
Scalable Sacrificial Templating to Increase Porosity and Platinum Utilisation in Graphene-Based Polymer Electrolyte Fuel Cell Electrodes

 , , ,
, , ,  , and
, and
Abstract
:
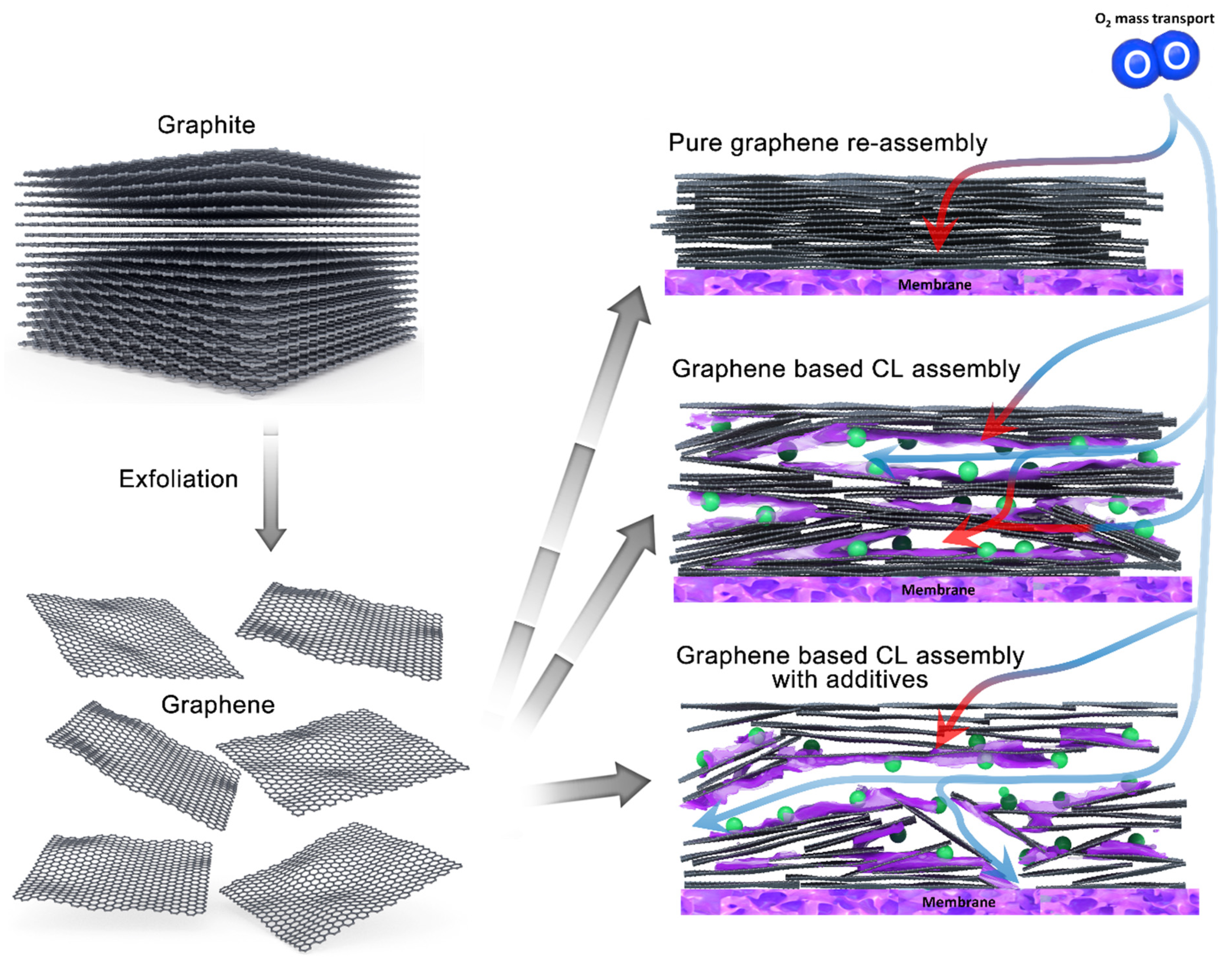
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis
2.3. Characterisation
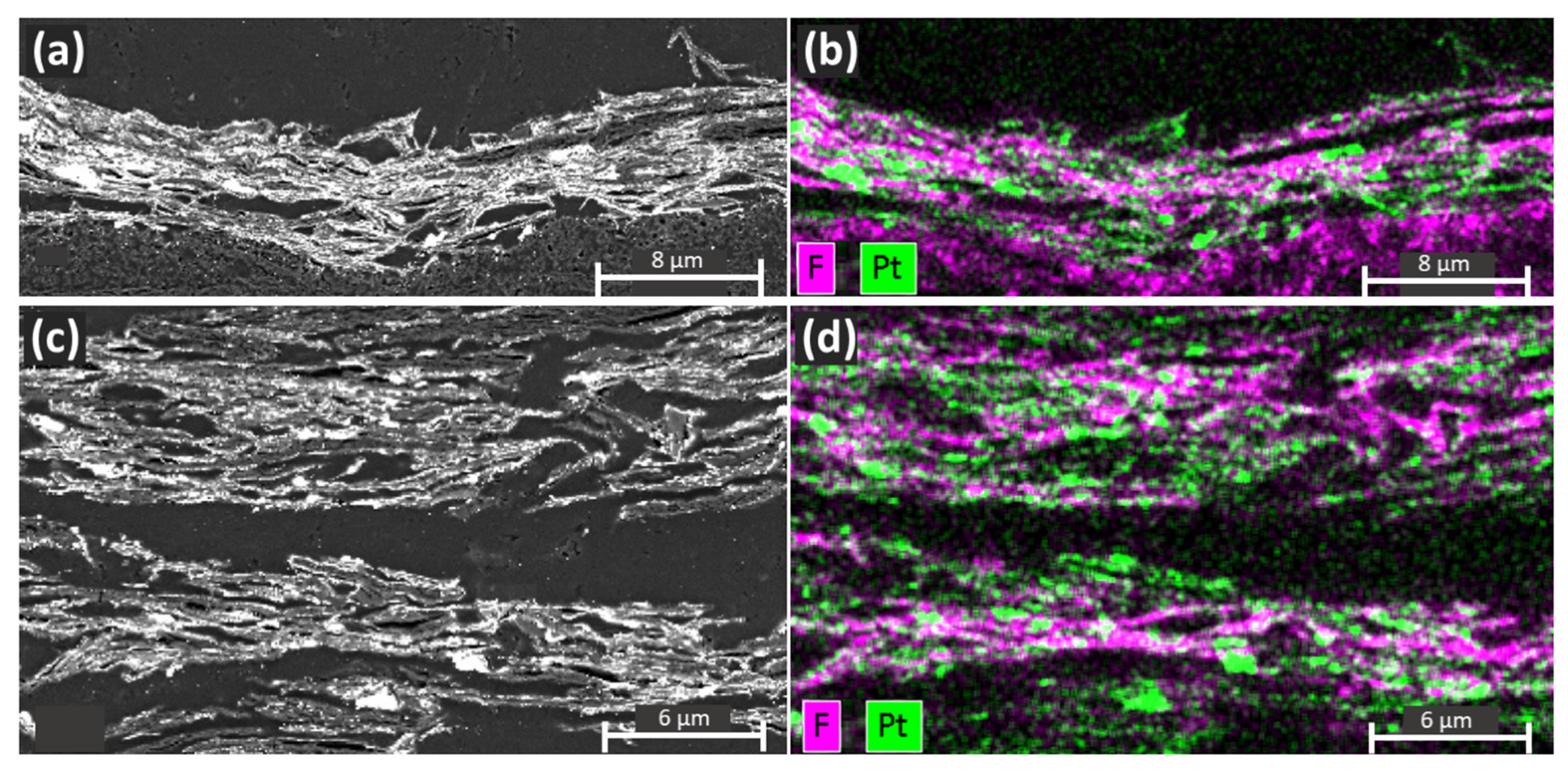
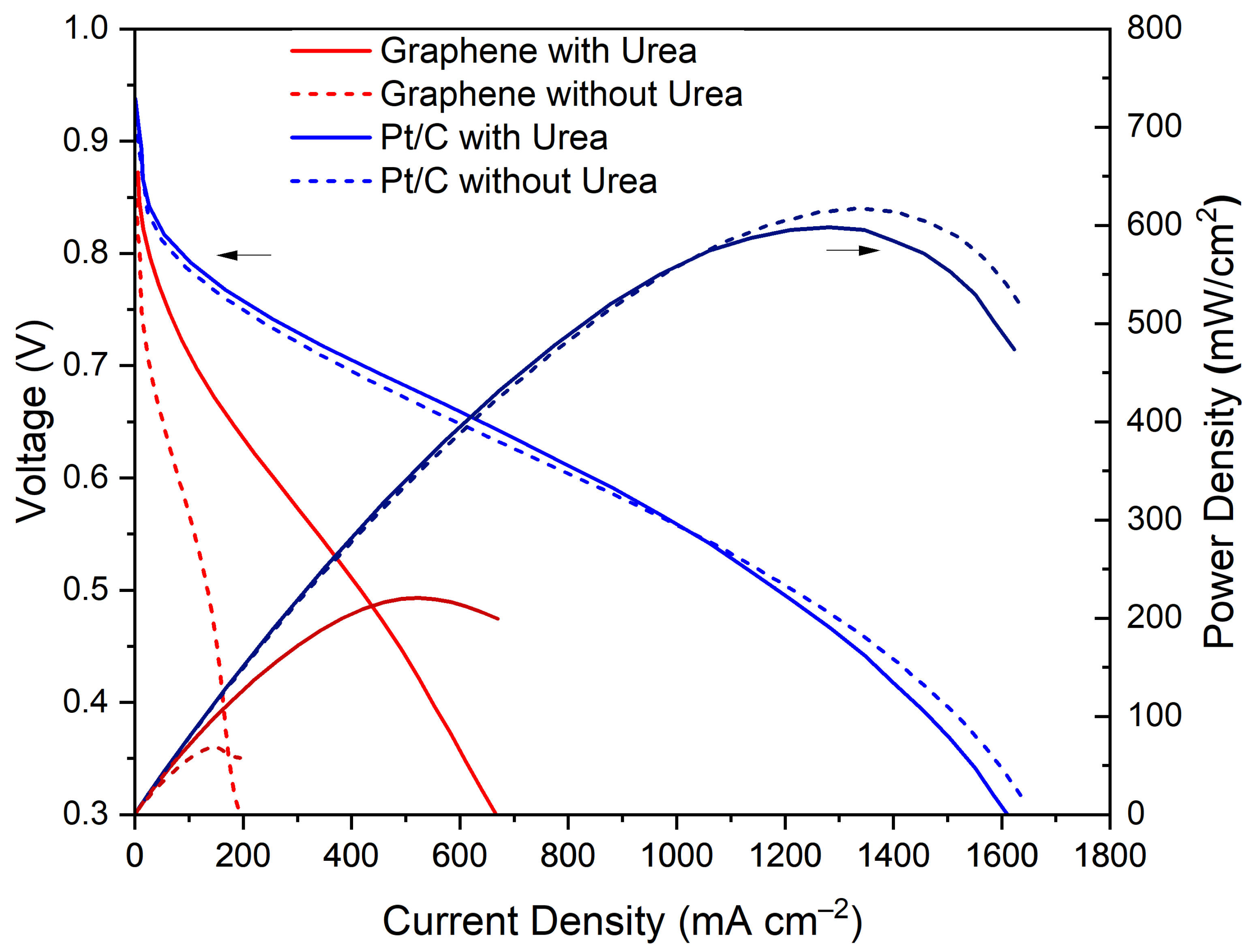
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Garche, J.; Jörissen, L. Applications of fuel cell technology: Status and perspectives. Electrochem. Soc. Interface 2015, 24, 39–43. [Google Scholar] [CrossRef]
- Staffell, I.; Scamman, D.; Abad, A.V.; Balcombe, P.; Dodds, P.E.; Ekins, P.; Shah, N.; Ward, K.R. The role of hydrogen and fuel cells in the global energy system. Energy Environ. Sci. 2019, 12, 463–491. [Google Scholar] [CrossRef] [Green Version]
- Hart, D.; Lehner, F.; Rose, R.; Lewis, J. The Fuel Cell Industry Review 2019. E4Tech 2019. Available online: https://www.californiahydrogen.org/wp-content/uploads/2019/01/TheFuelCellIndustryReview2018.pdf (accessed on 1 May 2021).
