
Membrane-Interacting DNA Nanotubes Induce Cancer Cell Death




Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Nanotube Design
2.2. Cytochrome c Conjugation
2.3. DNA Nanotube Assembly and Gel Analysis
2.4. Cell Culture
2.5. Flow Cytometry and Confocal Microscopy
2.6. Apoptosis Assay and Western Blotting
2.7. MTT Cell Viability Assay
2.8. Dye Efflux Analysis by Flow Cytometry
3. Results
3.1. Design and Self-Assembly of DNA Nanotubes
3.2. Cytochrome c Conjugation and Integration into the DNA Nanotubes
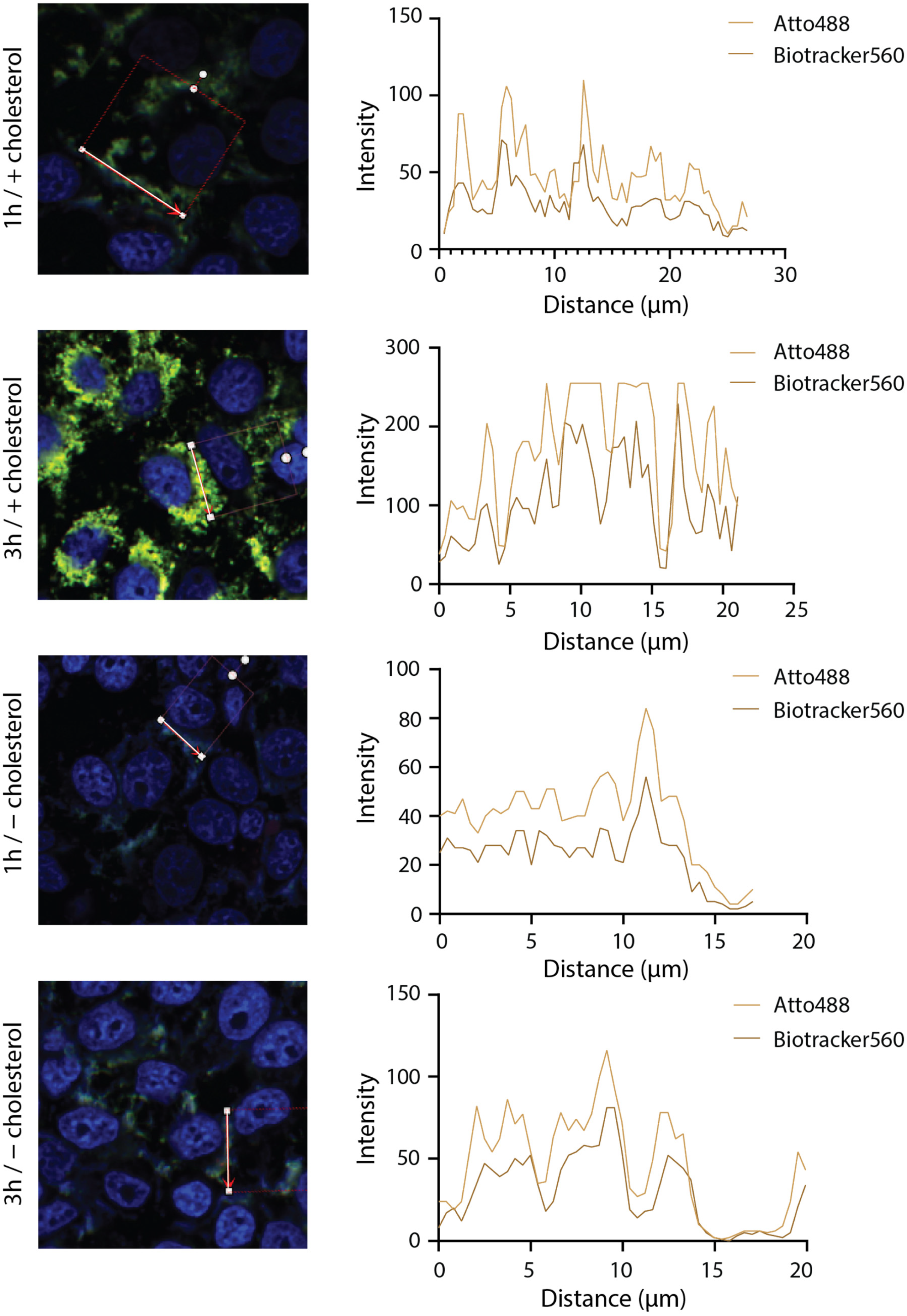
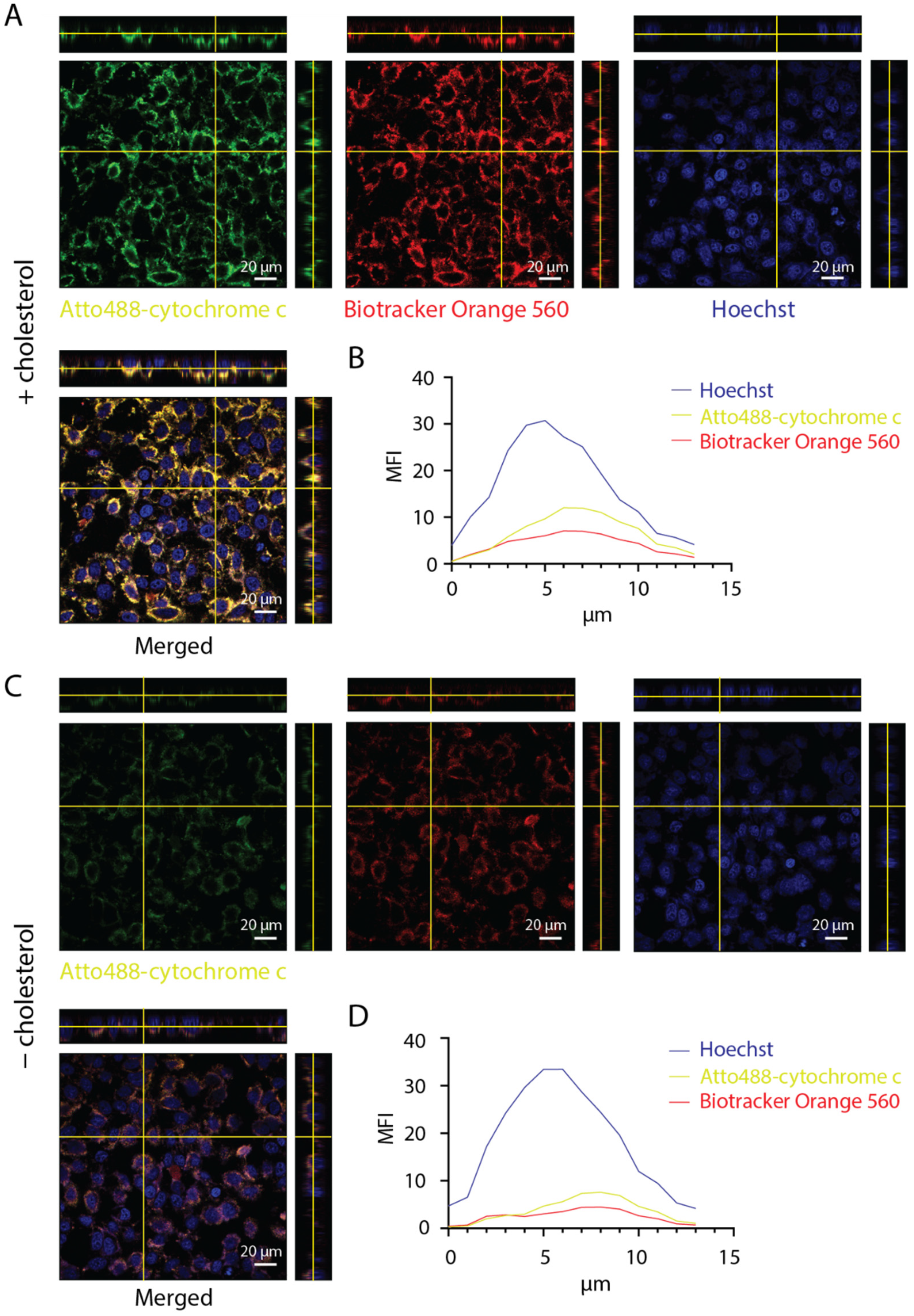
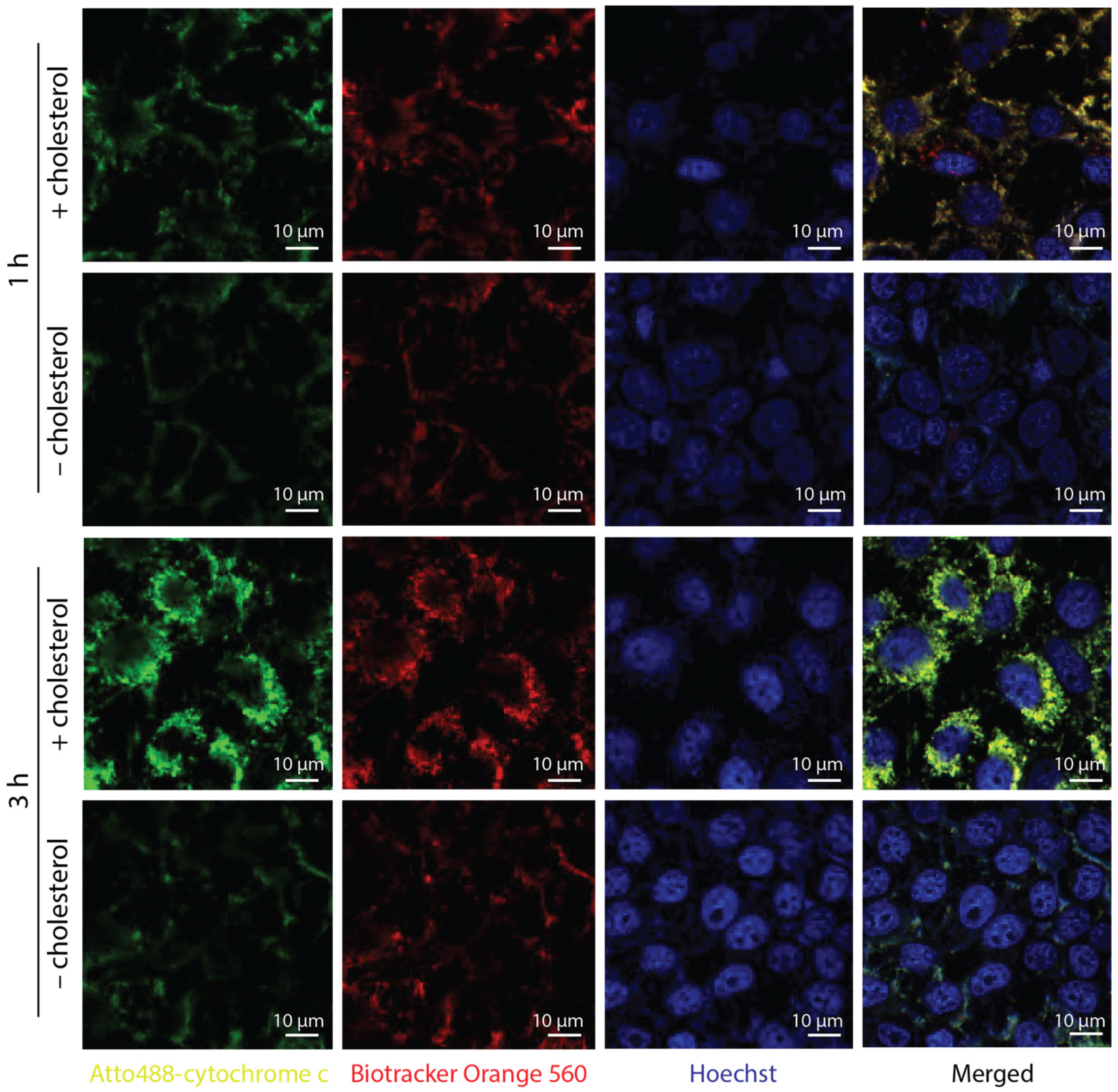
3.3. Interaction of DNA Nanotubes with Cancer Cell Membranes
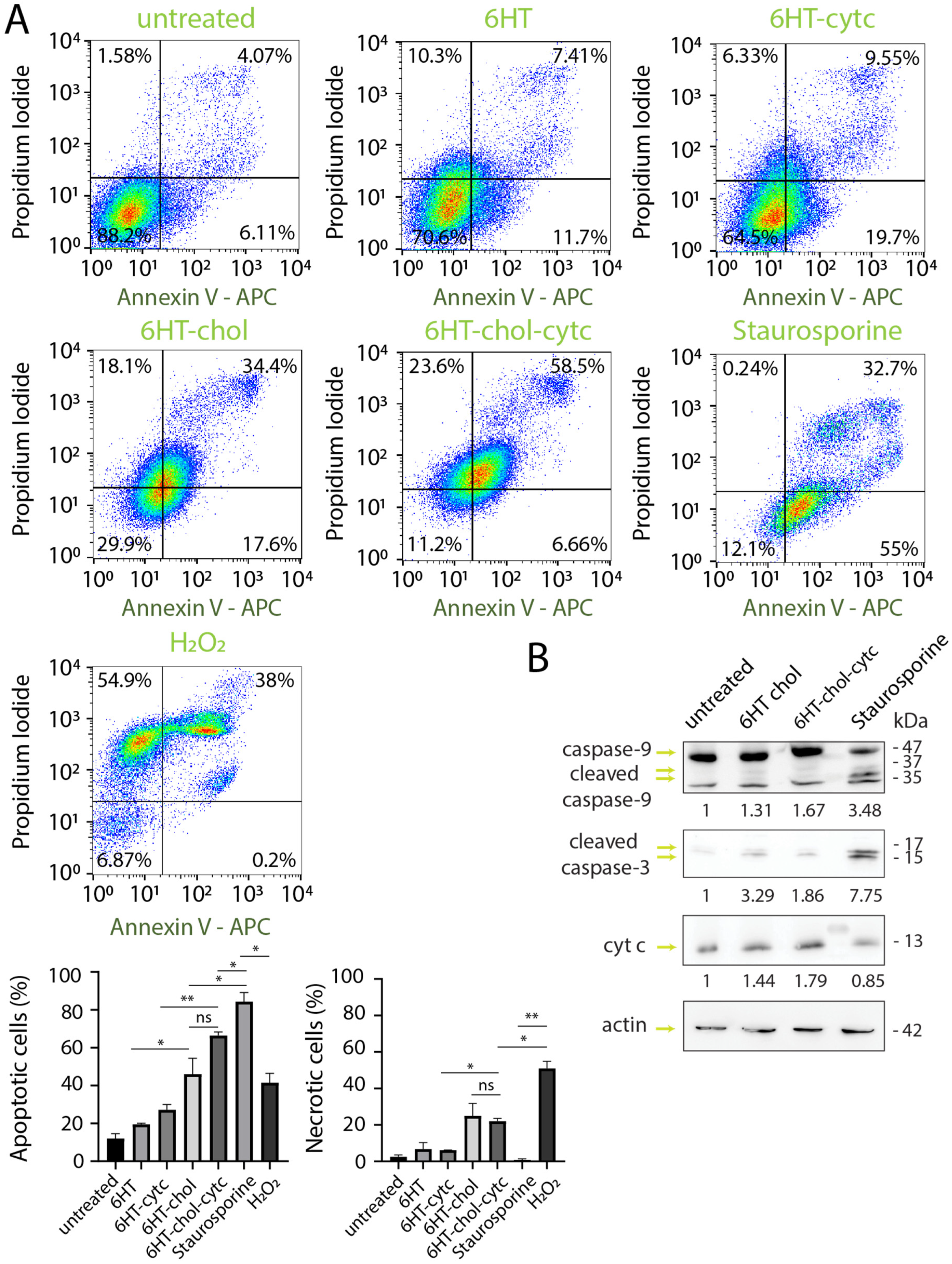
3.4. Induction of Cell Death by DNA Nanotubes
4. Discussion and Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Supporting Figures & Tables

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligonucleotide | Sequence | Modification |
|---|---|---|
| U1R1 | AAAAACTTACTGAGGATATTGCCTGAAGCTGTACCGTTTTAGGGGAAA | 5′ azide |
| U1R2 | ACGTACCTGAACTCCAGACTCGGGGCGAAAAAAAAGTCTTAGTACCGC | 3′ chol |
| U2R1 | CCCCTAAAACGGTACAGCTTCGGTACGTGCGGTACTAAGACTGGGGCGAATAGACAGGCTCCCCTCTCACTCGCTAGGAGGCAA | |
| U3R1 | AAATTGCCTCCTAGCGAGTGAGAGGTTTCCCGCATATTAACGCCTAAA | |
| U3R2 | CAGCGTCGGGAGCCTGTCTATTCGCCCCAAAAAATACCGTGGTTGAGT | 3′ chol |
| U4R1 | AGGCGTTAATATGCGGGAAACGACGCTGACTCAACCACGGTACGTTAGATGCCTCGCTGTACTAATAGTTGTCGACAGATCGTC | |
| U5R1 | AAAGACGATCTGTCGACAACTATTGGTCGGATCTGAGTCGACCAAAAA | 5′ Atto647 |
| U5R2 | ATGGAGCAGTACAGCGAGGCATCTAACGAAAAAAGCTCTTTGAGTATC | 3′ chol |
| U6R1 | TTGGTCGACTCAGATCCGACCGCTCCATGATACTCAAAGAGCTCGCCCCGAGTCTGGAGTTCAAGGCAATATCCTCAGTAAGTT |
| Antibody | Source | Dilution |
|---|---|---|
| Anti-caspase 9 | Cell Signaling, Danvers, MA, USA (#9502)—rabbit | 1:1000 |
| Anti-cleaved caspase 3 (Asp175) | Cell Signaling, Danvers, MA, USA (#9661)—rabbit | 1:1000 |
| Anti-cytochrome c | Biolegend, San Diego, CA, USA (#612503)—mouse | 1:500 |
| β-actin | Sigma Aldrich, Buchs, Switzerland (#A3853)—mouse | 1:10,000 |
| Goat-anti mouse Ig/HRP | Dako, Glostrup, Denmark (#P0447) | 1:10,000 |
| Goat-anti rabbit Ig/HRP | Dako, Glostrup, Denmark (#P0448) | 1:10,000 |







References
