Abstract
Generating clean and sustainable hydrogen from water splitting processes represent a practical alternative to solve the energy crisis. Ultrathin two-dimensional materials exhibit attractive properties as catalysts for hydrogen production owing to their large surface-to-volume ratios and effective chemisorption sites. However, the catalytically inactive surfaces of the transition metal dichalcogenides (TMD) possess merely small areas of active chemical sites on the edge, thus decreasing their possibilities for practical applications. Here, we propose a new class of out-of-plane deformed TMD (cTMD) monolayer to anchor transition metal atoms for the activation of the inert surface. The calculated adsorption energy of metals (e.g., Pt) on curved MoS2 (cMoS2) can be greatly decreased by 72% via adding external compressions, compared to the basal plane. The enlarged diffusion barrier energy indicates that cMoS2 with an enhanced fixation of metals could be a potential candidate as a single atom catalyst (SAC). We made a well-rounded assessment of the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER), which are two key processes in water splitting. The optimized Gibbs free energy of 0.02 for HER and low overpotential of 0.40 V for OER can be achieved when the proper compression and supported metals are selected. Our computational results provide inspiration and guidance towards the experimental design of TMD-based SACs.
1. Introduction
Hydrogen gas is considered the most plausible alternative to solve the energy crisis due to its green products, high heat of combustion, and sustainable properties [1,2,3]. Generally, electrolytic water is believed to be a clean and simple way to generate hydrogen compared with many other methods. Proper catalysts are usually used to speed this process. The selection of traditional catalysts focuses on noble metals (like bulk Pt), for their high stability, chemical activity and selectivity, and even inter-atomic cooperation [4,5,6]. But the low rate of exposed active sites, high cost, and limited storage create obstacles for the widespread application of traditional catalysts.
Mono- or few-layer two-dimensional (2D) materials, with plenty of exposed chemisorption sites, easy, low-cost fabrication, and high conductivity show great potential as potential candidates to replace the traditional catalysts for water splitting [7,8,9,10]. Transition metal dichalcogenides have aroused attention among 2D materials due to their wide range of bandgaps, strong chemical stability, and sandwich structure X–M–X since the first reported HER with MoS2 in 2005 [11]. At that time, only TMD edges were believed to act as effective sites for catalytic processes, because the bond-free surface was inert. But the exposed sites on edges are much less than that on the surface. Hence, kinds of methods are proposed to activate the surface, such as introducing strain and defects, as well as tuning phase and heterostructure construction [12,13,14,15]. These proposed methods can promote the catalytic performance of 2D materials to some extent, but low efficiency on a per atom basis still needs to be tackled [11,16].
The atomic efficiency and low unsaturated active sites quickly caught the attention of the catalysis community since the concept of single atom catalysts (SACs) was launched in 2011 [16]. Dispersing or anchoring metal atoms/clusters on TMD thin layers greatly improved the catalytic ability of TMD-based SACs [17,18]. However, the widely adopted doping and adsorption for anchoring single atoms on graphene cannot work well in a TMD system. The formation energy of doping in MoS2 is generally over 1.5 eV, and adsorption energy is much larger than that of doping on its basal plane, due to the weak adsorption of the S atom layer [14,19,20,21,22,23,24]. For example, Deng et al. systemically studied the possible pathways towards Pt anchoring on the surface of MoS2, that is, the doping and adsorption of Pt atoms to promote electrolytic activity [24]. They proved that the formation energy (Ef) of Pt doping is much lower than that of Pt adsorption on the MoS2 because it was hard to realize 3S-Pt structure in basal plane. That is why, until now, most efforts focus on the doping manner rather than adsorption (generally Ef > 2 eV in calculations) to promote the catalyst performance of TMD for water splitting. We also noticed that the successful realization of 3N- or 4N- Metal structure in graphene could promote the fixation of single atoms to facilitate HER, oxygen reduction reaction (ORR), and oxygen evolution reaction (OER) [25,26,27,28]. Therefore, it is necessary to figure out a suitable method to both promote the Ef and the fixation of single atoms on TMD thin layers.
Hence, we proposed a unique structure of MoS2 monolayers to support transition metals with the combination of strain engineering and transition metals’ decoration. We chose noble metal Pt and non-noble metal Fe for the assessment in our work. To evaluate the performance of metals on cMoS2, the adsorption energy Eads and diffusion barrier were calculated as a function of compressions. The great reduction (72% Max) of Eads at large curvatures indicates the more possible formation of M@cMoS2 than that on basal plane. The single atom would experience a strong fixation as the compression increases, due to the enlarged diffusion barrier up to three-fold of basal plane. We also find that Eads would be further decreased under charging the situation with 1e− or 2e−. The binding energy (Eb) of H-Fe@cMoS2 and H-Pt@cMoS2 is clearly affected by curvatures. The strongest binding was achieved a at curvature of 4% for Pt@cMoS2 and 16% for Fe@cMoS2. Correspondingly, the optimized Gibbs free energy values were −0.02 eV at 16% for Fe@cMoS2 and 0.03 eV at 4% for Pt@cMoS2. The HER performance could be further improved with charging one or two electrons. We also demonstrate that the large dissociation barrier of H2O in alkaline solution resulted in a slow reaction rate of HER of the M@cMoS2 in alkaline environment. At last, the OER performances of Pt@cMoS2 and Fe@cMoS2 were evaluated at different curvatures. The overpotential of 16%-Pt@cMoS2 could be comparable to basal one but has much lower thermal energy for O2 desorption. The promoted HER and OER of Pt or Fe anchored on MoS2 would be expand to other cTMDs and transition metals. The computational results pave the way towards the development of very efficient cTMD-based water splitting electrocatalysts.
2. Computational Methods
All the optimization and energy calculations are carried out with density-functional theory (DFT) to investigate the effect of surface curvature on the catalytic performance of cMoS2. The Perdew-Burke-Eznerhof generalized gradient approximation (PBE-GGA) [29] is used for the analysis of exchange and correlation potential. The projector augmented wave (PAW) method in the Vienna ab initio simulation package (VASP) is employed in this work [30,31]. The integration of the first Brillouin zone is carried out with the Gama scheme of k point sampling. The used supercell is about 33 Å × 10 Å × 20 Å (6 × 3 × 1 unit cells). A vacuum region of ≥15 Å is applied along the z axis to avoid the interaction between adjacent interlayers. All the calculation uses 5 × 10−6 eV/atom as the total energy convergence condition and 0.04 eV/Å as the maximum force convergence criteria. All the parameters are carefully tested before further calculations. Spin-polarization is considered in our calculation and the cutoff energy is set to 500 eV. For the evaluation of the kinetic analysis, we adopt the climbing image nudged elastic band (CINEB) method to figure out the diffusion energy barrier of metal atoms on cMoS2 and water dissociation of H2O molecular. The model construction of curved MoS2 monolayers is similar to our previous study [32].
The adsorption energy of metals on cMoS2 is calculated as:
where, the E (cMoS2 + M), E(cMoS2) and E(M) represent the total energy of monolayer curved MoS2 with one absorbed metal atom, the pure curved MoS2 and the metal, respectively. Since the calculation of E(M) is not unified in current research, we prefer to use the calculation method, that is, the energy of bulk metal divided by the number of metal atoms. The smaller the adsorption energy, the more stable the TMs would be on the curved structures.
△Eabs = E (cMoS2 + M) − E(cMoS2) − E(M)
The binding energy of bond H-M is calculated:
where, E (M@cMoS2 + H) stands for the energy of one H atom absorbed on the metal site. E(M@cMoS2) is defined as the energy of one metal atom anchored at the center site of the crest of cMoS2. E(H2) describes the total energy of the H2 molecule in its gas phase. The large binding energy indicates the strong adsorption capacity of the active site for hydrogen.
△Eb = E(M@cMoS2) + 1/2E(H2) − E (M@cMoS2 + H)
The process of hydrogen evolution reaction is considered in acidic solution [2,3,33]. The first step is that a proton from the solution absorbs on the slab and becomes an intermediate adsorbed hydrogen atom Had.
H+ + e− → Had
The adsorbed H atoms then combine into a hydrogen molecule.
2Had → H2
Therefore, it is a simple calculation of the Gibbs free energy in acidic solution through the computational hydrogen electrode (CHE) method [34],
where E(cMoS2 + H) and E(cMoS2) represent the total energies of cMoS2 with and without one adsorbed H, respectively. △Ezpe is the difference in zero-point energies between the adsorbed H and H in the gas phase of hydrogen molecules. △Ezpe − T△SH is calculated to be 0.24 eV [34].
