
Atomistic Assessment of Solute-Solute Interactions during Grain Boundary Segregation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Thermodynamics of Grain Boundary Segregation
2.1. Free Energy vs. Enthalpy of Segregation
2.2. Classical Segregation Models
2.3. Spectral Segregation Models
3. Atomistic Simulation Methods
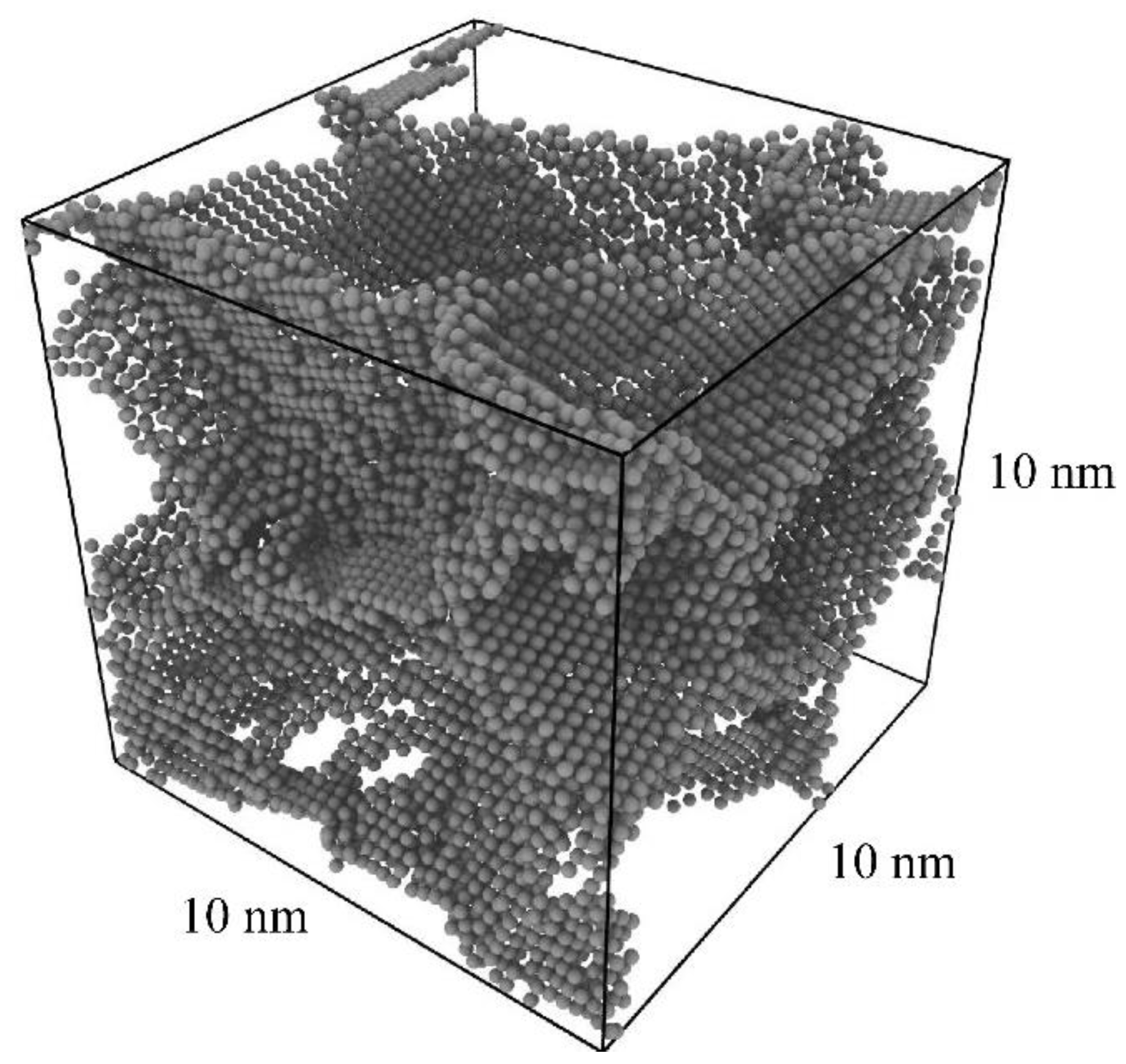
3.1. Production of Pure Al Polycrystal
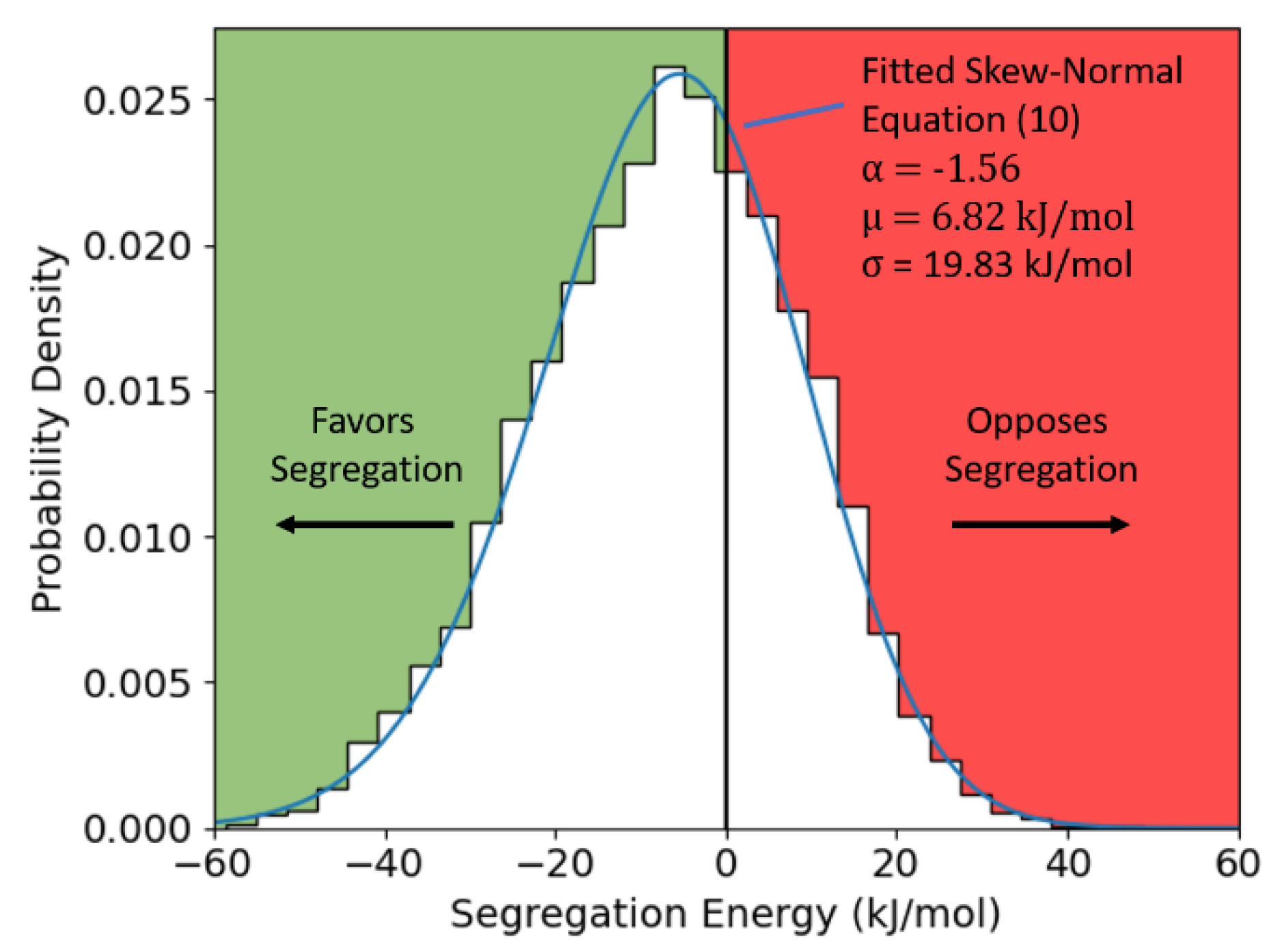
3.2. Dilute Limit Segregation Energy Distributions
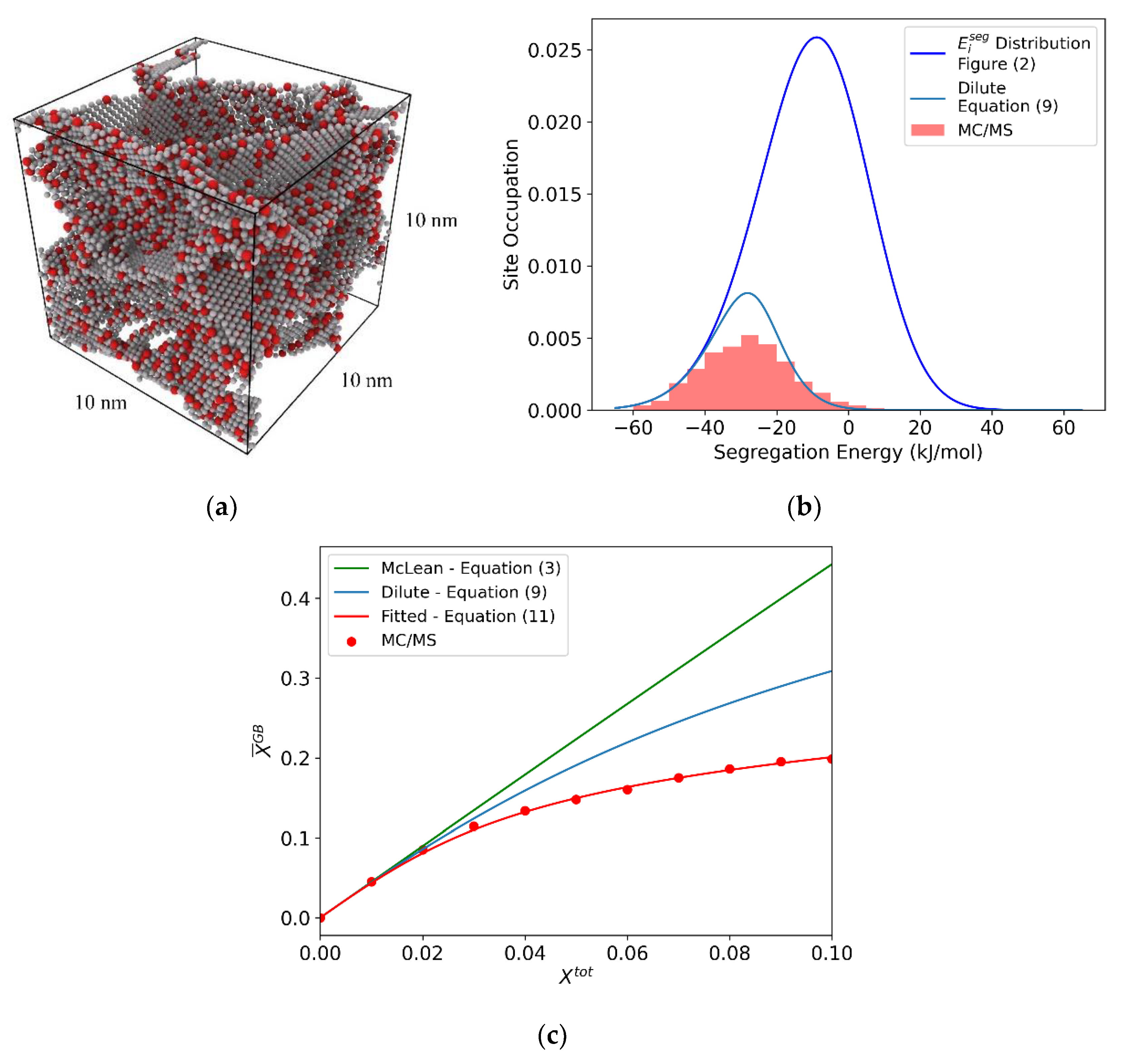
3.3. The True Equilibrium Segregation State: Hybrid MC/MS
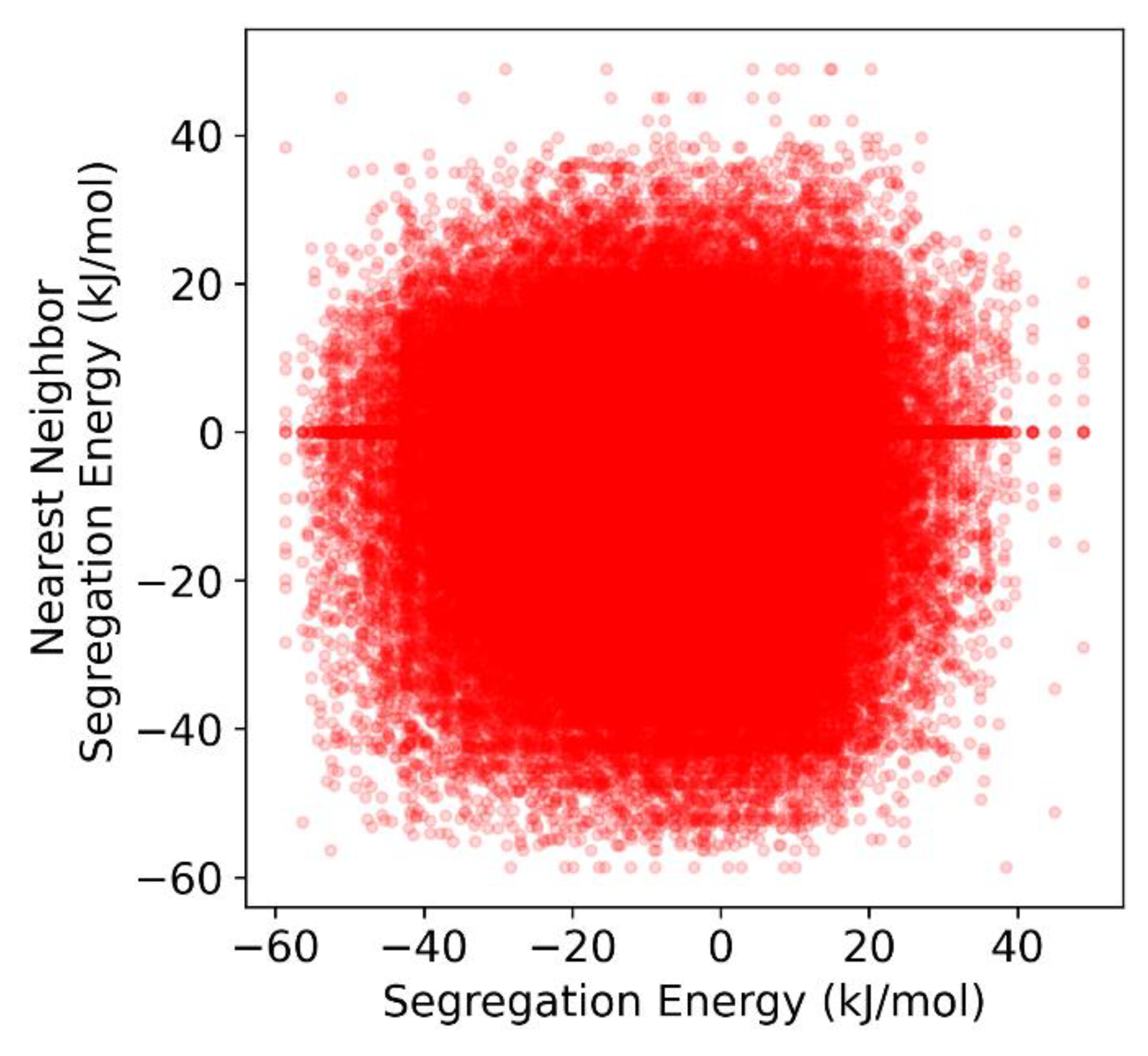
3.4. Grain Boundary Heat of Mixing Distributions
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Darling, K.A.; Rajagopalan, M.; Komarasamy, M.; Bhatia, M.A.; Hornbuckle, B.C.; Mishra, R.S.; Solanki, K.N. Extreme Creep Resistance in a Microstructurally Stable Nanocrystalline Alloy. Nature 2016, 537, 378–381. [Google Scholar] [CrossRef]
- Kotan, H.; Saber, M.; Koch, C.C.; Scattergood, R.O. Effect of Annealing on Microstructure, Grain Growth, and Hardness of Nanocrystalline Fe–Ni Alloys Prepared by Mechanical Alloying. Mater. Sci. Eng. A 2012, 552, 310–315. [Google Scholar] [CrossRef]
- Kumar, K.S.; Van Swygenhoven, H.; Suresh, S. Mechanical Behavior of Nanocrystalline Metals and Alloys. Acta Mater. 2003, 51, 5743–5774. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, G.J.; Jiang, F.; Ding, X.D.; Sun, Y.J.; Sun, J.; Ma, E. Nanostructured High-Strength Molybdenum Alloys with Unprecedented Tensile Ductility. Nat. Mater. 2013, 12, 344–350. [Google Scholar] [CrossRef]
- Meyers, M.A.; Mishra, A.; Benson, D.J. Mechanical Properties of Nanocrystalline Materials. Prog. Mater. Sci. 2006, 51, 427–556. [Google Scholar] [CrossRef]