- Rothemund, P.W.K. Folding DNA to create nanoscale shapes and patterns. Nat. Cell Biol. 2006, 440, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Kocabey, S.; Kempter, S.; List, J.; Xing, Y.; Bae, W.; Schiffels, D.; Shih, W.M.; Simmel, F.C.; Liedl, T. Membrane-Assisted Growth of DNA Origami Nanostructure Arrays. ACS Nano 2015, 9, 3530–3539. [Google Scholar] [CrossRef]
- Journot, C.M.A.; Ramakrishna, V.; Wallace, M.I.; Turberfield, A.J. Modifying Membrane Morphology and Interactions with DNA Origami Clathrin-Mimic Networks. ACS Nano 2019, 13, 9973–9979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howorka, S. Building membrane nanopores. Nat. Nanotechnol. 2017, 12, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.; Lundin, V.; Petrova, E.; Fördős, F.; Benson, E.; Al-Amin, R.A.; Herland, A.; Blokzijl, A.; Högberg, B.; Teixeira, A. Spatial control of membrane receptor function using ligand nanocalipers. Nat. Methods 2014, 11, 841–846. [Google Scholar] [CrossRef]
- Huang, D.; Patel, K.; Perez-Garrido, S.; Marshall, J.F.; Palma, M. DNA Origami Nanoarrays for Multivalent Investigations of Cancer Cell Spreading with Nanoscale Spatial Resolution and Single-Molecule Control. ACS Nano 2019, 13, 728–736. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Baars, I.; Fördös, F.; Högberg, B. Clustering of Death Receptor for Apoptosis Using Nanoscale Patterns of Peptides. ACS Nano 2021, 15, 9614–9626. [Google Scholar] [CrossRef]
- Berger, R.M.L.; Weck, J.M.; Kempe, S.M.; Hill, O.; Liedl, T.; Rädler, J.O.; Monzel, C.; Heuer-Jungemann, A. Nanoscale FasL Organization on DNA Origami to Decipher Apoptosis Signal Activation in Cells. Small 2021, 17, 2101678. [Google Scholar] [CrossRef] [PubMed]
- Hellmeier, J.; Platzer, R.; Eklund, A.S.; Schlichthaerle, T.; Karner, A.; Motsch, V.; Schneider, M.C.; Kurz, E.; Bamieh, V.; Brameshuber, M.; et al. DNA origami demonstrate the unique stimulatory power of single pMHCs as T cell antigens. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Liu, J.; Guo, L.; Yao, G.; Li, Q.; Wang, L.; Li, J.; Fan, C. Programming Cell-Cell Communications with Engineered Cell Origami Clusters. J. Am. Chem. Soc. 2020, 142, 8800–8808. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.M.; Bachelet, I.; Church, G.M. A logic-gated nanorobot for targeted transport of molecular payloads. Science 2012, 335, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Ora, A.; Jarvihaavisto, E.; Zhang, H.; Auvinen, H.; Santos, H.A.; Kostiainen, M.A.; Linko, V. Cellular delivery of enzyme-loaded DNA origami. Chem. Commun. 