△EH = E(M@cMoS2 + H) − E(M@cMoS2) − 1/2E(H2)
△GH = △EH + △Ezpe − T△SH
3. Results and Discussion
3.1. Anchoring-Activity of Metal on cMoS2
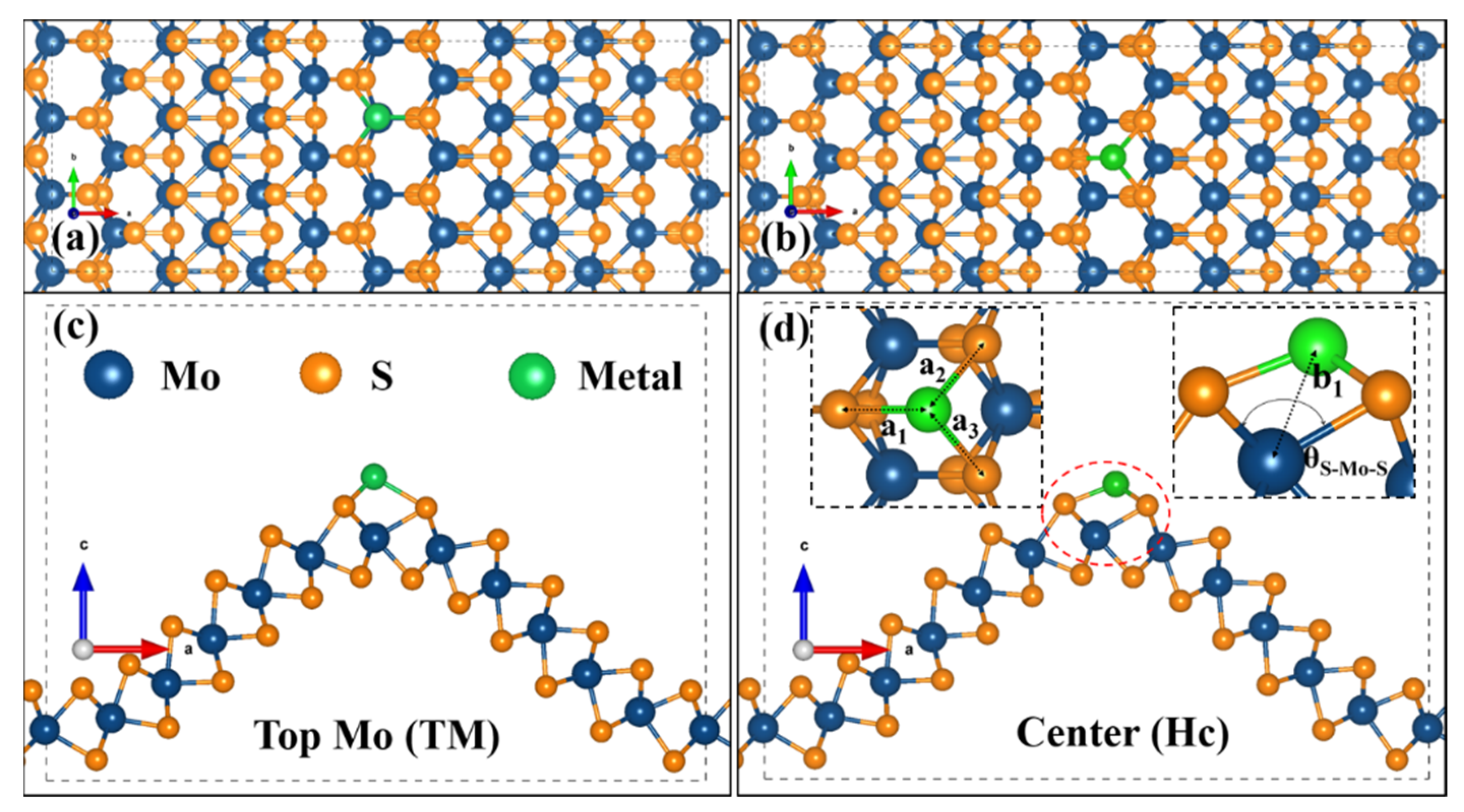
Our previous study indicated that the curved MoS could promote the surface activation due to the low adsorption energy of H atoms, but the optimized Gibbs free energy of 1.25 eV is still high for practical applications [32]. The active surface of cMoS2 at large curvatures and the theoretically 100% efficacy of SACs catalysts could be combined to fabricate highly active catalysts. Hence, we chose noble metal (Pt) and non-noble metal (Fe) atoms to evaluate the possible adsorption of single atoms in our unique curved structures. Figure 1 shows the schematic diagram of the possible adsorption sites of metals at the crest of cMoS2, top Mo site (TM) and the center site (Hc) of honeycomb. The increasing curvature leads to a dramatic structure change along the armchair direction (Tables S1 and S2). For S-Fe (S-Pt), the a1 is changed from 2.10 Å (2.35 Å) to 2.59 Å (3.28 Å), but the a2 and a3 show a limited increase, from 2.010 Å (2.35 Å) to 2.27 Å (2.38 Å). Then, the enlarged space allows metal atoms to close the inner site of Mo, which can be proved by the decreasing distance (b1) of Fe-Mo (Pt-Mo). The b1 changes from 2.94 Å (3.54 Å) to 2.4 Å (2.76 Å). We also noticed that even though the θS-Mo-S are still increasing, the b1 keeps almost static after δ = 8%, which could be attributed to the repel effect between metal atoms.
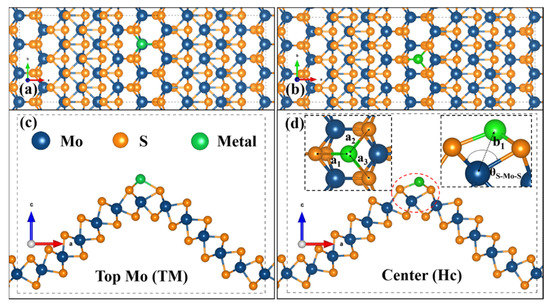
Figure 1.
Representative configuration of the metal atom adsorbed on the crest of cMoS2. (a,b) are the top view of the TM site and Hc site; (c,d) are the corresponding side views.
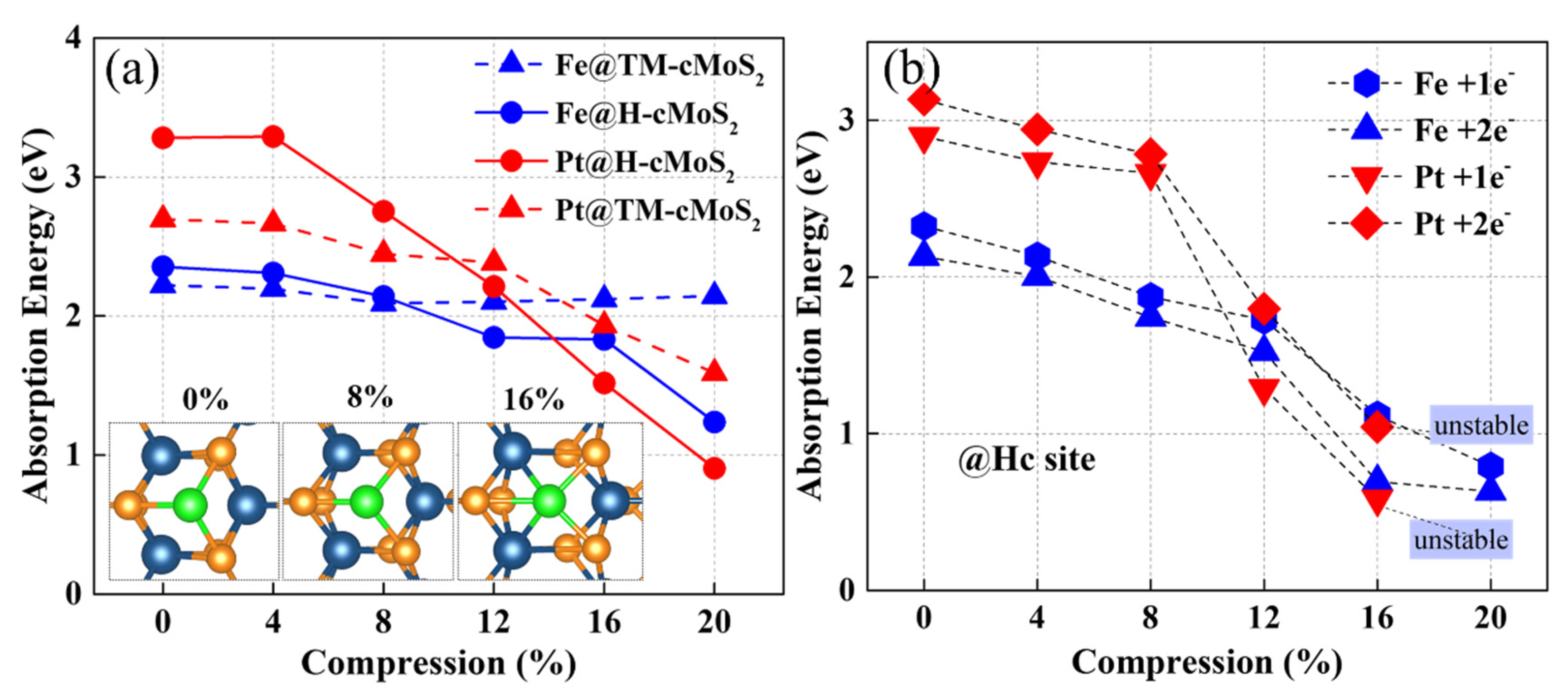
The adsorption energy of metal on TM and Hc are clearly affected with the change of curvature (Figure 2). The Eabs of Pt on the basal plane (0%) of MoS2 is around 3.2 eV at Hc site and 2.6 eV at TM site, which is in agreement with previous studies [24]. As the compression increases, Eabs gradually reduces for Fe and Pt, but with different degrees. The Pt and Fe atoms prefer the Hc sites at larger curvatures (δ ≥ 12%) rather than the TM site due to the low calculated Eabs at Hc sites. When the compression gets to 20%, the Eabs at the Hc site could be much lower to 0.9 eV (1.2 eV) with nearly 72% (49%) decrement for Pt (Fe). Remarkably, we found that Eabs at TM sites show a divergence between Pt and Fe when δ ≥ 8%, that is, Eabs of Fe, keeps almost unchanged while Pt shows a continuous decrease. Our calculation found that Top S (tS) is the most impossible to adsorb metal due to the much larger adsorption energy than that of the other two sites, which is consistent with previous reports [24]. Besides, the curvature has no positive effect on the Fe or Pt adsorption activity at tS on cMoS2 (Figure S1).