- Dresselhaus, M.S.; Chen, G.; Tang, M.Y.; Yang, R.G.; Lee, H.; Wang, D.Z.; Ren, Z.F.; Fleurial, J.-P.; Gogna, P. New Directions for Low-Dimensional Thermoelectric Materials. Adv. Mater. 2007, 19, 1043–1053. [Google Scholar] [CrossRef]
- Herzer, G. Grain Size Dependence of Coercivity and Permeability in Nanocrystalline Ferromagnets. IEEE Trans. Magn. 1990, 26, 1397–1402. [Google Scholar] [CrossRef]
- Manaf, A.; Buckley, R.A.; Davies, H.A. New Nanocrystalline High-Remanence Nd-Fe-B Alloys by Rapid Solidification. J. Magn. Magn. Mater. 1993, 128, 302–306. [Google Scholar] [CrossRef]
- Tuller, H. Ionic Conduction in Nanocrystalline Materials. Solid State Ionics 2000, 131, 143–157. [Google Scholar] [CrossRef] [Green Version]
- Xijuan, Y.; Pingbo, X.; Qingde, S. Size-Dependent Optical Properties of Nanocrystalline CeO2: Er Obtained by Combustion Synthesis. Phys. Chem. Chem. Phys. 2001, 3, 5266–5269. [Google Scholar] [CrossRef]
- Darling, K.A.; VanLeeuwen, B.K.; Koch, C.C.; Scattergood, R.O. Thermal Stability of Nanocrystalline Fe–Zr Alloys. Mater. Sci. Eng. A 2010, 527, 3572–3580. [Google Scholar] [CrossRef]
- Gertsman, V.Y.; Birringer, R. On the Room-Temperature Grain Growth in Nanocrystalline Copper. Scr. Metall. Mater. 1994, 30, 577–581. [Google Scholar] [CrossRef]
- Natter, H.; Schmelzer, M.; Hempelmann, R. Nanocrystalline Nickel and Nickel-Copper Alloys: Synthesis, Characterization, and Thermal Stability. J. Mater. Res. 1998, 13, 1186–1197. [Google Scholar] [CrossRef]
- Thuvander, M.; Abraham, M.; Cerezo, A.; Smith, G.D.W. Thermal Stability of Electrodeposited Nanocrystalline Nickel and Iron–Nickel Alloys. Mater. Sci. Technol. 2001, 17, 961–970. [Google Scholar] [CrossRef]