2016, 52, 14161–14164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.-X.; Shaw, A.; Zeng, X.; Benson, E.; Nyström, A.M.; Högberg, B. DNA Origami Delivery System for Cancer Therapy with Tunable Release Properties. ACS Nano 2012, 6, 8684–8691. [Google Scholar] [CrossRef] [Green Version]
- Ijäs, H.; Shen, B.; Heuer-Jungemann, A.; Keller, A.; Kostiainen, M.A.; Liedl, T.; Ihalainen, J.A.; Linko, V. Unraveling the interaction between doxorubicin and DNA origami nanostructures for customizable chemotherapeutic drug release. Nucleic Acids Res. 2021, 49, 3048–3062. [Google Scholar] [CrossRef]
- Schaffert, D.H.; Okholm, A.H.; Sorensen, R.S.; Nielsen, J.S.; Torring, T.; Rosen, C.B.; Kodal, A.L.; Mortensen, M.R.; Gothelf, K.V.; Kjems, J. Intracellular Delivery of a Planar DNA Origami Structure by the Transferrin-Receptor Internalization Pathway. Small 2016, 12, 2634–2640. [Google Scholar] [CrossRef]
- Kocabey, S.; Meinl, H.; MacPherson, I.S.; Cassinelli, V.; Manetto, A.; Rothenfusser, S.; Liedl, T.; Lichtenegger, F.S. Cellular Uptake of Tile-Assembled DNA Nanotubes. Nanomaterials 2015, 5, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Raniolo, S.; Vindigni, G.; Ottaviani, A.; Unida, V.; Iacovelli, F.; Manetto, A.; Figini, M.; Stella, L.; Desideri, A.; Biocca, S. Selective targeting and degradation of doxorubicin-loaded folate-functionalized DNA nanocages. Nanomedicine 2018, 14, 1181–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Song, P.; Marcher, A.; Kjems, J.; Yang, C.; Gothelf, K.V. Selective Delivery of Doxorubicin to EGFR(+) Cancer Cells by Cetuximab-DNA Conjugates. ChemBioChem 2019, 20, 1014–1018. [Google Scholar] [CrossRef]
- Li, S.; Jiang, Q.; Liu, S.; Zhang, Y.; Tian, Y.; Song, C.; Wang, J.; Zou, Y.; Anderson, G.; Han, J.-Y.; et al. A DNA nanorobot functions as a cancer therapeutic in response to a molecular trigger in vivo. Nat. Biotechnol. 2018, 36, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Zhan, Y.; Zhang, Y.; Shao, X.; Xie, X.; Mao, C.; Cui, W.; Li, Q.; Shi, J.; Li, J.; et al. An Intelligent DNA Nanorobot with in Vitro Enhanced Protein Lysosomal Degradation of HER2. Nano Lett. 2019, 19, 4505–4517. [Google Scholar] [CrossRef] [Green Version]
