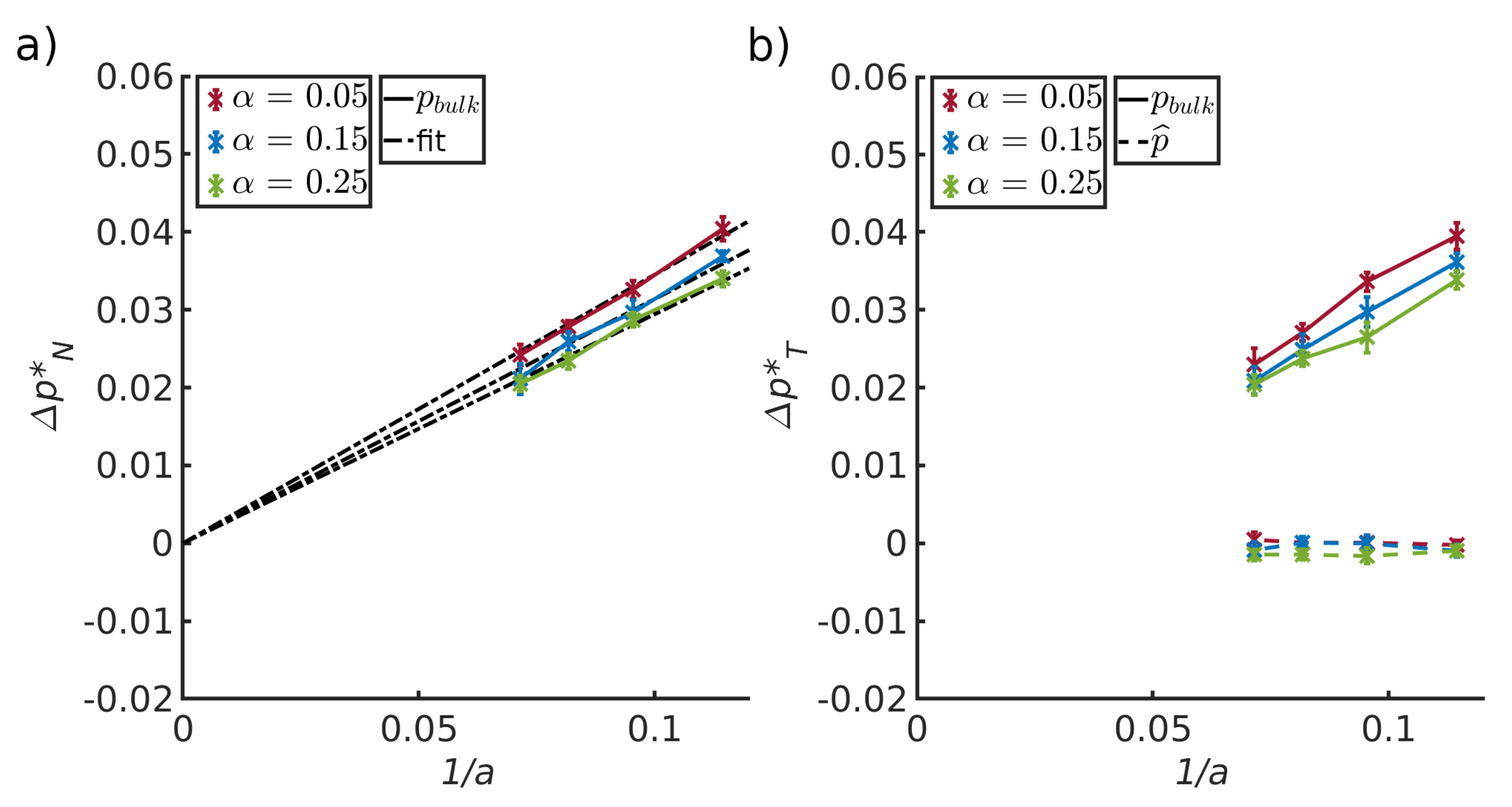
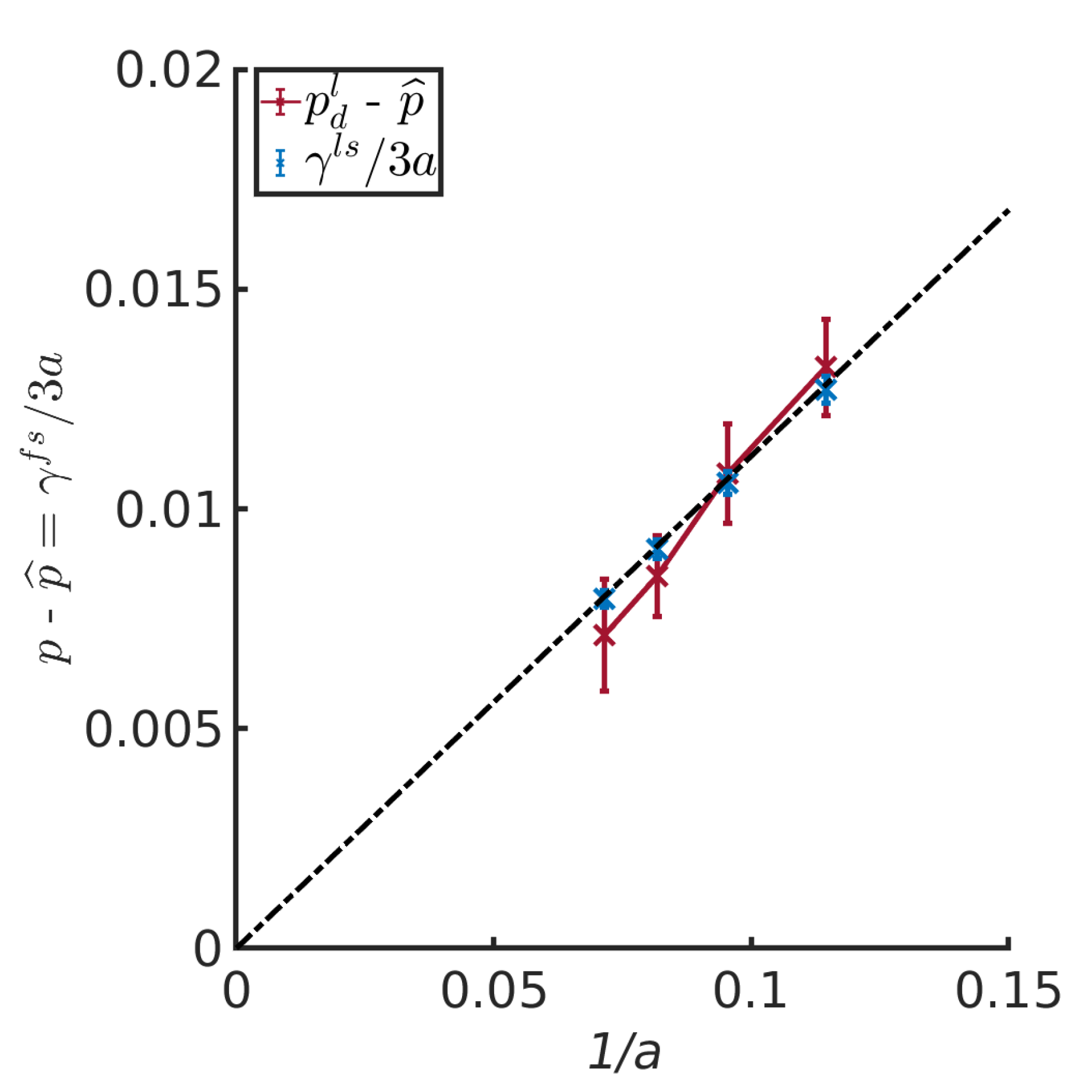
4.4. Pressure Differences Across the Liquid-Vapor Interface
4.5. The Small System Scaling Law and the Subdivison Potential
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Keshavarzi, E.; Kamalvand, M. Energy effects on the structure and thermodynamic properties of nanoconfined fluids (a density functional theory study). J. Phys. Chem. B 2009, 113, 5493–5499. [Google Scholar] [CrossRef] [PubMed]
- Braun, E.; Chen, J.J.; Schnell, S.K.; Lin, L.C.; Reimer, J.A.; Smit, B. Nanoporous Materials Can Tune the Critical Point of a Pure Substance. Angew. Chem. 2015, 127, 14557–14560. [Google Scholar] [CrossRef]
- Pharoah, J.; Karan, K.; Sun, W. On effective transport coefficients in PEM fuel cell electrodes: Anisotropy of the porous transport layers. J. Power Sources 2006, 161, 214–224. [Google Scholar] [CrossRef]
- Newman, J.; Tiedemann, W. Porous-electrode theory with battery applications. AIChE J. 1975, 21, 25–41. [Google Scholar] [CrossRef]
- Guo, H.; Wyart, Y.; Perot, J.; Nauleau, F.; Moulin, P. Low-pressure membrane integrity tests for drinking water treatment: A review. Water Res. 2010, 44, 41–57. [Google Scholar] [CrossRef]
- Alklaibi, A.M.; Lior, N. Transport analysis of air-gap membrane distillation. J. Membr. Sci. 2005, 255, 239–253. [Google Scholar] [CrossRef]
- Kuipers, N.; Hanemaaijer, J.H.; Brouwer, H.; van Medevoort, J.; Jansen, A.; Altena, F.; van der Vleuten, P.; Bak, H. Simultaneous production of high-quality water and electrical power from aqueous feedstock’s and waste heat by high-pressure membrane distillation. Desalin. Water Treat. 2015, 55, 2766–2776. [Google Scholar] [CrossRef]
- Schnell, S.K.; Vlugt, T.J.H.; Simon, J.M.; Bedeaux, D.; Kjelstrup, S. Thermodynamics of small systems embedded in a reservoir: A detailed analysis of finite size effects. Mol. Phys. 2012, 110, 1069–1079. [Google Scholar] [CrossRef]