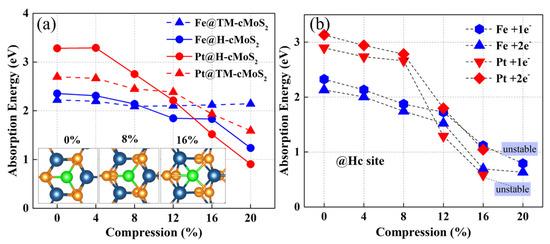
Figure 2.
(a) The adsorption energy of metal atoms on neutral cMoS2 as a function of compressions. The insets represent the top view of M@cMoS2 at different compressions. (b) The adsorption energy of metal atoms on the H site of charged cMoS2 as a function of compressions.
The systems charged with one electron and two electrons were also calculated for simulating the real situation in experiments. Figure 2b shows that the trend of Eb at the H site is similar to the neutral situation. The increasing number of electrons into the system results in a promotion of the metal adsorption of cMoS2, that is, the Eads with 1e− is lower than that with 2e−. The difference of Eads between 1e− and 2e− is enlarged at δ ≥ 12%, which would mean that the large curvature helps adsorbents attract more extra charge. The unstable structure of Pt-cMoS2 is observed when δ > 16%, which would result from the weak interaction of S-Pt induced by the enlarged a1 in a charged system (Tables S2–S4).
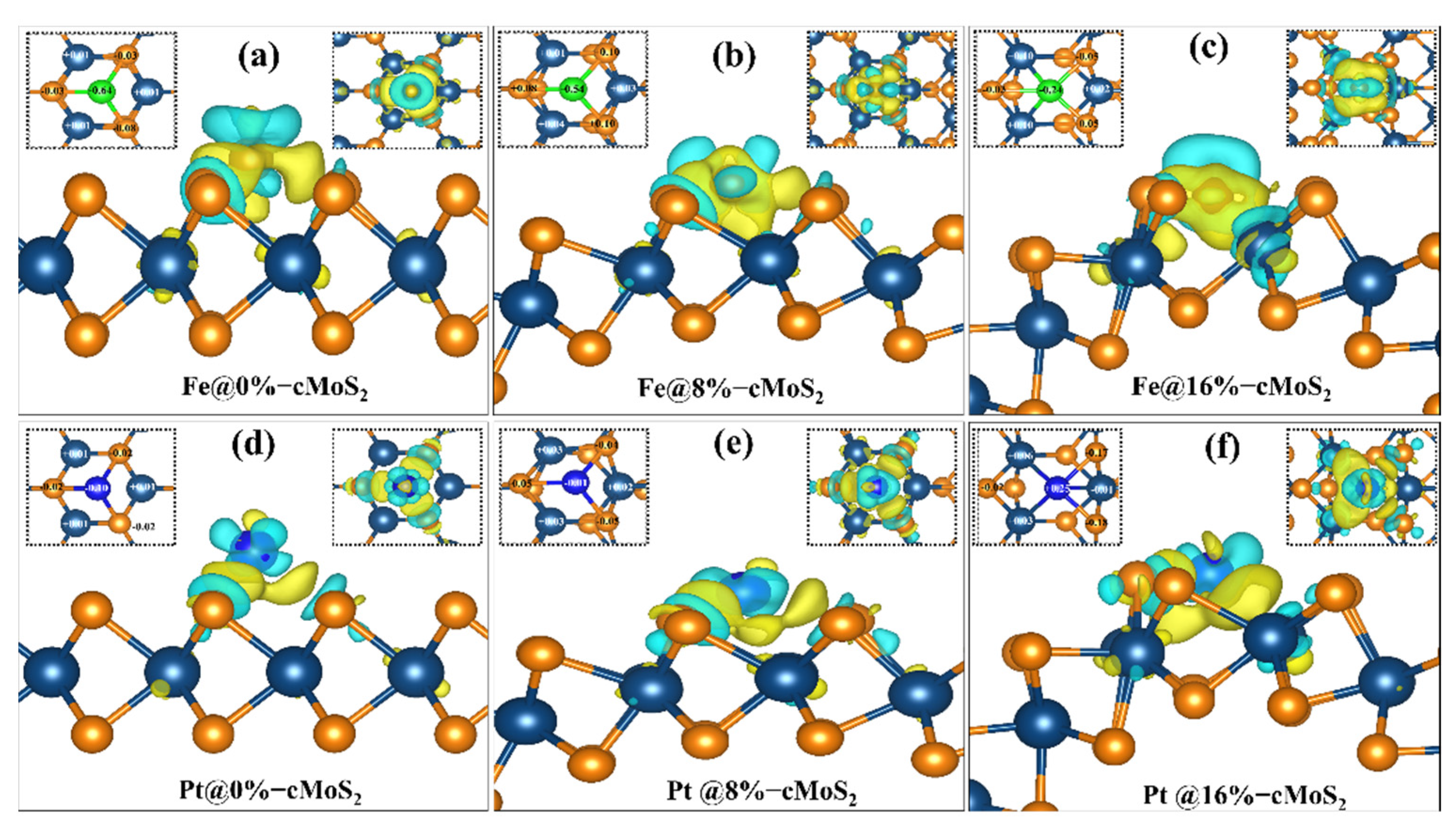
To dig out the relationship between the adsorption energy and the curvature of cMoS2, the charge density difference isosurface and Bader charge transfer are calculated (Figure 3). In the sandwiched structure of MoS2, generally, the middle layer of Mo atoms is screened by outer S atoms, which makes the adsorbate hard to interact with the Mo layer. Moreover, the saturated surface bonds of the S atom layer show a weak adsorption of most adsorbates, leading to an inert chemical activity [35]. For the basal plane (0%) MoS2, the Pt and Fe atoms show less effect on the charge density of Mo (Figure 3a,d). As the curvature increases, the distance of Pt/Fe-Mo decreases (b1 in Tables S1 and S2). Then, a strong interaction between Pt/Fe and Mo appears at a large compression, which is indicated by the large charge density transfer (Figure 3c,f). The calculated Bader transfer also proves the electronic redistribution. For example, the Bader charges of Fe and Pt are −0.64 eV and −0.1 eV at 0%-cMoS2, which increase to −0.24 eV and +0.25 eV at 16%-cMoS2 (Table S3). The higher charge transfer indicates a stronger interaction. In addition, the charged electrons accumulate around the absorbates to further increase the ability of the adsorption. For example, the increasing electrons accumulate around Pt after more electrons charged into the system (Figure S2). Therefore, the more electrons accumulate around adsorbates, the stronger adsorption cMoS2 has. Because of the strong ability to adsorb single atoms, monolayer MoS2 with its curved deformation has the potential to act as a single-atom catalyst, which could be further proved by the AIMD simulation at room temperature (Figure S3).