- Kiviaho, J.K.; Linko, V.; Ora, A.; Tiainen, T.; Jarvihaavisto, E.; Mikkila, J.; Tenhu, H.; Nonappa; Kostiainen, M.A. Cationic polymers for DNA origami coating-examining their binding efficiency and tuning the enzymatic reaction rates. Nanoscale 2016, 8, 11674–11680. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.T.; Gray, M.A.; Xuan, S.; Lin, Y.; Byrnes, J.; Nguyen, A.I.; Todorova, N.; Stevens, M.M.; Bertozzi, C.R.; Zuckermann, R.N.; et al. DNA origami protection and molecular interfacing through engineered sequence-defined peptoids. Proc. Natl. Acad. Sci. USA 2020, 117, 6339–6348. [Google Scholar] [CrossRef] [Green Version]
- Ponnuswamy, N.; Bastings, M.M.C.; Nathwani, B.; Ryu, J.H.; Chou, L.Y.T.; Vinther, M.; Li, W.A.; Anastassacos, F.M.; Mooney, D.J.; Shih, W.M. Oligolysine-based coating protects DNA nanostructures from low-salt denaturation and nuclease degradation. Nat. Commun. 2017, 8, 15654. [Google Scholar] [CrossRef]
- Perrault, S.D.; Shih, W.M. Virus-Inspired Membrane Encapsulation of DNA Nanostructures To Achieve In Vivo Stability. ACS Nano 2014, 8, 5132–5140. [Google Scholar] [CrossRef]
- Langecker, M.; Arnaut, V.; Martin, T.G.; List, J.; Renner, S.; Mayer, M.; Dietz, H.; Simmel, F.C. Synthetic lipid membrane channels formed by designed DNA nanostructures. Science 2012, 338, 932–936. [Google Scholar] [CrossRef] [Green Version]
- Gopfrich, K.; Zettl, T.; Meijering, A.E.; Hernandez-Ainsa, S.; Kocabey, S.; Liedl, T.; Keyser, U.F. DNA-Tile Structures Induce Ionic Currents through Lipid Membranes. Nano Lett. 2015, 15, 3134–3138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, J.R.; Seifert, A.; Fertig, N.; Howorka, S. A biomimetic DNA-based channel for the ligand-controlled transport of charged molecular cargo across a biological membrane. Nat. Nanotechnol. 2016, 11, 152–156. [Google Scholar] [CrossRef]
- Lanphere, C.; Arnott, P.M.; Jones, S.F.; Korlova, K.; Howorka, S. A Biomimetic DNA-Based Membrane Gate for Protein-Controlled Transport of Cytotoxic Drugs. Angew. Chem. Int. Ed. Engl. 2021, 60, 1903–1908. [Google Scholar] [CrossRef] [PubMed]
- Howorka, S.; Siwy, Z. Nanopore analytics: Sensing of single molecules. Chem. Soc. Rev. 2009, 38, 2360–2384. [Google Scholar] [CrossRef] [PubMed]