- Yoon, W.; Weber, A.Z. Modeling Low-Platinum-Loading Effects in Fuel-Cell Catalyst Layers. J. Electrochem. Soc. 2011, 158, B1007. [Google Scholar] [CrossRef]
- Ono, Y.; Mashio, T.; Takaichi, S.; Ohma, A.; Kanesaka, H.; Shinohara, K. The Analysis of Performance Loss with Low Platinum Loaded Cathode Catalyst Layers. ECS Trans. 2019, 28, 69–78. [Google Scholar] [CrossRef]
- Kongkanand, A.; Mathias, M.F. The Priority and Challenge of High-Power Performance of Low-Platinum Proton-Exchange Membrane Fuel Cells. J. Phys. Chem. Lett. 2016, 7, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Dodelet, J.P.; Wu, G.; Zelenay, P. PGM-Free Cathode Catalysts for PEM Fuel Cells: A Mini-Review on Stability Challenges. Adv. Mater. 2019, 31, 1807615. [Google Scholar] [CrossRef] [PubMed]
- Jaouen, F.; Jones, D.; Coutard, N.; Artero, V.; Strasser, P.; Kucernak, A. Toward platinum group metal-free catalysts for hydrogen/air proton-exchange membrane fuel cells. Johns. Matthey Technol. Rev. 2018, 62, 231–255. [Google Scholar] [CrossRef]
- Banham, D.; Ye, S.Y. Current Status and Future Development of Catalyst Materials and Catalyst Layers for Proton Exchange Membrane Fuel Cells: An Industrial Perspective. Acs Energy Lett. 2017, 2, 629–638. [Google Scholar] [CrossRef]
- Bing, Y.; Liu, H.; Zhang, L.; Ghosh, D.; Zhang, J. Nanostructured Pt-alloy electrocatalysts for PEM fuel cell oxygen reduction reaction. Chem. Soc. Rev. 2010, 39, 2184–2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, H.; Henning, S.; Herranz, J.; Eychmüller, A.; Uchida, M.; Schmidt, T.J. Analysis and Modeling of Polymer Electrolyte Fuel Cell Unsupported Catalyst Layers. J. Electrochem. Soc. 2018, 165, F7–F16. [Google Scholar] [CrossRef] [Green Version]
- Henning, S.; Ishikawa, H.; Kühn, L.; Herranz, J.; Müller, E.; Eychmüller, A.; Schmidt, T.J. Unsupported Pt-Ni Aerogels with Enhanced High Current Performance and Durability in Fuel Cell Cathodes. Angew. Chem. Int. Ed. 2017, 56, 10707–10710. [Google Scholar] [CrossRef] [PubMed]
- Karuppanan, K.K.; Raghu, A.V.; Panthalingal, M.K.; Thiruvenkatam, V.; Karthikeyan, P.; Pullithadathil, B. 3D-porous electrocatalytic foam based on Pt@N-doped graphene for high performance and durable polymer electrolyte membrane fuel cells. Sustain. Energy Fuels 2019, 3, 996–1011. [Google Scholar] [CrossRef]
- Kim, O.H.; Cho, Y.H.; Kang, S.H.; Park, H.Y.; Kim, M.; Lim, J.W.; Chung, D.Y.; Lee, M.J.; Choe, H.; Sung, Y.E. Ordered macroporous platinum electrode and enhanced mass transfer in fuel cells using inverse opal structure. Nat. Commun. 2013, 4, 2473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusoglu, A.; Weber, A.Z. New Insights into Perfluorinated Sulfonic-Acid Ionomers. Chem. Rev. 2017, 117, 987–1104. [Google Scholar] [CrossRef] [PubMed]
- Karan, K. Recent advances in materials, microstructural characterization, and modeling. Curr. Opin. Electrochem. 2017, 5, 27–35. [Google Scholar] [CrossRef]
- Uchida, M. Effect of support microstructure on both distributions of Pt and ionomer and cell performance and durability. Curr. Opin. Electrochem. 2020, 21, 209–218. [Google Scholar] [CrossRef]