- Choi, P.; Dasilva, M.; Klement, U.; Alkassab, T.; Kirchheim, R. Thermal Stability of Electrodeposited Nanocrystalline Co-1.1at.%P. Acta Mater. 2005, 53, 4473–4481. [Google Scholar] [CrossRef]
- Darling, K.A.; VanLeeuwen, B.K.; Semones, J.E.; Koch, C.C.; Scattergood, R.O.; Kecskes, L.J.; Mathaudhu, S.N. Stabilized Nanocrystalline Iron-Based Alloys: Guiding Efforts in Alloy Selection. Mater. Sci. Eng. A 2011, 528, 4365–4371. [Google Scholar] [CrossRef]
- Detor, A.; Schuh, C. Grain Boundary Segregation, Chemical Ordering and Stability of Nanocrystalline Alloys: Atomistic Computer Simulations in the Ni–W System. Acta Mater. 2007, 55, 4221–4232. [Google Scholar] [CrossRef]
- Detor, A.J.; Schuh, C.A. Microstructural Evolution during the Heat Treatment of Nanocrystalline Alloys. J. Mater. Res. 2007, 22, 3233–3248. [Google Scholar] [CrossRef]
- Hentschel, T.; Isheim, D.; Kirchheim, R.; Müller, F.; Kreye, H. Nanocrystalline Ni–3.6 at.% P and Its Transformation Sequence Studied by Atom-Probe Field-Ion Microscopy. Acta Mater. 2000, 48, 933–941. [Google Scholar] [CrossRef] [Green Version]
- Saber, M.; Kotan, H.; Koch, C.C.; Scattergood, R.O. Thermal Stability of Nanocrystalline Fe–Cr Alloys with Zr Additions. Mater. Sci. Eng. A 2012, 556, 664–670. [Google Scholar] [CrossRef]
- Mehta, S.C.; Smith, D.A.; Erb, U. Study of Grain Growth in Electrodeposited Nanocrystalline Nickel-1.2 Wt.% Phosphorus Alloy. Mater. Sci. Eng. A 1995, 204, 227–232. [Google Scholar] [CrossRef]
- Xing, W.; Kube, S.A.; Kalidindi, A.R.; Amram, D.; Schroers, J.; Schuh, C.A. Stability of Ternary Nanocrystalline Alloys in the Pt–Pd–Au System. Materialia 2019, 8, 100449. [Google Scholar] [CrossRef]