- Franquelim, H.G.; Khmelinskaia, A.; Sobczak, J.-P.; Dietz, H.; Schwille, P. Membrane sculpting by curved DNA origami scaffolds. Nat. Commun. 2018, 9, 811. [Google Scholar] [CrossRef]
- Xu, W.; Nathwani, B.; Lin, C.; Wang, J.; Karatekin, E.; Pincet, F.; Shih, W.; Rothman, J.E. A Programmable DNA Origami Platform to Organize SNAREs for Membrane Fusion. J. Am. Chem. Soc. 2016, 138, 4439–4447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehouse, W.L.; Noble, J.E.; Ryadnov, M.G.; Howorka, S. Cholesterol Anchors Enable Efficient Binding and Intracellular Uptake of DNA Nanostructures. Bioconjug. Chem. 2019, 30, 1836–1844. [Google Scholar] [CrossRef]
- Arulkumaran, N.; Lanphere, C.; Gaupp, C.; Burns, J.R.; Singer, M.; Howorka, S. DNA Nanodevices with Selective Immune Cell Interaction and Function. ACS Nano 2021, 15, 4394–4404. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wijesekara, P.; Kumar, S.; Wang, W.; Ren, X.; Taylor, R.E. The effects of overhang placement and multivalency on cell labeling by DNA origami. Nanoscale 2021, 13, 6819–6828. [Google Scholar] [CrossRef]
- Lv, C.; Gu, X.; Li, H.-W.; Zhao, Y.; Yang, D.; Yu, W.; Han, D.; Li, J.; Tan, W. Molecular Transport through a Biomimetic DNA Channel on Live Cell Membranes. ACS Nano 2020, 14, 14616–14626. [Google Scholar] [CrossRef]
- Jürgensmeier, J.M.; Xie, Z.; Deveraux, Q.; Ellerby, L.; Bredesen, D.; Reed, J.C. Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. USA 1998, 95, 4997–5002. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Henzel, W.; Liu, X.; Lutschg, A.; Wang, X. Apaf-1, a Human Protein Homologous to C. elegans CED-4, Participates in Cytochrome c–Dependent Activation of Caspase-3. Cell 1997, 90, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of Apoptotic Program in Cell-Free Extracts: Requirement for dATP and Cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Antonsson, B.; Conti, F.; Ciavatta, A.; Montessuit, S.; Lewis, S.; Martinou, I.; Bernasconi, L.; Bernard, A.; Mermod, J.-J.; Mazzei, G.; et al. Inhibition of Bax Channel-Forming Activity by Bcl-2. Science 1997, 277, 370–372. [Google Scholar] [CrossRef]
- Monni, O.; Joensuu, H.; Franssila, K.; Klefstrom, J.; Alitalo, K.; Knuutila, S. BCL2 overexpression associated with chromosomal amplification in diffuse large B-cell lymphoma. Blood 1997, 90, 1168–1174. [Google Scholar] [CrossRef] [Green Version]