- Padgett, E.; Yarlagadda, V.; Holtz, M.E.; Ko, M.; Levin, B.D.A.; Kukreja, R.S.; Ziegelbauer, J.M.; Andrews, R.N.; Ilavsky, J.; Kongkanand, A.; et al. Mitigation of PEM Fuel Cell Catalyst Degradation with Porous Carbon Supports. J. Electrochem. Soc. 2019, 166, F198–F207. [Google Scholar] [CrossRef]
- Sneed, B.T.; Cullen, D.A.; Reeves, K.S.; Dyck, O.E.; Langlois, D.A.; Mukundan, R.; Borup, R.L.; More, K.L. 3D Analysis of Fuel Cell Electrocatalyst Degradation on Alternate Carbon Supports. ACS Appl. Mater. Interfaces 2017, 9, 29839–29848. [Google Scholar] [CrossRef]
- Takahashi, K.; Koda, R.; Kakinuma, K.; Uchida, M. Improvement of Cell Performance in Low-Pt-Loading PEFC Cathode Catalyst Layers with Pt/Ta-SnO 2 Prepared by the Electrospray Method. J. Electrochem. Soc. 2017, 164, F235–F242. [Google Scholar] [CrossRef]
- Kim, T.; Xie, T.; Jung, W.S.; Popov, B.N. Development of ultra–low highly active and durable hybrid compressive platinum lattice cathode catalysts for polymer electrolyte membrane fuel cells. Int. J. Hydrogen Energy 2017, 42, 12507–12520. [Google Scholar] [CrossRef]
- Suter, T.A.M.; Smith, K.; Hack, E.; Rasha, L.; Rana, Z.; Angel, G.M.A.; Shearing, P.R.; Miller, T.S.; Brett, D.J.L. Engineering Catalyst Layers for Next-Generation Polymer Electrolyte Fuel Cells: A Review of Design, Materials, and Methods. Adv. Energy Mater. 2021, 2101025. [Google Scholar] [CrossRef]
- Yarlagadda, V.; Carpenter, M.K.; Moylan, T.E.; Kukreja, R.S.; Koestner, R.; Gu, W.; Thompson, L.; Kongkanand, A. Boosting Fuel Cell Performance with Accessible Carbon Mesopores. ACS Energy Lett. 2018, 3, 618–621. [Google Scholar] [CrossRef] [Green Version]
- Soboleva, T.; Zhao, X.; Malek, K.; Xie, Z.; Navessin, T.; Holdcroft, S. On the micro-, meso-, and macroporous structures of polymer electrolyte membrane fuel cell catalyst layers. ACS Appl. Mater. Interfaces 2010, 2, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Forouzandeh, F.; Li, X.; Banham, D.W.; Feng, F.; Ye, S.; Birss, V. Understanding the Corrosion Resistance of Meso- and Micro-Porous Carbons for Application in PEM Fuel Cells. J. Electrochem. Soc. 2018, 165, F3230–F3240. [Google Scholar] [CrossRef] [Green Version]
- Fang, Z.; Lee, M.S.; Kim, J.Y.; Kim, J.H.; Fuller, T.F. The Effect of Carbon Support Surface Functionalization on PEM Fuel Cell Performance, Durability, and Ionomer Coverage in the Catalyst Layer. J. Electrochem. Soc. 2020, 167, 064506. [Google Scholar] [CrossRef]
- Lee, G.; Choi, H.; Tak, Y. In situ durability of various carbon supports against carbon corrosion during fuel starvation in a PEM fuel cell cathode. Nanotechnology 2019, 30, 085402. [Google Scholar] [CrossRef]
- Oh, H.S.; Oh, J.G.; Haam, S.; Arunabha, K.; Roh, B.; Hwang, I.; Kim, H. On-line mass spectrometry study of carbon corrosion in polymer electrolyte membrane fuel cells. Electrochem. Commun. 2008, 10, 1048–1051. [Google Scholar] [CrossRef]