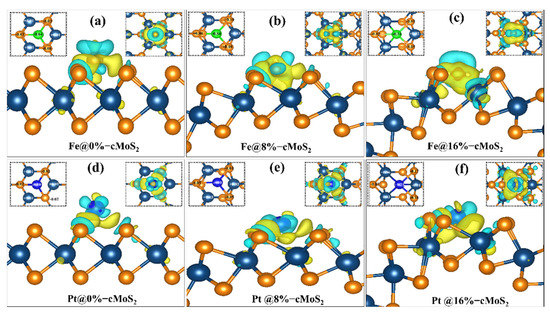
Figure 3.
(a–f) The charge density difference between cMoS2 and Fe@cMoS2 (the upper)/Pt@cMoS2 (the lower). The yellow and blue represent the charge accumulation and dissociation. The left and right insets indicate the Bader charge change around absorbents and the top view of charge density difference for each situation. The isosurface value is set to 0.004 e/Å3.
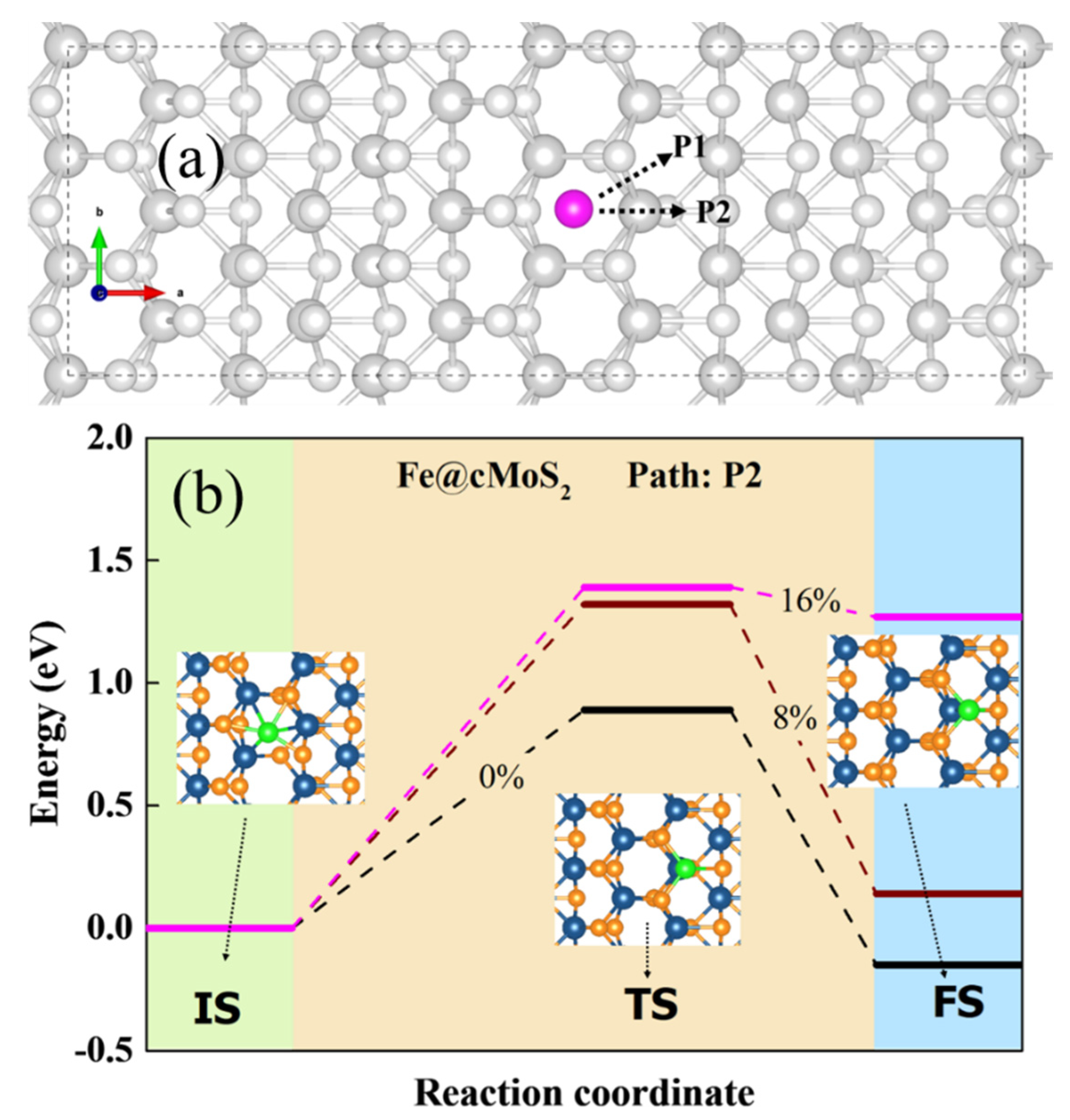
Another important issue of potential SACs focuses on their stability on the surface of the slab, meaning the single atom would not diffuse arbitrarily. Hence, we calculated the diffusion barrier of Pt and Fe on the crest of MoS2. Figure 4a shows the possible diffusion pathway for a single atom: P1 and P2. The results show that P2 is more possible than P1, because the Eb of top Mo along the P2 is lower than that of the bridge of S-Mo along P1. From Figure 4b, we can see the energy and optimized structures of three states when the Fe atom would diffuse on the surface of cMoS2. For a basal plane, the adsorption energy of Fe is slightly different (Figure 2a). The Fe atom needs to conquer an energy barrier of 0.89 eV to arrive to the TM site. The energy of finale state (FS) is a negative value, due to the lower Eads at TM than that at Tc. But for Pt atoms, the large difference of Eads between TM and Tc leads to a barrierless movement from Hc to TM on basal MoS2 (Figure S4).
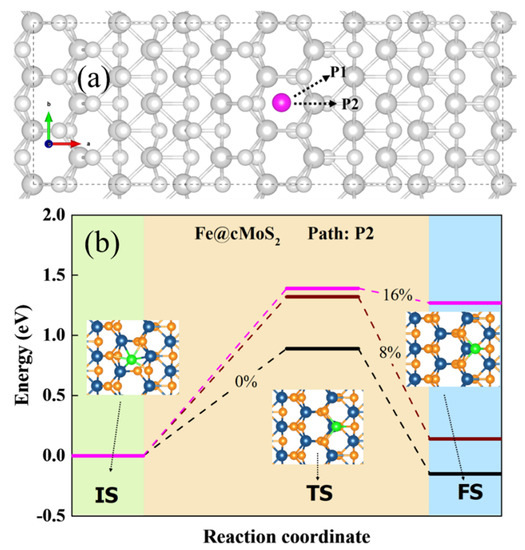
Figure 4.
(a) The possible pathway of M diffusion in cMoS2. (b) The diffusion coordinates of the Fe atom in cMoS2 at different compressions. The inset is the corresponding top view of different states.
As the curvature enlarges, Fe and Pt have difficulty going down the curved surface due to the increasing energy of the transition state (TS) (Figure 4b and Figure S4). In comparison with basal plane, the diffusion barrier experiences 1.6-fold (0.89 eV → 1.4 eV) and 3-fold (0.45 eV → 1.5 eV) increments for Fe and Pt, respectively. Additionally, atoms are difficult to anchor at other sites when δ ≥ 16%, since the energy of FS increases sharply from δ = 8% to δ = 16%, which would strengthen the fixation selectivity of metals adsorbed on cMoS2. Overall, the curvature-promoted single-atom-anchored on cMoS2 indicates a great potential as SACs.
