
Miktoarm Star Copolymers Prepared by Transformation from Enhanced Spin Capturing Polymerization to Nitroxide-Mediated Polymerization (ESCP-Ŧ-NMP) toward Nanomaterials

 , , ,
, , , 
_Lin.png)

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization
2.3. Synthesis of (1Z,1′Z)-1,1′-(1,4-Phenylene) bis (N-Tert-Butylmethanimine Oxide) (PBBN)
2.4. ESCPs of St and tBA and Thermolysis of the (PSt)4 Homo-Star
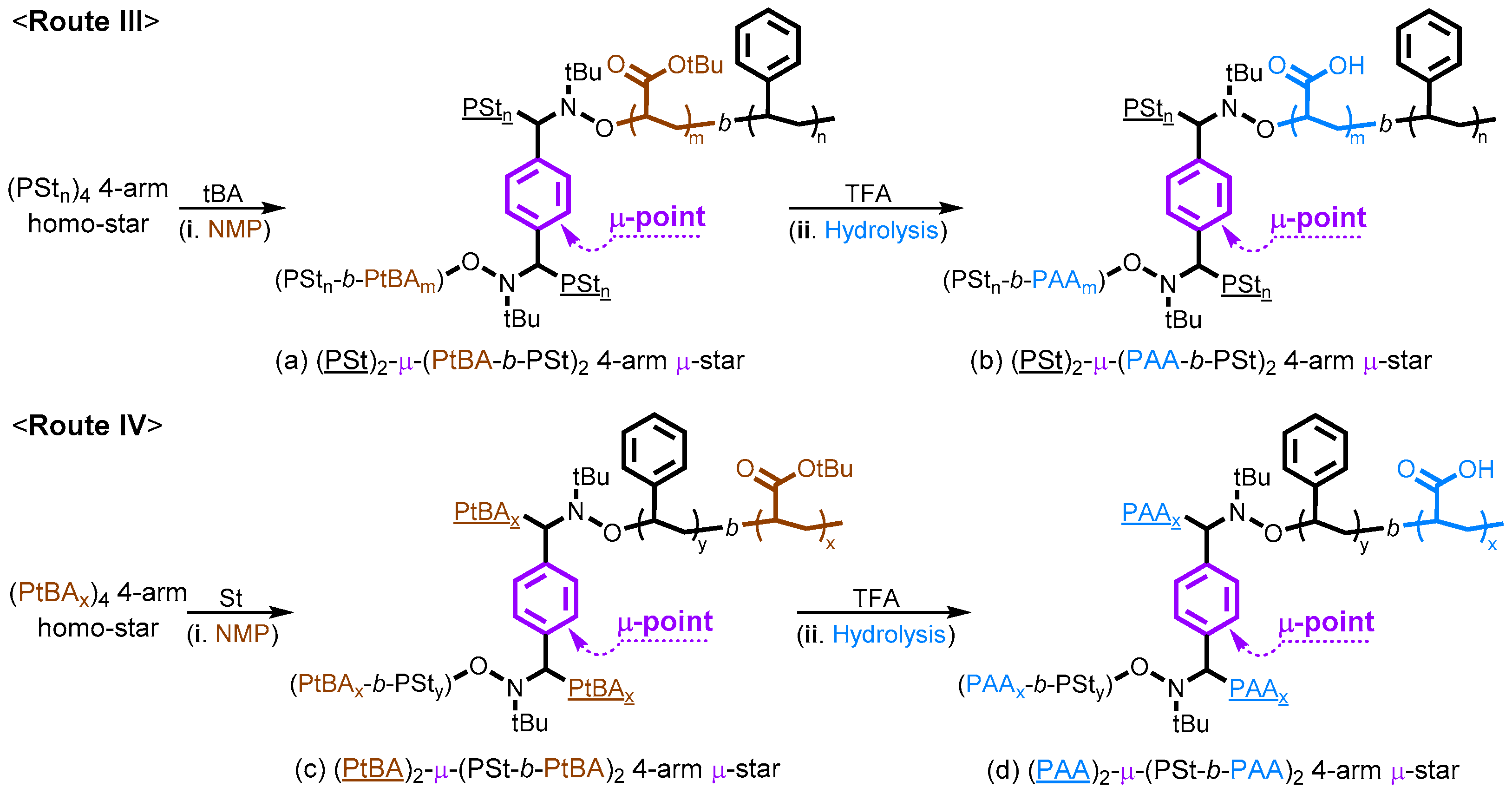
2.5. Synthesis of Miktoarm Star (μ-Star) Copolymers via Nitroxide-Mediated Chain Extensions of Homo-Stars and Hydrolysis of PtBA Segments
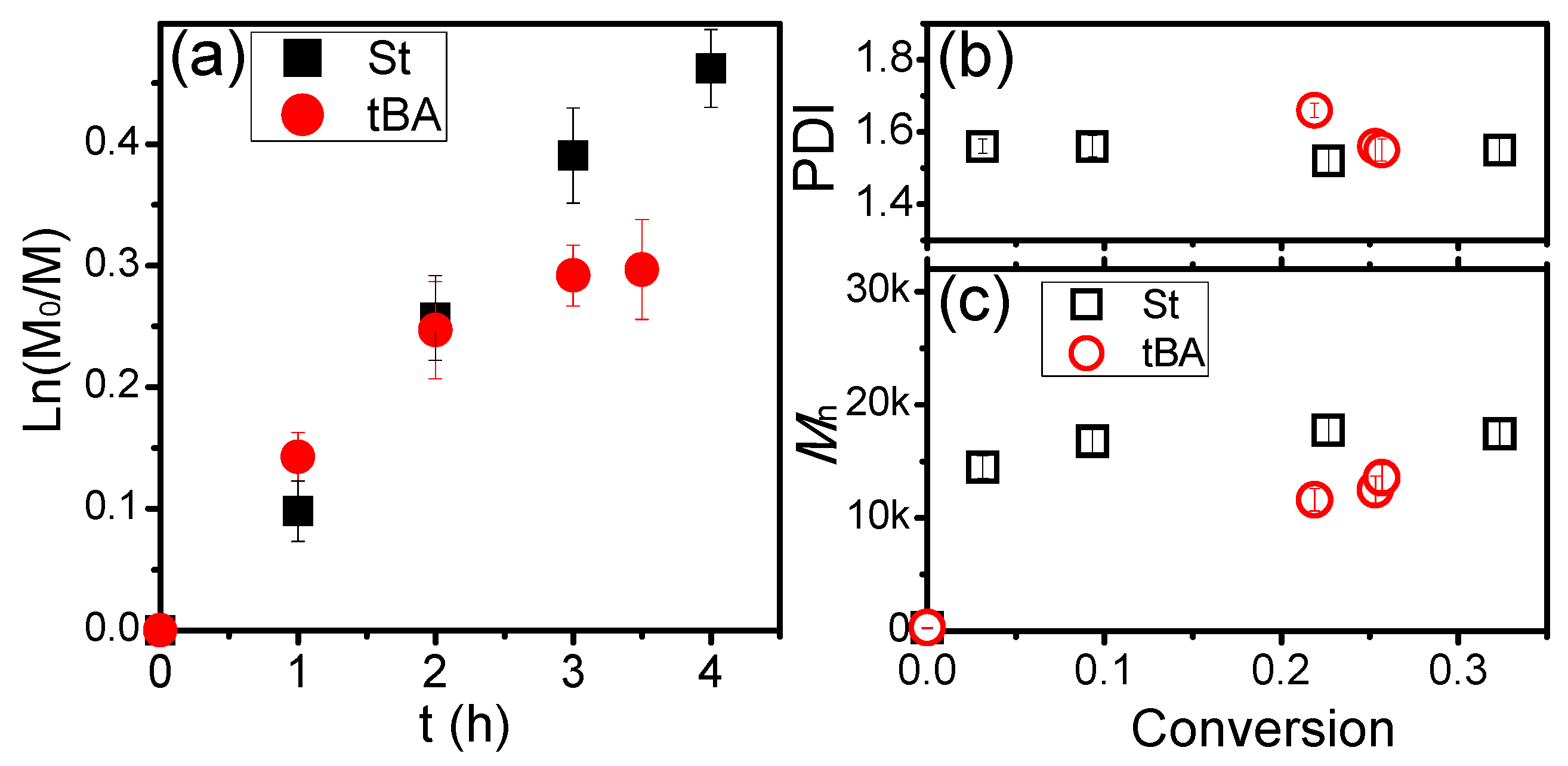
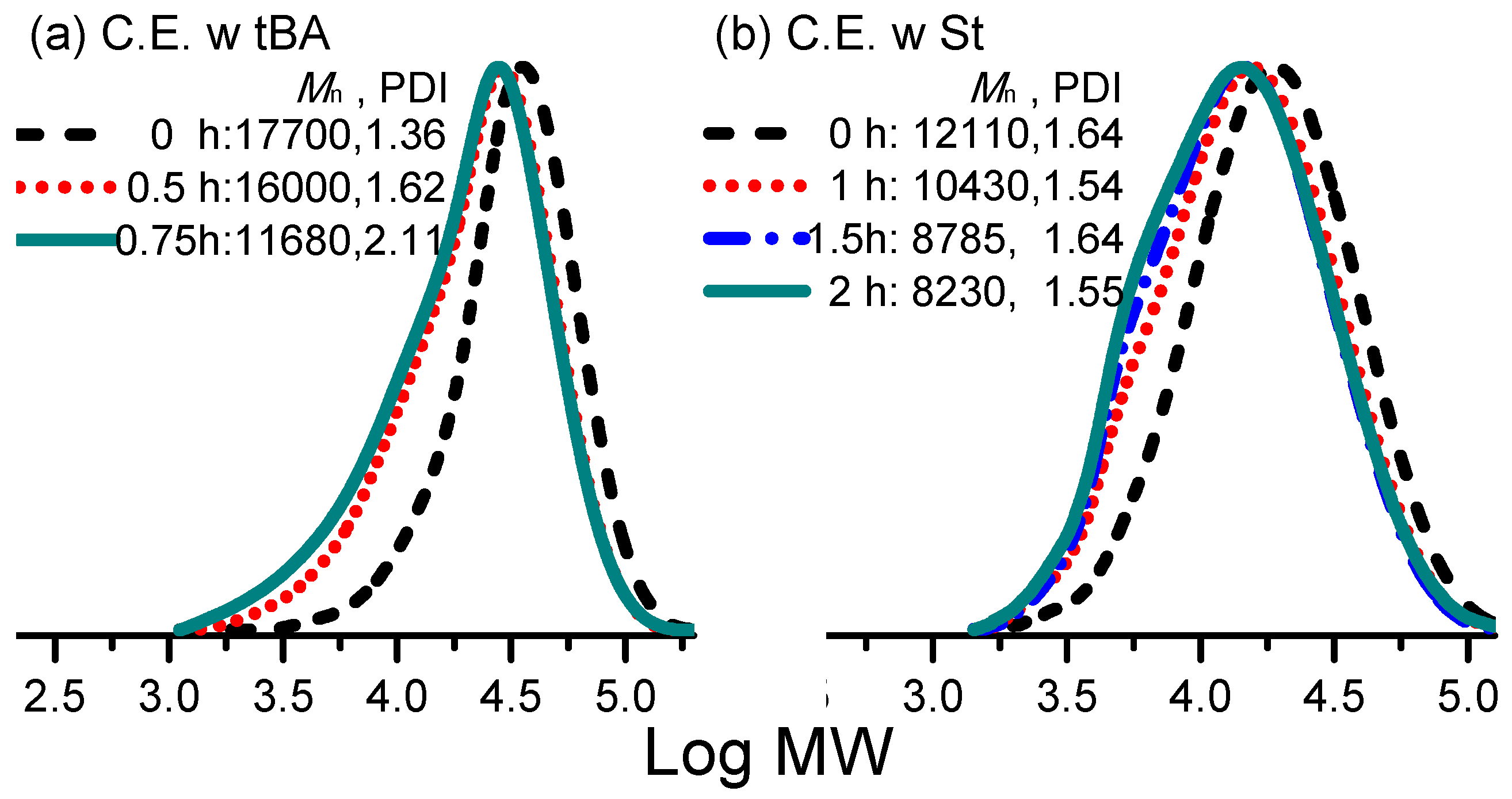
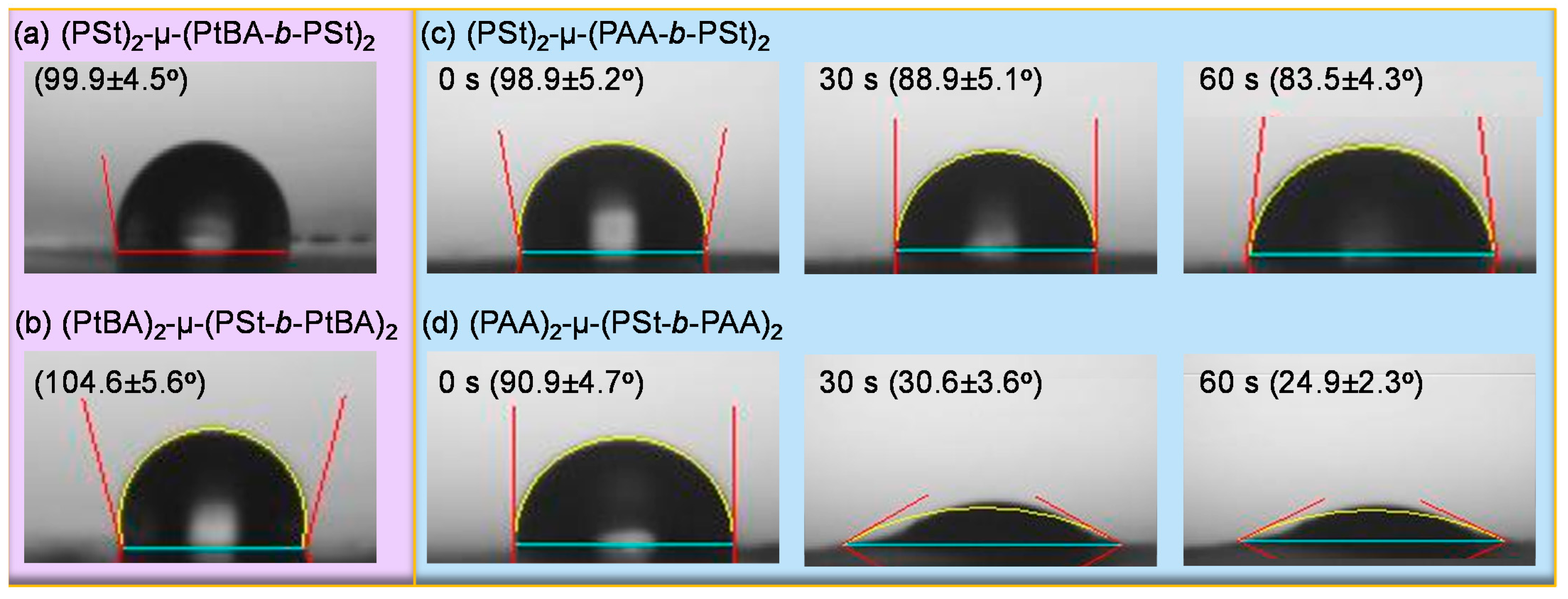
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Matyjaszewski, K. Controlled/Living Radical Polymerization. ACS Symp. Ser. 2009, 1023, 3–14. [Google Scholar]