- Meier, J.C.; Galeano, C.; Katsounaros, I.; Witte, J.; Bongard, H.J.; Topalov, A.A.; Baldizzone, C.; Mezzavilla, S.; Schüth, F.; Mayrhofer, K.J.J. Design criteria for stable Pt/C fuel cell catalysts. Beilstein J. Nanotechnol. 2014, 5, 44–67. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Shao, Y.; Wan, H.; Rieke, P.C.; Viswanathan, V.V.; Towne, S.A.; Saraf, L.V.; Liu, J.; Lin, Y.; Wang, Y. Design of graphene sheets-supported Pt catalyst layer in PEM fuel cells. Electrochem. Commun. 2011, 13, 258–261. [Google Scholar] [CrossRef]
- Xia, Z.; Wang, S.; Jiang, L.; Sun, H.; Qi, F.; Jin, J.; Sun, G. Rational design of a highly efficient Pt/graphene-Nafion® composite fuel cell electrode architecture. J. Mater. Chem. A 2015, 3, 1641–1648. [Google Scholar] [CrossRef]
- Daş, E.; Kaplan, B.Y.; Gürsel, S.A.; Yurtcan, A.B. Graphene nanoplatelets-carbon black hybrids as an efficient catalyst support for Pt nanoparticles for polymer electrolyte membrane fuel cells. Renew. Energy 2019, 139, 1099–1110. [Google Scholar] [CrossRef]
- Malek, K.; Mashio, T.; Eikerling, M. Microstructure of Catalyst Layers in PEM Fuel Cells Redefined: A Computational Approach. Electrocatalysis 2011, 2, 141–157. [Google Scholar] [CrossRef]
- Inoue, G.; Kawase, M. Effect of porous structure of catalyst layer on effective oxygen diffusion coefficient in polymer electrolyte fuel cell. J. Power Sources 2016, 327, 1–10. [Google Scholar] [CrossRef]
- Şanlı, L.I.; Yarar, B.; Bayram, V.; Gürsel, S.A. Electrosprayed catalyst layers based on graphene–carbon black hybrids for the next-generation fuel cell electrodes. J. Mater. Sci. 2017, 52, 2091–2102. [Google Scholar] [CrossRef] [Green Version]
- Ott, S.; Orfanidi, A.; Schmies, H.; Anke, B.; Nong, H.N.; Hübner, J.; Gernert, U.; Gliech, M.; Lerch, M.; Strasser, P. Ionomer distribution control in porous carbon-supported catalyst layers for high-power and low Pt-loaded proton exchange membrane fuel cells. Nat. Mater. 2020, 19, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Haber, J. Manual on catalyst characterization. Pure Appl. Chem. 1991, 63, 1227–1246. [Google Scholar] [CrossRef]
- Kjelstrup, S.; Coppens, M.O.; Pharoah, J.G.; Pfeifer, P. Nature-inspired energy-and material-efficient design of a polymer electrolyte membrane fuel cell. Energy Fuels 2010, 24, 5097–5108. [Google Scholar] [CrossRef]
- Shinozaki, K.; Morimoto, Y.; Pivovar, B.S.; Kocha, S.S. Suppression of oxygen reduction reaction activity on Pt-based electrocatalysts from ionomer incorporation. J. Power Sources 2016, 325, 745–751. [Google Scholar] [CrossRef] [Green Version]
- Jinnouchi, R.; Kudo, K.; Kitano, N.; Morimoto, Y. Molecular Dynamics Simulations on O2 Permeation through Nafion Ionomer on Platinum Surface. Electrochim. Acta 2016, 188, 767–776. [Google Scholar] [CrossRef]
- Subbaraman, R.; Strmcnik, D.; Paulikas, A.P.; Stamenkovic, V.R.; Markovic, N.M. Oxygen reduction reaction at three-phase interfaces. ChemPhysChem 2010, 11, 2825–2833. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.C.; Tokiwa, H.; Kakinuma, K.; Watanabe, M.; Uchida, M. Effects of carbon supports on Pt distribution, ionomer coverage and cathode performance for polymer electrolyte fuel cells. J. Power Sources 2016, 315, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Sui, S.; Wang, X.; Zhou, X.; Su, Y.; Riffat, S.; Liu, C.J. A comprehensive review of Pt electrocatalysts for the oxygen reduction reaction: Nanostructure, activity, mechanism and carbon support in PEM fuel cells. J. Mater. Chem. A 2017, 5, 1808–1825. [Google Scholar] [CrossRef]