- Strøm, B.; Simon, J.M.; Schnell, S.; Kjelstrup, S.; He, J.; Bedeaux, D. Size and shape effects on the thermodynamic properties of nanoscale volumes of water. Phys. Chem. Chem. Phys. 2017, 19, 9016. [Google Scholar] [CrossRef]
- Erdős, M.; Galteland, O.; Bedeaux, D.; Kjelstrup, S.; Moultos, O.A.; Vlugt, T.J.H. Gibbs Ensemble Monte Carlo Simulation of Fluids in Confinement: Relation between the Differential and Integral Pressures. Nanomaterials 2020, 10, 293. [Google Scholar] [CrossRef]
- Hill, T.L. Thermodynamics of Small Systems; Dover Publications Inc.: New York, NY, USA, 1994. [Google Scholar]
- Dong, X.; Liu, H.; Hou, J.; Wu, K.; Chen, Z. Phase equilibria of confined fluids in nanopores of tight and shale rocks considering the effect of capillary pressure and adsorption film. Ind. Eng. Chem. Res. 2016, 55, 798–811. [Google Scholar] [CrossRef]
- Gubbins, K.E.; Long, Y.; Śliwinska-Bartkowiak, M. Thermodynamics of confined nano-phases. J. Chem. Thermodyn. 2014, 74, 169–183. [Google Scholar] [CrossRef]
- Giovambattista, N.; Rossky, P.J.; Debenedetti, P.G. Phase transitions induced by nanoconfinement in liquid water. Phys. Rev. Lett. 2009, 102, 050603. [Google Scholar] [CrossRef] [PubMed]
- Eslami, H.; Mehdipour, N. Local chemical potential and pressure tensor in inhomogeneous nanoconfined fluids. J. Chem. Phys. 2012, 137, 144702. [Google Scholar] [CrossRef] [PubMed]
- Bennethum, L.S.; Weinstein, T. Three pressures in porous media. Transp. Porous Med. 2004, 54, 1–34. [Google Scholar] [CrossRef]
- Kjelstrup, S.; Bedeaux, D.; Hansen, A.; Hafskjold, B.; Galteland, O. Non-isothermal transport of multi-phase fluids in porous media. Constitutive equations. Front. Phys. 2019, 6, 150. [Google Scholar] [CrossRef]
- Galteland, O.; Bedeaux, D.; Kjelstrup, S.; Hafskjold, B. Pressures inside a nano-porous medium. The case of a single phase fluid. Front. Phys. 2019, 7, 60. [Google Scholar] [CrossRef]
- Bedeaux, D.; Kjelstrup, S.; Schnell, S.K. Nanothermodynamics. General Theory; PoreLab, NTNU Grafisk: Trondheim, Norway, 2020. [Google Scholar]