- Chookajorn, T.; Schuh, C.A. Thermodynamics of Stable Nanocrystalline Alloys: A Monte Carlo Analysis. Phys. Rev. B 2014, 89, 064102. [Google Scholar] [CrossRef]
- Kalidindi, A.R.; Schuh, C.A. Stability Criteria for Nanocrystalline Alloys. Acta Mater. 2017, 132, 128–137. [Google Scholar] [CrossRef]
- Kirchheim, R. Reducing Grain Boundary, Dislocation Line and Vacancy Formation Energies by Solute Segregation. I. Theoretical Background. Acta Mater. 2007, 55, 5129–5138. [Google Scholar] [CrossRef]
- Murdoch, H.A.; Schuh, C.A. Stability of Binary Nanocrystalline Alloys against Grain Growth and Phase Separation. Acta Mater. 2013, 61, 2121–2132. [Google Scholar] [CrossRef] [Green Version]
- Saber, M.; Kotan, H.; Koch, C.C.; Scattergood, R.O. A Predictive Model for Thermodynamic Stability of Grain Size in Nanocrystalline Ternary Alloys. J. Appl. Phys. 2013, 114, 103510. [Google Scholar] [CrossRef]
- Trelewicz, J.R.; Schuh, C.A. Grain Boundary Segregation and Thermodynamically Stable Binary Nanocrystalline Alloys. Phys. Rev. B 2009, 79, 094112. [Google Scholar] [CrossRef]
- Weissmüller, J. Alloy Effects in Nanostructures. Nanostruct. Mater. 1993, 3, 261–272. [Google Scholar] [CrossRef]
- Kalidindi, A.R.; Chookajorn, T.; Schuh, C.A. Nanocrystalline Materials at Equilibrium: A Thermodynamic Review. JOM 2015, 67, 2834–2843. [Google Scholar] [CrossRef]
- Boylan, K.; Ostrander, D.; Erb, U.; Palumbo, G.; Aust, K.T. An In-Situ Tem Study of the Thermal Stability of Nanocrystalline NiP. Scr. Metall. Mater. 1991, 25, 2711–2716. [Google Scholar] [CrossRef]
- Humphreys, F.J.; Hatherly, M. Recrystallization and Related Annealing Phenomena; Pergamon Press: Oxford, UK, 1995; pp. 121–167. [Google Scholar]