- Şanlı, L.I.; Bayram, V.; Ghobadi, S.; Düzen, N.; Gürsel, S.A. Engineered catalyst layer design with graphene-carbon black hybrid supports for enhanced platinum utilization in PEM fuel cell. Int. J. Hydrogen Energy 2017, 42, 1085–1092. [Google Scholar] [CrossRef]
- Ghosh, A.; Basu, S.; Verma, A. Graphene and functionalized graphene supported platinum catalyst for PEMFC. Fuel Cells 2013, 13, 355–363. [Google Scholar] [CrossRef]
- Cho, S.H.; Yang, H.N.; Lee, D.C.; Park, S.H.; Kim, W.J. Electrochemical properties of Pt/graphene intercalated by carbon black and its application in polymer electrolyte membrane fuel cell. J. Power Sources 2013, 225, 200–206. [Google Scholar] [CrossRef]
- Marinkas, A.; Arena, F.; Mitzel, J.; Prinz, G.M.; Heinzel, A.; Peinecke, V.; Natter, H. The influences of carbon additives and catalyst preparation methods on the performance of PEM fuel cells. Carbon N. Y. 2013, 58, 139–150. [Google Scholar] [CrossRef]
- Kaplan, B.Y.; Haghmoradi, N.; Biçer, E.; Merino, C.; Gürsel, S.A. High performance electrocatalysts supported on graphene based hybrids for polymer electrolyte membrane fuel cells. Int. J. Hydrogen Energy 2018, 43, 23221–23230. [Google Scholar] [CrossRef]
- Şanli, L.I.; Bayram, V.; Yarar, B.; Ghobadi, S.; Gürsel, S.A. Development of graphene supported platinum nanoparticles for polymer electrolyte membrane fuel cells: Effect of support type and impregnation-reduction methods. Int. J. Hydrogen Energy 2016, 41, 3414–3427. [Google Scholar] [CrossRef]
- Arici, E.; Kaplan, B.Y.; Mert, A.M.; Gursel, S.A.; Kinayyigit, S. An effective electrocatalyst based on platinum nanoparticles supported with graphene nanoplatelets and carbon black hybrid for PEM fuel cells. Int. J. Hydrogen Energy 2019, 44, 14175–14183. [Google Scholar] [CrossRef]
- Antolini, E. Graphene as a new carbon support for low-temperature fuel cell catalysts. Appl. Catal. B Environ. 2012, 123–124, 52–68. [Google Scholar] [CrossRef]
- Joo, S.H.; Choi, S.J.; Oh, I.; Kwak, J.; Liu, Z.; Terasaki, O.; Ryoo, R. Ordered nanoporous arrays of carbon supporting high dispersions of platinum nanoparticles. Nature 2001, 412, 169–172. [Google Scholar] [CrossRef]
- Clancy, A.J.; Melbourne, J.; Shaffer, M.S.P. A one-step route to solubilised, purified or functionalised single-walled carbon nanotubes. J. Mater. Chem. A 2015, 3, 16708–16715. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; White, E.R.; Clancy, A.J.; Rubio, N.; Suter, T.; Miller, T.S.; McColl, K.; McMillan, P.F.; Brázdová, V.; Corà, F.; et al. Fast Exfoliation and Functionalisation of Two-Dimensional Crystalline Carbon Nitride by Framework Charging. Angew. Chem. Int. Ed. 2018, 57, 12656–12660. [Google Scholar] [CrossRef]
- Kepp, K.P. A Quantitative Scale of Oxophilicity and Thiophilicity. Inorg. Chem. 2016, 55, 9461–9470. [Google Scholar] [CrossRef]