- Slowing, I.I.; Trewyn, B.G.; Lin, V.S.-Y. Mesoporous Silica Nanoparticles for Intracellular Delivery of Membrane-Impermeable Proteins. J. Am. Chem. Soc. 2007, 129, 8845–8849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, M.; Delgado, Y.; Sharma, R.K.; Sharma, S.; Guzmán, S.L.P.D.L.; Tinoco, A.D.; Griebenow, K. Inducing cell death in vitro in cancer cells by targeted delivery of cytochrome c via a transferrin conjugate. PLoS ONE 2018, 13, e0195542. [Google Scholar] [CrossRef]
- Morales-Cruz, M.; Figueroa, C.M.; Gonzalez-Robles, T.; Delgado, Y.; Molina, A.; Mendez, J.; Morales, M.; Griebenow, K. Activation of caspase-dependent apoptosis by intracellular delivery of cytochrome c-based nanoparticles. J. Nanobiotechnol. 2014, 12, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcelo-Bovea, V.; Dominguez-Martinez, I.; Joaquin-Ovalle, F.; Amador, L.A.; Castro-Rivera, E.; Medina-Álvarez, K.; McGoron, A.; Griebenow, K.; Ferrer-Acosta, Y. Optimization and Characterization of Protein Nanoparticles for the Targeted and Smart Delivery of Cytochrome c to Non-Small Cell Lung Carcinoma. Cancers 2020, 12, 1215. [Google Scholar] [CrossRef]
- Wei, B.; Dai, M.; Yin, P. Complex shapes self-assembled from single-stranded DNA tiles. Nat. Cell Biol. 2012, 485, 623–626. [Google Scholar] [CrossRef] [Green Version]
- Schlichthaerle, T.; Eklund, A.S.; Schueder, F.; Strauss, M.T.; Tiede, C.; Curd, A.; Ries, J.; Peckham, M.; Tomlinson, D.C.; Jungmann, R. Site-Specific Labeling of Affimers for DNA-PAINT Microscopy. Angew. Chem. Int. Ed. 2018, 57, 11060–11063. [Google Scholar] [CrossRef] [Green Version]
- Haberland, M.E.; Reynolds, J.A. Self-association of Cholesterol in Aqueous Solution. Proc. Natl. Acad. Sci. USA 1973, 70, 2313–2316. [Google Scholar] [CrossRef] [Green Version]
- Hannibal, L.; Tomasina, F.; Capdevila, D.A.; Demicheli, V.; Tórtora, V.; Paggi, D.A.; Jemmerson, R.; Murgida, D.H.; Radi, R. Alternative Conformations of Cytochrome c: Structure, Function, and Detection. Biochemistry 2016, 55, 407–428. [Google Scholar] [CrossRef]
- Santra, S.; Kaittanis, C.; Perez, J.M. CytochromecEncapsulating Theranostic Nanoparticles: A Novel Bifunctional System for Targeted Delivery of Therapeutic Membrane-Impermeable Proteins to Tumors and Imaging of Cancer Therapy. Mol. Pharm. 2010, 7, 1209–1222. [Google Scholar] [CrossRef] [Green Version]