- Knauth, P.; Charaï, A.; Gas, P. Grain Growth of Pure Nickel and of a Ni-Si Solid Solution Studied by Differential Scanning Calorimetry on Nanometer-Sized Crystals. Scr. Metall. Mater. 1993, 28, 325–330. [Google Scholar] [CrossRef]
- Lücke, K.; Detert, K. A Quantitative Theory of Grain-Boundary Motion and Recrystallization in Metals in the Presence of Impurities. Acta Mater. 1957, 5, 628–637. [Google Scholar] [CrossRef]
- Perez, R.J.; Jiang, H.G.; Lavernia, E.J.; Dogan, C.P. Grain Growth of Nanocrystalline Cryomilled Fe-Al Powders. Metall. Mater. Trans. A 1998, 29, 2469–2475. [Google Scholar] [CrossRef]
- Shaw, L.; Luo, H.; Villegas, J.; Miracle, D. Thermal Stability of Nanostructured Al93Fe3Cr2Ti2 Alloys Prepared via Mechanical Alloying. Acta Mater. 2003, 51, 2647–2663. [Google Scholar] [CrossRef]
- Hondros, E.D.; Seah, M.P. The Theory of Grain Boundary Segregation in Terms of Surface Adsorption Analogues. MTA 1977, 8, 1363–1371. [Google Scholar] [CrossRef]
- McLean, D. Grain Boundaries in Metals; Clarendon Press: Oxford, UK, 1957; pp. 116–150. [Google Scholar]
- Fowler, R.H.; Guggenheim, E.A. Statistical Thermodynamics: A Version of Staistical Mechanics for Students of Physics and Chemistry; Cambridge University Press: Cambridge, MA, USA, 1939. [Google Scholar]
- Guttmann, M. Equilibrium Segregation in a Ternary Solution: A Model for Temper Embrittlement. Surf. Sci. 1975, 53, 213–227. [Google Scholar] [CrossRef]
- Wynblatt, P.; Chatain, D. Anisotropy of Segregation at Grain Boundaries and Surfaces. Metall. Mater. Trans. A 2006, 37, 2595–2620. [Google Scholar] [CrossRef]
- Abdeljawad, F.; Lu, P.; Argibay, N.; Clark, B.G.; Boyce, B.L.; Foiles, S.M. Grain Boundary Segregation in Immiscible Nanocrystalline Alloys. Acta Mater. 2017, 126, 528–539. [Google Scholar] [CrossRef] [Green Version]