- Bedeaux, D.; Kjelstrup, S. Hill’s thermodynamics is equivalent with Gibb’s thermodynamics for surfaces of constant curvatures. Chem. Phys. Lett. 2018, 707, 40–43. [Google Scholar] [CrossRef]
- Tsai, D. The virial theorem and stress calculation in molecular dynamics. J. Chem. Phys. 1979, 70, 1375–1382. [Google Scholar] [CrossRef]
- Chatterjee, J. Prediction of coupled menisci shapes by Young–Laplace equation and the resultant variability in capillary retention. J. Colloid Interface Sci. 2007, 314, 199–206. [Google Scholar] [CrossRef]
- Gras, J.P.; Delenne, J.Y.; El Youssoufi, M.S. Study of capillary interaction between two grains: A new experimental device with suction control. Granular Matter 2013, 15, 49–56. [Google Scholar] [CrossRef]
- Kirkwood, J.G.; Buff, F.P. The statistical mechanical theory of surface tension. J. Chem. Phys. 1949, 17, 338–343. [Google Scholar] [CrossRef]
- Hafskjold, B.; Travis, K.P.; Hass, A.B.; Hammer, M.; Aasen, A.; Wilhelmsen, Ø. Thermodynamic properties of the 3D Lennard-Jones/spline model. Mol. Phys. 2019, 117, 3754–3769. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Braga, C.; Travis, K.P. A configurational temperature Nosé-Hoover thermostat. J. Chem. Phys. 2005, 123, 134101. [Google Scholar] [CrossRef]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications; Elsevier: Amsterdam, The Netherlands, 2001; Volume 1. [Google Scholar]
- Hafskjold, B.; Ikeshoji, T. Microscopic pressure tensor for hard-sphere fluids. Phys. Rev. E 2002, 66, 011203. [Google Scholar] [CrossRef]
- Ikeshoji, T.; Hafskjold, B.; Furuholt, H. Molecular-level calculation scheme for pressure in inhomogeneous systems of flat and spherical layers. Mol. Simul. 2003, 29, 101–109. [Google Scholar] [CrossRef]
- Todd, B.; Evans, D.J.; Daivis, P.J. Pressure tensor for inhomogeneous fluids. Phys. Rev. E 1995, 52, 1627. [Google Scholar] [CrossRef]