3.2. Hydrogen Evolution Reaction
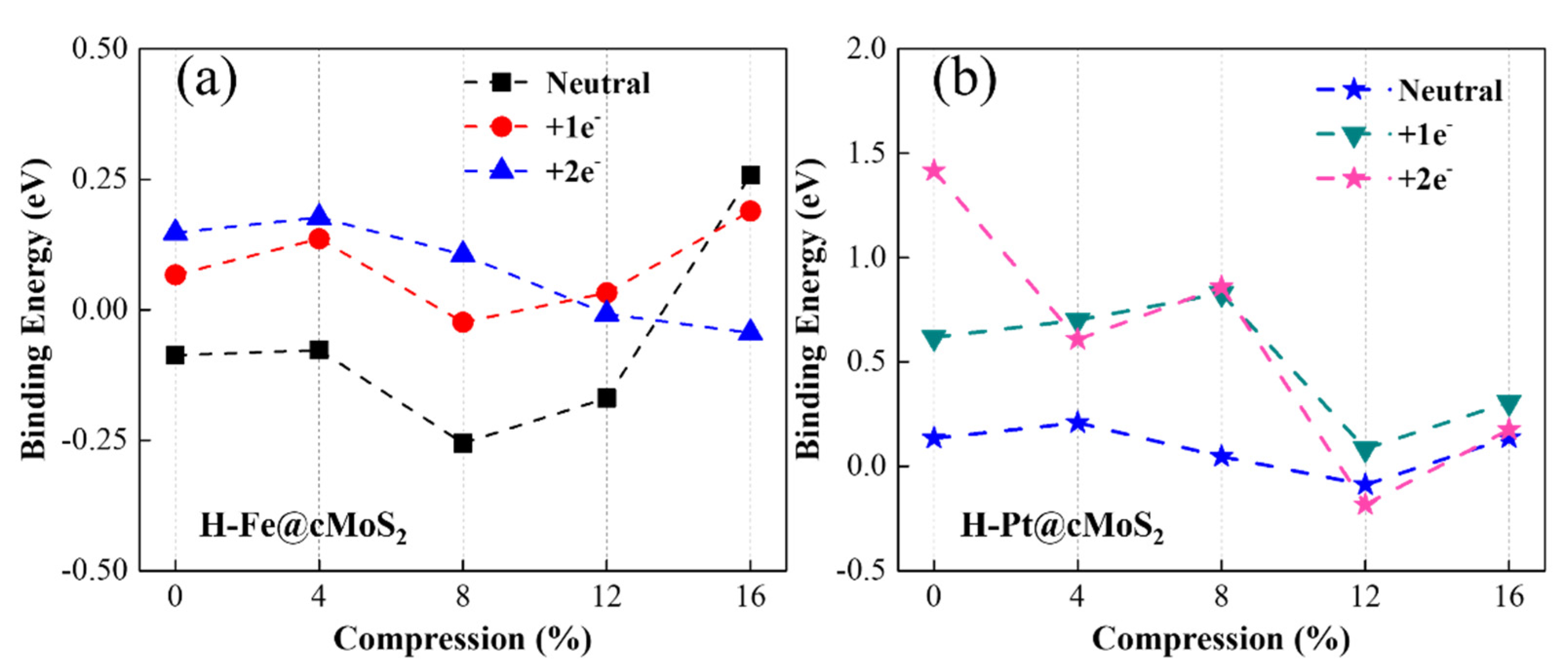
To evaluate the ability of water splitting based on metal-decorated cMoS2, we first investigated the hydrogen evolution reaction based on Fe@cMoS2 and Pt@cMoS2. The HER reaction mechanism depends on the pH of the solution. In acid solution, a sufficient H+ became indeterminates of Had after getting electrons from the electrode [2]. The key issue is that the surface of catalysts provides active sites to combine two adsorbed Had into H2 gas. Hence, we first calculate the binding energy (Eb) of an H-M bond to evaluate the adsorption of H on Pt@cMoS2 and Fe@cMoS2. From Figure 5a,b, the basal plane of Pt@MoS2 is more active for the H adsorption than that of Fe@MoS2, due to the high binding energy 0.18 eV than 0.1 eV of Fe. The positive value (Figure 5b) indicates it is an exothermic process in a Pt-based catalyst, which is in agreement with other reports [36]. The Eb is fluctuating within the range of 0.5 eV along with the increment of the compression. The Eb of H-Pt initially goes up to 0.21 eV by adding small stress, and then linearly decreases to −0.08 eV (δ = 12%), indicating a weak trend of H adsorption. A dominant improvement of binding appears at large curvatures. H-Fe shows a similar trend, but the turning point (8%) is earlier than Pt, which is attributed to the magnetic influence. All in all, we find that the strongest binging of H-Pt appears at a small compression of 4%, while that of H-Fe is larger (δ = 16%). It should be noted that the structure of M@cMoS2 becomes unstable after one H atom absorbs on it under a large compression (>16%).
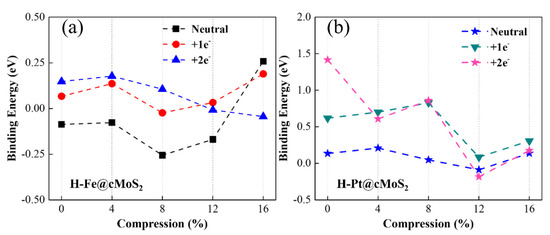
Figure 5.
Binding energy of Fe@cMoS2 (a) and Pt@cMoS2 (b) as a function of compressions at neutral, +1e− and 2e− situations.
We then considered the charging situation. With one extra electron, the trend of binding energy is similar to the neutral one as compression increases (Figure 5a,b), except for the H-Pt@cMoS2 with 2e− (Figure 5b). Because the extra electron leads to the shifting of the Pt adsorption position from Hc to TM and then induces a strange binding energy of H-Pt on 0%-cMoS2. Compared with a neutral situation, charging one electron brings a stronger binding between the H atom and the Fe/Pt atom at the small curvature (δ ≤ 8%), due to the much higher binding energy. For example, Eb of H-Fe is above zero with one extra electron, which means a spontaneous adsorption of the H atom. When the compression gets large, the binding energy is distinguished. The Eb with one electron is smaller than that in a neutral system for H-Fe after 16% and for H-Pt after 12%. This finding indicates the adsorption of H becomes weak, which would result in more 3d empty orbitals of metals being occupied by extra electrons, since the overlap of charge density between Mo and Fe or Pt gets larger at a high compression (Figure 3c,f). This conclusion can be further proved by the adsorption results with two electrons charging [37]. The curvature facilitates the injection of electrons to the adatoms (Fe, Pt) via the exposed Mo atom at a large compression, and then fewer empty orbitals lead to a weak adsorption.
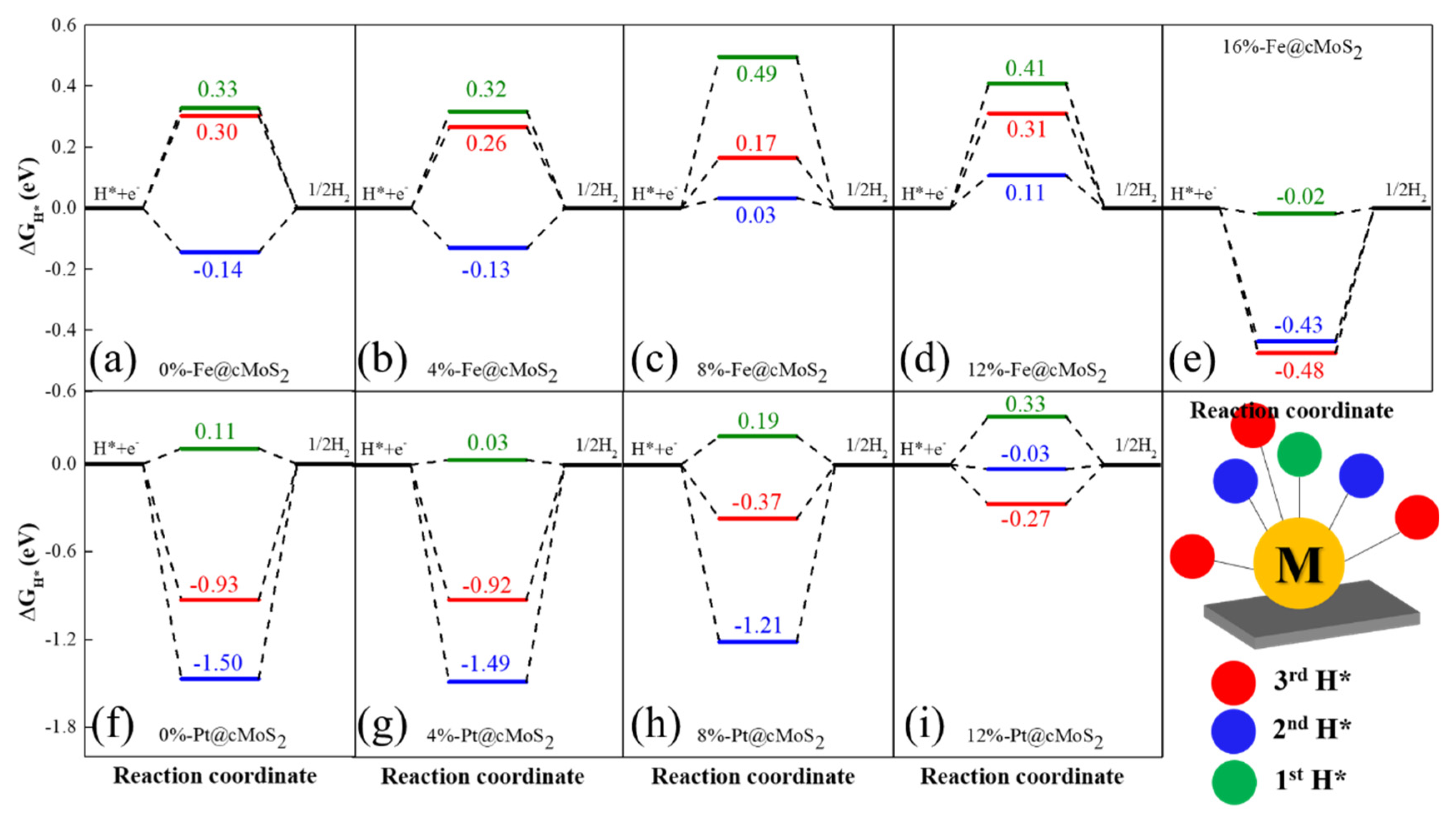
The Gibbs free energy (ΔGH) is widely used to judge the HER performance of a catalyst. Since HER involves two steps (H adsorption and H2 desorption), the optimized ΔGH is equal to zero. For the specific site of unsaturated single atoms supported on cMoS2, we calculate the Gibbs free energy for the first (green), second (blue), and third hydrogen atoms (red) at different curvatures (Figure 6). For Fe@cMoS2 from 0% to 12%, we find that the high reaction activity appears when the second H atom is adsorbed, and the low HER activity with one H, due to the low reaction barrier at each compression (the blue line closes to 0 eV). The optimized ΔGH of 0.03 eV is achieved at δ = 8%. But at large curvatures (16%), the high HER activity (ΔG = −0.02 eV) would be achieved when Fe@cMoS2 adsorbs one hydrogen, because the strong binding (ΔG = −0.43 eV) of H-Fe-H needs energy for H2 desorption with one more hydrogen adsorbed. Meanwhile, for Pt@cMoS2, the strong interaction between 2H/3H and Pt indicates the difficult desorption of H2 at small curvatures. Instead, the one electron is possible due to the lower reaction barrier. The optimized Gibbs free energy (0.03 eV) is located at 8%-Pt@cMoS2. But when compression gets to 12%, the Pt with two electrons owns high activity (ΔG = −0.03 eV). After 12%, Pt@cMoS2 shows an unstable structure with one more hydrogen absorbed. Therefore, the curved MoS2 with metals anchored have a great potential for HER under a proper compression force.