- Yin, V.Y.; Shaw, G.; Konermann, L. Cytochrome c as a Peroxidase: Activation of the Precatalytic Native State by H2O2-Induced Covalent Modifications. J. Am. Chem. Soc. 2017, 139, 15701–15709. [Google Scholar] [CrossRef]
- Kagan, V.E.; Tyurin, V.; Jiang, J.; Tyurina, Y.; Ritov, V.B.; Amoscato, A.; Osipov, A.N.; Belikova, N.A.; Kapralov, O.; Kini, V.; et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, X. CytochromeC-Mediated Apoptosis. Annu. Rev. Biochem. 2004, 73, 87–106. [Google Scholar] [CrossRef]
- van Engeland, M.; Nieland, L.J.; Ramaekers, F.C.; Schutte, B.; Reutelingsperger, C.P. Annexin V-affinity assay: A review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry 1998, 31, 1–9. [Google Scholar] [CrossRef]
- Wallberg, F.; Tenev, T.; Meier, P. Time-Lapse Imaging of Cell Death. Cold Spring Harb. Protoc. 2016, 2016, 087395. [Google Scholar] [CrossRef] [Green Version]
- Burns, J.R.; Al-Juffali, N.; Janes, S.M.; Howorka, S. Membrane-spanning DNA nanopores with cytotoxic effect. Angew. Chem. Int. Ed. Engl. 2014, 53, 12466–12470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surre, J.; Saint-Ruf, C.; Collin, V.; Orenga, S.; Ramjeet, M.; Matic, I. Strong increase in the autofluorescence of cells signals struggle for survival. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Belmokhtar, C.A.; Hillion, J.; Ségal-Bendirdjian, E. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene 2001, 20, 3354–3362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saint, N.; Marri, L.; Marchini, D.; Molle, G. The antibacterial peptide ceratotoxin A displays alamethicin–like behavior in lipid bilayers. Peptides 2003, 24, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Fennouri, A.; List, J.; Ducrey, J.; Dupasquier, J.; Sukyte, V.; Mayer, S.F.; Vargas, R.D.; Fernandez, L.P.; Bertani, F.; Gonzalo, S.R.; et al. Tuning the Diameter, Stability, and Membrane Affinity of Peptide Pores by DNA-Programmed Self-Assembly. ACS Nano 2021, 15, 11263–11275. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Tweten, R.K.; Johnson, A.E. Membrane-dependent conformational changes initiate cholesterol–dependent cytolysin oligomerization and intersubunit β-strand alignment. Nat. Struct. Mol. Biol. 2004, 11, 697–705. [Google Scholar] [CrossRef]