- Todd, B.D.; Daivis, P.J. Nonequilibrium Molecular Dynamics: Theory, Algorithms and Applications; Cambridge University Press: Cambridge, UK, 2017. [Google Scholar]
- Skorpa, R.; Simon, J.M.; Bedeaux, D.; Kjelstrup, S. The reaction enthalpy of hydrogen dissociation calculated with the Small System Method from simulation of molecular fluctuations. Phys. Chem. Chem. Phys. 2014, 16, 19681. [Google Scholar] [CrossRef]
- Daivis, P.J.; Evans, D.J. Comparison of constant pressure and constant volume nonequilibrium simulations of sheared model decane. J. Chem. Phys. 1994, 100, 541–547. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pore Size | Normal Pressure, | Bulk Fluid Pressure, |
|---|---|---|
| 5 | ||
| 6 | ||
| 9 | ||
| 12 | ||
| 15 | ||
| 21 | ||
| 27 | ||
| 35 | ||
| 5 | ||
| 6 | ||
| 9 | ||
| 12 | ||
| 15 | ||
| 21 | ||
| 27 | ||
| 35 |
| 0.05 | 0.36 ± 0.02 | 0.0067 ± 0.0026 |
| 0.15 | 0.33 ± 0.01 | 0.005 ± 0.0035 |
| 0.25 | 0.31 ± 0.01 | 0.004 ± 0.0045 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rauter, M.T.; Galteland, O.; Erdős, M.; Moultos, O.A.; Vlugt, T.J.H.; Schnell, S.K.; Bedeaux, D.; Kjelstrup, S. Two-Phase Equilibrium Conditions in Nanopores. Nanomaterials 2020, 10, 608. https://doi.org/10.3390/nano10040608
Rauter MT, Galteland O, Erdős M, Moultos OA, Vlugt TJH, Schnell SK, Bedeaux D, Kjelstrup S. Two-Phase Equilibrium Conditions in Nanopores. Nanomaterials. 2020; 10(4):608. https://doi.org/10.3390/nano10040608
Chicago/Turabian StyleRauter, Michael T., Olav Galteland, Máté Erdős, Othonas A. Moultos, Thijs J. H. Vlugt, Sondre K. Schnell, Dick Bedeaux, and Signe Kjelstrup. 2020. "Two-Phase Equilibrium Conditions in Nanopores" Nanomaterials 10, no. 4: 608. https://doi.org/10.3390/nano10040608
APA StyleRauter, M. T., Galteland, O., Erdős, M., Moultos, O. A., Vlugt, T. J. H., Schnell, S. K., Bedeaux, D., & Kjelstrup, S. (2020). Two-Phase Equilibrium Conditions in Nanopores. Nanomaterials, 10(4), 608. https://doi.org/10.3390/nano10040608