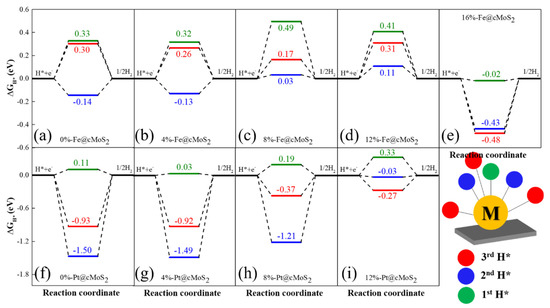
Figure 6.
(a–i) The free energy diagram of Fe@cMoS2 and Pt@cMoS2 for hydrogen evolution reactions at different compressions. The Gibbs free energy of an ideal catalyst for the HER should be close to 0.
Additionally, the catalytic performance of Pt@MoS2 and Fe@MoS2 in neutral or alkaline solutions are also investigated. Different from the acid electrolyte, the proton is the minority in alkaline one. The first step is the water dissociation, in which the H2O molecule would adsorb on an active surface and then dissociate into OH− and H+ [34]. The calculated dissociation barrier is larger than that of a traditional Pt/C electrode due to the weak binding between H and S atoms. All the details are discussed in the supporting information (Figures S5 and S6).
3.3. Oxygen Evolution Reaction
The oxygen evolution reaction (OER) is another key half reaction that involves four electrons transfer steps in the electrocatalytic water splitting, the same importance to HER. Therefore, a correct assessment of OER is beneficial for an in-depth understanding of the catalyst for water splitting. Up to now, the metal-based catalysts for OER mainly focus on the edge effect and edge-supported single atoms [38,39,40]. For instance, Xu et al. reported a Pt anchored at the edge of MoS2 demonstrated an ultra-low overpotential (η) of 0.46 V, which delivered a performance close to the bulk noble metal [38]. Similarly, the limited area of the edge restricted its massive application. The recent experimental results show that co-doping MoS2 exhibited a similar result (ηmin = 0.48 V) to the edge MoS2 but provided sufficient active reaction sites [41]. This provides a clear direction to the TMD-based SACs. However, the large doping energy (>2 eV, in calculation) makes doping difficult to some extent [42,43]. The above discussed adsorption promotion of metals in our system would alleviate the problem to a more possible degree. Here, we calculate the four electron transfer steps and make an assessment of OER in the M@cMoS2. The OER is a four-electron water oxidation process, and the probable reaction mechanism steps are as follows [2,3]:
* + H2O → HO* + (H+ + e−)
HO* → O* + (H+ + e−)
O* + H2O → HOO* + (H+ + e−)
HOO* → * + O2 + (H+ + e−)
The Gibbs free energy of each step is calculated as the HER reaction in the previous part.
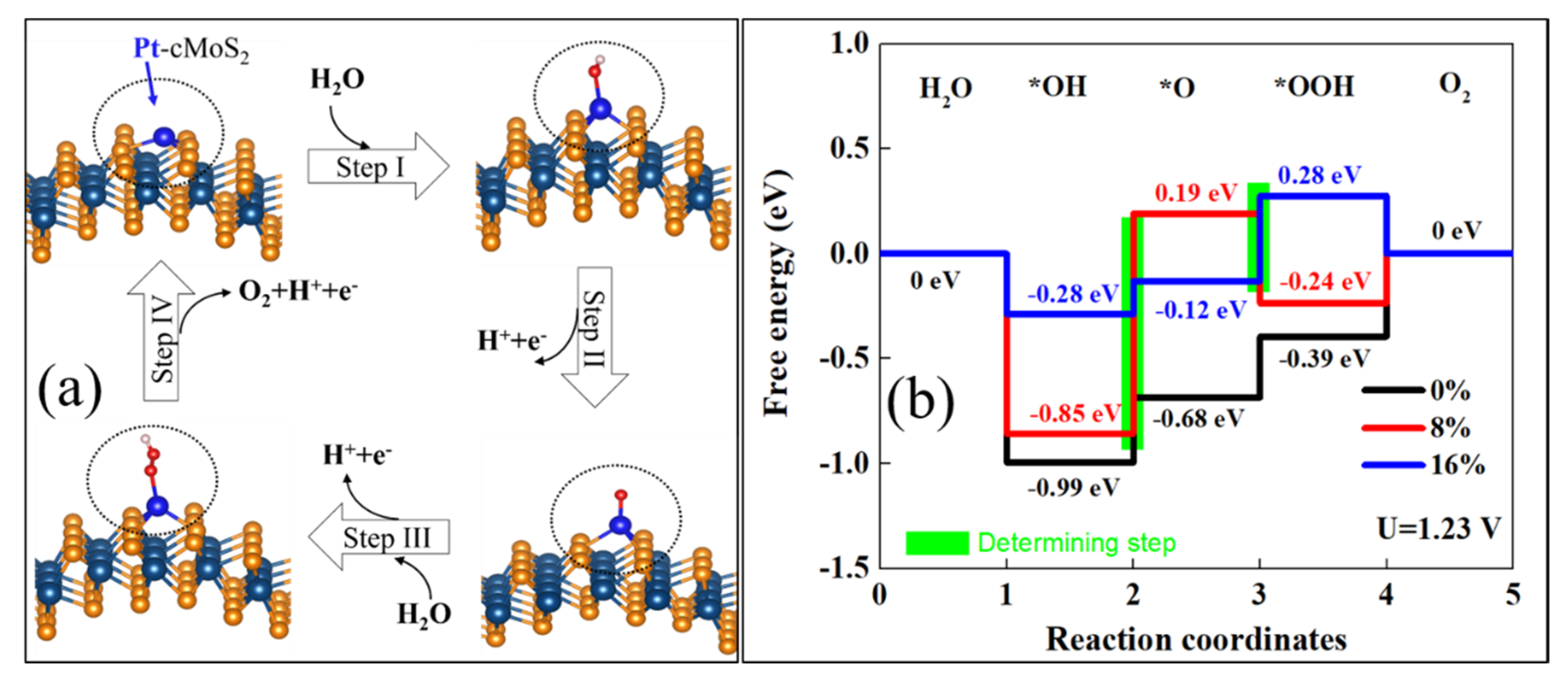
Figure 7a shows the optimized structure for each reaction step. The OER reaction is clearly tuned by the curvature at equilibrium hydrolysis potential (Figure 7b). We see that the enlarged curvature leads to a weak *OH adsorption of Pt@cMoS2. The potential determining step (PDS) is *OH oxidation (Step II) at a small curvature (≤ 8%), while PDS locates at the step *O → *OOH (Step III) at δ = 16%. The reaction rate would be 0%-Pt@cMoS2 > 16%-Pt@cMoS2 > 8%-Pt@cMoS2, which PDS are 0.31 eV, 0.40 eV and 1.04 eV. But the last step contains a thermal desorption of O2 from the slab, the production of O2 on 16%-Pt@cMoS2 would be more than that on 0%-Pt@cMoS2, due to the different trend between the downhill step and uphill step at Step IV for 16% and 0%, respectively. For Fe@cMoS2, the strong binding between *O and Fe leads to an easy oxidation of *OH (Step II, Figure S7). The determining steps are all located at Step III, and are 0.93 eV, 1.01 eV, and 1.13 eV for 0%, 8%, and 16%, respectively. The effect of curvatures on the OER performance of Fe@cMoS2 is similar to that of Pt@cMoS2. But the much larger PDS of Fe@cMoS2 obstacles its practical application. We also consider another pathway for *O → *OOH. For example, the *O and *OH adsorb on the metal at the same time, but the large formation energy and physical adsorption format make the pathway impossible. It means the metal decorated-cMoS2 has a good selectivity as an OER catalyst. Therefore, it is believed that M@cMoS2 would be used as a potential SAC candidate for OER if the proper anchored metals and curvatures are selected.