- Clancy, A.J.; Bayazit, M.K.; Hodge, S.A.; Skipper, N.T.; Howard, C.A.; Shaffer, M.S.P. Charged Carbon Nanomaterials: Redox Chemistries of Fullerenes, Carbon Nanotubes, and Graphenes. Chem. Rev. 2018, 118, 7363–7408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, L.; Yang, F.; Rasouli, S.; Qiu, Y.; Li, Z.F.; Uzunoglu, A.; Sun, C.J.; Liu, Y.; Ferreira, P.; Li, W.; et al. Understanding Pt Nanoparticle Anchoring on Graphene Supports through Surface Functionalization. ACS Catal. 2016, 6, 2642–2653. [Google Scholar] [CrossRef]
- Ahmadi, R.; Amini, M.K. Synthesis and characterization of Pt nanoparticles on sulfur-modified carbon nanotubes for methanol oxidation. Int. J. Hydrogen Energy 2011, 36, 7275–7283. [Google Scholar] [CrossRef]
- Shahgaldi, S.; Alaefour, I.; Li, X. Impact of manufacturing processes on proton exchange membrane fuel cell performance. Appl. Energy 2018, 225, 1022–1032. [Google Scholar] [CrossRef]
- Yurtcan, A.B.; Daş, E. Chemically synthesized reduced graphene oxide-carbon black based hybrid catalysts for PEM fuel cells. Int. J. Hydrogen Energy 2018, 43, 18691–18701. [Google Scholar] [CrossRef]
- Sun, C.-N.; More, K.L.; Veith, G.M.; Zawodzinski, T.A. Composition Dependence of the Pore Structure and Water Transport of Composite Catalyst Layers for Polymer Electrolyte Fuel Cells. J. Electrochem. Soc. 2013, 160, F1000–F1005. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Weight% | C 1s | N 1s | O 1s | F 1s | S 2p | Pt 3d |
|---|---|---|---|---|---|---|
| No Urea | 24.9 | 0.5 | 6.6 | 60.8 | 2.6 | 4.5 |
| Urea | 25.2 | 2.1 | 6.4 | 48.4 | 1.4 | 16.6 |
| Urea and washed | 27.2 | 0.9 | 6.4 | 55.9 | 2.8 | 6.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suter, T.A.M.; Clancy, A.J.; Rubio Carrero, N.; Heitzmann, M.; Guetaz, L.; Shearing, P.R.; Mattevi, C.; Gebel, G.; Howard, C.A.; Shaffer, M.S.P.; et al. Scalable Sacrificial Templating to Increase Porosity and Platinum Utilisation in Graphene-Based Polymer Electrolyte Fuel Cell Electrodes. Nanomaterials 2021, 11, 2530. https://doi.org/10.3390/nano11102530
Suter TAM, Clancy AJ, Rubio Carrero N, Heitzmann M, Guetaz L, Shearing PR, Mattevi C, Gebel G, Howard CA, Shaffer MSP, et al. Scalable Sacrificial Templating to Increase Porosity and Platinum Utilisation in Graphene-Based Polymer Electrolyte Fuel Cell Electrodes. Nanomaterials. 2021; 11(10):2530. https://doi.org/10.3390/nano11102530
Chicago/Turabian StyleSuter, Theo A. M., Adam J. Clancy, Noelia Rubio Carrero, Marie Heitzmann, Laure Guetaz, Paul R. Shearing, Cecilia Mattevi, Gérard Gebel, Christopher A. Howard, Milo S. P. Shaffer, and et al. 2021. "Scalable Sacrificial Templating to Increase Porosity and Platinum Utilisation in Graphene-Based Polymer Electrolyte Fuel Cell Electrodes" Nanomaterials 11, no. 10: 2530. https://doi.org/10.3390/nano11102530
APA StyleSuter, T. A. M., Clancy, A. J., Rubio Carrero, N., Heitzmann, M., Guetaz, L., Shearing, P. R., Mattevi, C., Gebel, G., Howard, C. A., Shaffer, M. S. P., McMillan, P. F., & Brett, D. J. L. (2021). Scalable Sacrificial Templating to Increase Porosity and Platinum Utilisation in Graphene-Based Polymer Electrolyte Fuel Cell Electrodes. Nanomaterials, 11(10), 2530. https://doi.org/10.3390/nano11102530