- Wagih, M.; Schuh, C.A. Spectrum of Grain Boundary Segregation Energies in a Polycrystal. Acta Mater. 2019, 181, 228–237. [Google Scholar] [CrossRef]
- White, C.L.; Stein, D.F. Sulfur Segregation to Grain Boundaries in Ni3Al and Ni3(AI,Ti) Alloys. MTA 1978, 9, 13–22. [Google Scholar] [CrossRef]
- Kirchheim, R. Hydrogen Solubility and Diffusivity in Defective and Amorphous Metals. Prog. Mater. Sci. 1988, 32, 261–325. [Google Scholar] [CrossRef]
- Mütschele, T.; Kirchheim, R. Segregation and Diffusion of Hydrogen in Grain Boundaries of Palladium. Scr. Metall. 1987, 21, 135–140. [Google Scholar] [CrossRef]
- Wagih, M.; Schuh, C.A. Grain Boundary Segregation beyond the Dilute Limit: Separating the Two Contributions of Site Spectrality and Solute Interactions. Acta Mater. 2020, 199, 63–72. [Google Scholar] [CrossRef]
- Steigerwald, D.A.; Wynblatt, P. Calculation of the Anisotropy of Equilibrium Surface Composition in Metallic Solid Solutions Using the Embedded Atom Method. Surf. Sci. 1988, 193, 287–303. [Google Scholar] [CrossRef]
- Creuze, J.; Berthier, F.; Tétot, R.; Legrand, B. Intergranular Segregation and Ordering Effect: A Mixed Monte Carlo Mean-Field Approach. Phys. Rev. B 2000, 62, 2813–2824. [Google Scholar] [CrossRef]
- Lejček, P.; Zheng, L.; Hofmann, S.; Šob, M. Applied Thermodynamics: Grain Boundary Segregation. Entropy 2014, 16, 1462–1483. [Google Scholar] [CrossRef] [Green Version]
- Lejcek, P. Grain Boundary Segregation in Metals; Springer Series in Materials Science; Springer: Berlin/Heidelberg, Germany, 2010; Volume 136, pp. 51–101. [Google Scholar]
- Ishida, K. Effect of Grain Size on Grain Boundary Segregation. J. Alloys Compd. 1996, 235, 244–249. [Google Scholar] [CrossRef]