- Song, L.; Hobaugh, M.R.; Shustak, C.; Cheley, S.; Bayley, H.; Gouaux, J.E. Structure of Staphylococcal alpha-Hemolysin, a Heptameric Transmembrane Pore. Science 1996, 274, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Su, Z.; Simmons, C.R.; Li, J.; Jiang, S.; Li, W.; Yang, Y.; Liu, Y.; Chiu, W.; Fan, C.; et al. Redox Engineering of Cytochrome c using DNA NanostructureBased Charged Encapsulation and Spatial Control. ACS Appl. Mater. Interfaces 2019, 11, 13874–13880. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, D.; Arumugam, S.; Nasilowski, M.; Joshi, H.; Wunder, C.; Chambon, V.; Prakash, V.; Grazon, C.; Nadal, B.; Maiti, P.K.; et al. Quantum dot-loaded monofunctionalized DNA icosahedra for single-particle tracking of endocytic pathways. Nat. Nanotechnol. 2016, 11, 1112–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- Hilvo, M.; Denkert, C.; Lehtinen, L.; Müller, B.; Brockmöller, S.; Seppänen-Laakso, T.; Budczies, J.; Bucher, E.; Yetukuri, L.; Castillo, S.; et al. Novel Theranostic Opportunities Offered by Characterization of Altered Membrane Lipid Metabolism in Breast Cancer Progression. Cancer Res. 2011, 71, 3236–3245. [Google Scholar] [CrossRef] [Green Version]
- Rysman, E.; Brusselmans, K.; Scheys, K.; Timmermans, L.; Derua, R.; Munck, S.; Van Veldhoven, P.P.; Waltregny, D.; Daniels, V.; Machiels, J.; et al. De novo Lipogenesis Protects Cancer Cells from Free Radicals and Chemotherapeutics by Promoting Membrane Lipid Saturation. Cancer Res. 2010, 70, 8117–8126. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.R.; Cheng, K.H.; Huang, J. Assess the nature of cholesterol-lipid interactions through the chemical potential of cholesterol in phosphatidylcholine bilayers. Proc. Natl. Acad. Sci. USA 2007, 104, 5372–5377. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kocabey, S.; Ekim Kocabey, A.; Schneiter, R.; Rüegg, C. Membrane-Interacting DNA Nanotubes Induce Cancer Cell Death. Nanomaterials 2021, 11, 2003. https://doi.org/10.3390/nano11082003
Kocabey S, Ekim Kocabey A, Schneiter R, Rüegg C. Membrane-Interacting DNA Nanotubes Induce Cancer Cell Death. Nanomaterials. 2021; 11(8):2003. https://doi.org/10.3390/nano11082003
Chicago/Turabian StyleKocabey, Samet, Aslihan Ekim Kocabey, Roger Schneiter, and Curzio Rüegg. 2021. "Membrane-Interacting DNA Nanotubes Induce Cancer Cell Death" Nanomaterials 11, no. 8: 2003. https://doi.org/10.3390/nano11082003