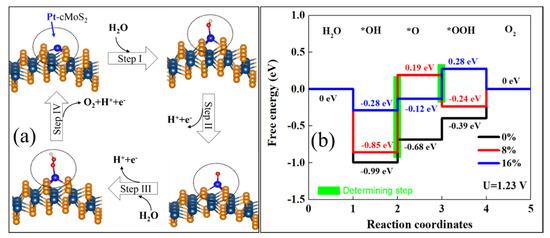
Figure 7.
(a) Proposed 4e-mechanism of oxygen evolution reaction on Pt@cMoS2. (b) Gibbs free-energy diagram for the four steps of OER on Pt@cMoS2 at different curvatures. The green box step is the rate determining.
4. Conclusions
In summary, we have proposed an effective structure with an aim to activate the inert surface of TMD monolayers, which are believed to be activated by an edge effect or prior doping. We took the noble metal Pt and nonnoble metal Fe as examples. Compared with the basal MoS2, the curved one demonstrated a promoted ability to hold metal atoms, especially at center sites of the honeycomb structure on the crest cMoS2 with large curvatures. The promotion can be up to 72% for Pt@cMoS2. Additionally, the CINEB calculation showed the diffusion barrier increased to the point that the metal atom was stable at the H site under large compressions. Moreover, we made a series of assessments of water splitting based on the single atom anchored at cMoS2. The excellent HER appeared at 4%-cMoS2 for Pt and at 16%-cMoS2 for Fe, and are comparable to the traditional bulk electrode Pt/C. However, the ability of water dissociation was still inferior in our system, which indicated a limited HER performance in alkane solution. At last, the calculated OER at 16%-Pt@cMoS2 showed a fast reaction, due to the low PDS of 0.40 eV and the downhill step of O2 desorption compared with the basal plane at the equilibrium potential. Our calculations provide insightful explanations for the design of TMD-based SACs, suggesting that the curved-TMD represents a feasible approach to fabricate efficient SA water splitting electrocatalysts.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/nano11123173/s1, Figure S1: Adsorption energy of Fe and Pt atoms at top S site on neutral cMoS2 as a function of compressions, Figure S2: Charge density difference of Pt@cMoS2 in neutral (a,d), 1e− (b,e) and 2e− (c,f) situations. The isosurface value is set to 0.004 e/Å3, Figure S3: AIMD simulation of Pt-12%cMoS2 as the function of simulation time. The timespan is over 4 ps, Figure S4: Diffusion coordinates of Pt atom in cMoS2 at different compressions, Figure S5: Optimized structures for the initial (IS, leftmost panels), transition (TS, center panels), and final (FS, rightmost panels) states of the most favorable path for the H2O → OH− +H+ reaction on Fe@cMoS2, Figure S6: Reaction coordinate of water dissociation of Fe@cMoS2 at δ = 16% and the comparison with Pt bulk, Figure S7: Gibbs free-energy diagram for the four steps of OER on Fe@cMoS2 at different curvatures. The green box step is the rate determining, Table S1: Structure parameters of cMoS2 absorbed Fe atom as compressions increases, Table S2: Structure parameters of cMoS2 absorbed Pt atom as compressions increases, Table S3: Bader charge of Fe@cMoS2 at different compressions, Table S4: Bader charge of Pt@cMoS2 at different compressions.
Author Contributions
Conceptualization, W.L. and Y.K.; methodology, Y.K.; software, P.L.; validation, B.W. and T.H.; formal analysis, Y.K.; investigation, W.L. and Y.K.; resources, X.L.; data curation, Y.K.; writing—original draft preparation, W.L.; writing—review and editing, A.R.P.S., T.H.; supervision, X.L.; funding acquisition, B.W. and X.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 61504118, 51902226, 52002288), the Natural Science Foundation of Jiangsu Province (No. BK20130423), and the National Natural Science Foundation of Guangdong Province (No.2019A1515012072), the research and development fund of Wuyi University joint Hong Kong-Macao (No. 2019WGALH04, No. 2019WGALH09).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Momirlan, M.; Veziroglu, T. Current status of hydrogen energy, Renew. Sustain. Energy Rev. 2002, 6, 141–179. [Google Scholar] [CrossRef]
- Murthya, A.; Madhavana, J.; Muruganb, K. Recent advances in hydrogen evolution reaction catalysts on carbon/carbon-based supports in acid media. J. Power Sources 2018, 398, 9–26. [Google Scholar] [CrossRef]
- Wu, Y.; Yao, J.; Gao, J. Interface Chemistry of Platinum-Based Materials for Electrocatalytic Hydrogen Evolution in Alkaline Conditions. In Methods for Electrocatalysis; Springer: Cham, Switzerland, 2020. [Google Scholar] [CrossRef]
- Zou, X.X.; Zhang, Y. Noble metal-free hydrogen evolution catalysts for water splitting. Chem. Soc. Rev. 2015, 44, 5148–5180. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, F.; Jin, H.; Chen, Y.; Wang, Y. Non-Noble Metal-based Carbon Composites in Hydrogen Evolution Reaction: Fundamentals to Applications. Adv. Mater. 2017, 29, 1605838. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Stambula, S.; Wang, D.; Banis, M.N.; Liu, J.; Riese, A.; Xiao, B.; Li, R.; Sham, T.-K.; Liu, L.-M.; et al. Platinum single-atom and cluster catalysis of the hydrogen evolution reaction. Nat. Commun. 2016, 7, 13638. [Google Scholar] [CrossRef]
- Voiry, D.; Yang, J.; Chhowalla, M. Recent Strategies for Improving the Catalytic Activity of 2D TMD Nanosheets Toward the Hydrogen Evolution Reaction. Adv. Mater. 2016, 28, 6197–6206. [Google Scholar] [CrossRef] [PubMed]
- Di, J.; Yan, C.; Handoko, A.D.; Seh, Z.W.; Li, H.; Liu, Z. Ultrathin two-dimensional materials for photo- and electrocatalytic hydrogen evolution. Mater. Today 2018, 21, 749–770. [Google Scholar] [CrossRef]
- Lu, Q.; Yu, Y.; Ma, Q.; Chen, B.; Zhang, H. 2D Transition-Metal-Dichalcogenide-Nanosheet-Based Composites for Photocatalytic and Electrocatalytic Hydrogen Evolution Reactions. Adv. Mater. 2016, 28, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Novoselov, K.S.; Mishchenko, A.; Carvalho, A.; Neto, A.H.C. 2D materials and van der Waals heterostructures. Science 2016, 353, aac9439. [Google Scholar] [CrossRef] [Green Version]
- Hinnemann, B.; Moses, P.G.; Bonde, J.; Jørgensen, K.P.; Nielsen, J.H.; Horch, S.; Chorkendorff, I.; Nørskov, J.K. Biomimetic Hydrogen Evolution: MoS2 Nanoparticles as Catalyst for Hydrogen Evolution. J. Am. Chem. Soc. 2005, 127, 5308–5309. [Google Scholar] [CrossRef] [PubMed]
- Deng, D.; Novoselov, K.; Fu, Q.; Zheng, N.; Tian, N.Z.Z.; Bao, X. Catalysis with two-dimensional materials and their heterostructures. Nat. Nanotechnol. 2016, 11, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Z.; Wu, D.; Zhang, X.; Zhao, X.; Zhou, Z. Computational Screening of 2D Materials and Rational Design of Heterojunctions for Water Splitting Photocatalysts. Small Methods 2018, 2, 1700359. [Google Scholar] [CrossRef]