- Wagih, M.; Larsen, P.M.; Schuh, C.A. Learning Grain Boundary Segregation Energy Spectra in Polycrystals. Nat. Commun. 2020, 11, 6376. [Google Scholar] [CrossRef] [PubMed]
- Hirel, P. Atomsk: A Tool for Manipulating and Converting Atomic Data Files. Comput. Phys. Commun. 2015, 197, 212–219. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and Analysis of Atomistic Simulation Data with OVITO–the Open Visualization Tool. Modelling Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Mendelev, M.I.; Asta, M.; Rahman, M.J.; Hoyt, J.J. Development of Interatomic Potentials Appropriate for Simulation of Solid–Liquid Interface Properties in Al–Mg Alloys. Philos. Mag. 2009, 89, 3269–3285. [Google Scholar] [CrossRef]
- Huber, L.; Grabowski, B.; Militzer, M.; Neugebauer, J.; Rottler, J. Ab Initio Modelling of Solute Segregation Energies to a General Grain Boundary. Acta Mater. 2017, 132, 138–148. [Google Scholar] [CrossRef]
- Wagih, M.; Schuh, C.A. Thermodynamics and Design of Nanocrystalline Alloys Using Grain Boundary Segregation Spectra. Acta Mater. 2021, 217, 117177. [Google Scholar] [CrossRef]
- Gupta, A.; Zhou, X.; Thompson, G.B.; Tucker, G.J. Role of Grain Boundary Character and Its Evolution on Interfacial Solute Segregation Behavior in Nanocrystalline Ni-P. Acta Mater. 2020, 190, 113–123. [Google Scholar] [CrossRef]
- Sadigh, B.; Erhart, P.; Stukowski, A.; Caro, A.; Martinez, E.; Zepeda-Ruiz, L. Scalable Parallel Monte Carlo Algorithm for Atomistic Simulations of Precipitation in Alloys. Phys. Rev. B 2012, 85, 184203. [Google Scholar] [CrossRef] [Green Version]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef] [Green Version]
- Seki, A.; Seidman, D.N.; Oh, Y.; Foiles, S.M. Monte Carlo Simulations of Segregation at [001] Twist Boundaries in a Pt(Au) Alloy—I. Results. Acta Metall. Mater. 1991, 39, 3167–3177. [Google Scholar] [CrossRef]
- Seki, A.; Seidman, D.N.; Oh, Y.; Foiles, S.M. Monte Carlo Simulations of Segregation at [001] Twist Boundaries in a Pt(Au) Alloy—II. Discussion. Acta Metall. Mater. 1991, 39, 3179–3185. [Google Scholar] [CrossRef] [Green Version]
- Menyhard, M.; Yan, M.; Vitek, V. Atomistic vs. Phenomenological Approaches to Grain Boundary Segregation: Computer Modeling of Cu–Ag Alloys. Acta Metall. Mater. 1994, 42, 2783–2796. [Google Scholar] [CrossRef]
- Foiles, S.M. Calculation of Grain-Boundary Segregation in Ni-Cu Alloys. Phys. Rev. B 1989, 40, 11502–11506. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Adams, J.B. Grain-Boundary Segregation in Al–10%Mg Alloys at Hot Working Temperatures. Acta Mater. 1998, 46, 3467–3476. [Google Scholar] [CrossRef]
- Azzalini, A. The Multivariate Skew-Normal Distribution. Biometrika 1996, 83, 715–726. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matson, T.P.; Schuh, C.A. Atomistic Assessment of Solute-Solute Interactions during Grain Boundary Segregation. Nanomaterials 2021, 11, 2360. https://doi.org/10.3390/nano11092360
Matson TP, Schuh CA. Atomistic Assessment of Solute-Solute Interactions during Grain Boundary Segregation. Nanomaterials. 2021; 11(9):2360. https://doi.org/10.3390/nano11092360
Chicago/Turabian StyleMatson, Thomas P., and Christopher A. Schuh. 2021. "Atomistic Assessment of Solute-Solute Interactions during Grain Boundary Segregation" Nanomaterials 11, no. 9: 2360. https://doi.org/10.3390/nano11092360
APA StyleMatson, T. P., & Schuh, C. A. (2021). Atomistic Assessment of Solute-Solute Interactions during Grain Boundary Segregation. Nanomaterials, 11(9), 2360. https://doi.org/10.3390/nano11092360