- Shi, Y.; Zhou, Y.; Yang, D.-R.; Xu, W.-X.; Wang, C.; Wang, F.-B.; Xu, J.-J.; Xia, X.-H.; Chen, H.-Y. Energy Level Engineering of MoS2 by Transition-Metal Doping for Accelerating Hydrogen Evolution Reaction. J. Am. Chem. Soc. 2017, 139, 15479–15485. [Google Scholar] [CrossRef] [PubMed]
- Le, D.; Rawal, T.B.; Rahman, T.S. Single-Layer MoS2 with Sulfur Vacancies: Structure and Catalytic Application. J. Phys. Chem. C 2014, 118, 5346–5351. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Alarawi, A.A.; Ramalingam, V.; He, J.-H. Recent advances in emerging single atom confined two-dimensional materials for water splitting applications. Mater. Today Energy 2019, 11, 1–23. [Google Scholar] [CrossRef]
- He, T.; Zhang, C.; Du, A. Single-atom supported on graphene grain boundary as an efficient electrocatalyst for hydrogen evolution reaction. Chem. Eng. Sci. 2019, 194, 58–63. [Google Scholar] [CrossRef]
- Ma, D.; Li, T.; Zhang, X.; He, C.; Tang, Y.; Yang, Z. Modulating electronic, magnetic and chemical properties of MoS2 monolayer sheets by substitutional doping with transition metals. Appl. Surf. Sci. 2016, 364, 181–189. [Google Scholar] [CrossRef]
- Saab, M.; Raybaud, P. Tuning the Magnetic Properties of MoS2 Single Nanolayers by 3d Metals Edge Doping. J. Phys. Chem. C 2016, 120, 10691–10697. [Google Scholar] [CrossRef]
- Zhu, Y.; Peng, W.; Li, Y.; Zhang, G.; Zhang, F.; Fan, X. Modulating the Electronic Structure of Single-Atom Catalysts on 2D Nanomaterials for Enhanced Electrocatalytic Performance. Small Methods 2019, 3, 1800438. [Google Scholar] [CrossRef]
- Qi, K.; Yu, S.; Wang, Q.; Zhang, W.; Fan, J.; Zheng, W.; Cui, X. Decoration of the Inert Basal Plane of Defect-Rich MoS2 with Pd Atoms for Achieving Pt-Similar HER Activity. J. Mater. Chem. A 2016, 4, 4025–4031. [Google Scholar] [CrossRef]
- Lin, L.; Sherrell, P.; Liu, Y.; Lei, W.; Zhang, S.; Zhang, H.; Wallace, G.G.; Chen, J. Engineered 2D Transition Metal Dichalcogenides—A Vision of Viable Hydrogen Evolution Reaction Catalysis. Adv. Energy Mater. 2020, 10, 1903870. [Google Scholar] [CrossRef]
- Deng, J.; Li, H.; Xiao, J.; Tu, Y.; Deng, D.; Yang, H.; Tian, H.; Li, J.; Ren, P.; Bao, X. Triggering the electrocatalytic hydrogen evolution activity of the inert two-dimensional MoS2 surface via single-atom metal doping. Energy Environ. Sci. 2015, 8, 1594–1601. [Google Scholar] [CrossRef]
- Chung, H.T.; Cullen, D.A.; Higgins, D.; Sneed, B.T.; Holby, E.F.; More, K.L.; Zelenay, P. Direct atomic-level insight into the active sites of a high-performance PGM-free ORR catalyst. Science 2017, 357, 479–484. [Google Scholar] [CrossRef] [Green Version]
- Fei, H.; Dong, J.; Arellano-Jiménez, M.J.; Ye, G.; Kim, N.D.; Samuel, E.L.G.; Peng, Z.; Zhu, Z.; Qin, F.; Bao, J.; et al. Atomic cobalt on nitrogen-doped graphene for hydrogen generation. Nat. Commun. 2015, 6, 8668. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Matta, S.; Will, G.; Du, A. Transition-metal single atoms anchored on graphdiyne as high-efficiency electrocatalysts for water splitting and oxygen reduction. Small Methods 2019, 3, 1800419. [Google Scholar] [CrossRef]
- Cheng, N.; Zhang, L.; Doyle-Davis, K.; Sun, X. Single-Atom Catalysts: From Design to Application. Electrochem. Energy Rev. 2019, 2, 539–573. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector Augmented-wave Method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kong, Y.; Ai, H.; Wang, W.; Xie, X.; Lo, K.H.; Wang, S.; Pan, H. Waved 2D Transition-Metal Disulfides for Nanodevices and Catalysis: A First-Principle Study. ACS Appl. Nano Mater. 2020, 3, 2804–2812. [Google Scholar] [CrossRef]
- Laursen, A.B.; Varela, A.S.; Dionigi, F.; Fanchiu, H.; Miller, C.; Trinhammer, O.L.; Rossmeisl, J.; Dahl, S. Electrochemical Hydrogen Evolution: Sabatier’s Principle and the Volcano Plot. J. Chem. Educ. 2012, 89, 1595–1599. [Google Scholar] [CrossRef]
- Nørskov, J.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jonsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Joshi, Y.V.; Ghosh, P.; Venkataraman, P.S.; Delgass, W.N.; Thomson, K.T. Electronic Descriptors for the Adsorption Energies of Sulfur-Containing Molecules on Co/MoS2, Using DFT Calculations. J. Phys. Chem. C 2009, 113, 9698–9709. [Google Scholar] [CrossRef]
- Fajín, J.L.C.; Cordeiro, M.N.D.S.; Gomes, J.R.B. Density Functional Theory Study of the Water Dissociation on Platinum Surfaces: General Trends. J. Phys. Chem. A 2014, 118, 5832–5840. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Li, Z.; Searles, D.; Chen, Y.; Lu, G.; Du, A. Charge-Controlled Switchable CO2 Capture on Boron Nitride Nanomaterials. J. Am. Chem. Soc. 2013, 135, 8246–8253. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xu, H.; Cheng, D. Design of high-performance MoS2 edge supported single-metal atom bifunctional catalysts for overall water splitting via a simple equation. Nanoscale 2019, 11, 20228–20237. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Yu, Z.G.; Seng, H.L.; Zhang, N.; Liu, X.; Zhang, Y.-W.; Yang, W.; Gong, H. Simultaneous edge and electronic control of MoS2 nanosheets through Fe doping for an efficient oxygen evolution reaction. Nanoscale 2018, 10, 20113–20119. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, B.; Ghorbani-Asl, M.; Kretschmer, S.; Ghosh, A.; Guha, P.; Panda, S.K.; Jena, B.; Krasheninnikov, A.V.; Jena, B.K. MoS2 Quantum Dots as Efficient Catalyst Materials for the Oxygen Evolution Reaction. ACS Catal. 2018, 8, 1683–1689. [Google Scholar] [CrossRef]
- Xiong, Q.; Wang, Y.; Liu, P.F.; Zheng, L.R.; Wang, G.; Yang, H.G.; Wong, P.K.; Zhang, H.; Zhao, H. Cobalt Covalent Doping in MoS2 to Induce Bifunctionality of Overall Water Splitting. Adv. Mater. 2018, 30, e1801450. [Google Scholar] [CrossRef]
- Li, Q.; Zhao, X.; Deng, L.; Shi, Z.; Liu, S.; Wei, Q.; Zhang, L.; Cheng, Y.; Zhang, L.; Lu, H.; et al. Enhanced Valley Zeeman Splitting in Fe-Doped Monolayer MoS2. ACS Nano 2020, 14, 4636–4645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mombrú, D.; Faccio, R.; Mombrú, Á.W. Possible doping of single-layer MoS2 with Pt: A DFT study. Appl. Surf. Sci. 2018, 462, 409–416. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).