- Corrigan, N.; Jung, K.; Moad, G.; Hawker, C.J.; Matyjaszewski, K.; Boyer, C. Reversible-deactivation radical polymerization (controlled/living radical polymerization): From discovery to materials design and applications. Prog. Polym. Sci. 2020, 111, 10131. [Google Scholar] [CrossRef]
- Benoit, D.; Chaplinski, V.; Braslau, R.; Hawker, C.J. Development of a Universal Alkoxyamine for “Living” Free Radical Polymerizations. J. Am. Chem. Soc. 1999, 121, 3904–3920. [Google Scholar] [CrossRef]
- Hawker, C.J.; Bosman, A.W.; Harth, E. New polymer synthesis by nitroxide mediated living radical polymerizations. Chem. Rev. 2001, 101, 3661–3688. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.-F.; Han, Y.-M.; Chen, W.-H.; Chu, Y.-L.; Lin, C.-H.; Huang, Y.-S.; Nakamura, Y.; Huang, C.-F. Synthesis of thiophene-containing acyclic alkoxyamine for nitroxide-mediated radical polymerization of acrylates and styrene. Polymer 2021, 230, 124062. [Google Scholar] [CrossRef]
- Wang, J.S.; Matyjaszewski, K. Controlled living radical polymerization-Atom-transfer radical polymerization in the presence of transition-metal complexes. J. Am. Chem. Soc. 1995, 117, 5614–5615. [Google Scholar] [CrossRef]
- Tang, W.; Kwak, Y.; Braunecker, W.; Tsarevsky, N.V.; Coote, M.L.; Matyjaszewski, K. Understanding atom transfer radical polymerization: Effect of ligand and initiator structures on the equilibrium constants. J. Am. Chem. Soc. 2008, 130, 10702–10713. [Google Scholar] [CrossRef]
- Huang, Y.-S.; Hsueh, H.-Y.; Aimi, J.; Chou, L.-C.; Lu, Y.-C.; Kuo, S.-W.; Wang, C.-C.; Chen, K.-Y.; Huang, C.-F. Effects of various Cu(0), Fe(0), and Proanthocyanidin Reducing Agents on Fe(III)-catalysed ATRP for the Synthesis of PMMA Block Copolymers and their Self-assembly Behaviours. Polym. Chem. 2020, 11, 5147. [Google Scholar] [CrossRef]
- Moad, G.; Chong, Y.K.; Postma, A.; Rizzardo, E.; Thang, S.H. Advances in RAFT polymerization: The synthesis of polymers with defined end-groups. Polymer 2005, 46, 8458–8468. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living radical polymerization by the RAFT process. Aust. J. Chem. 2005, 58, 379–410. [Google Scholar] [CrossRef]
- Perrier, S.; Takolpuckdee, P. Macromolecular design via reversible addition-fragmentation chain transfer (RAFT)/Xanthates (MADIX) polymerization. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 5347–5393. [Google Scholar] [CrossRef]
- Huang, C.-F.; Hsieh, Y.-A.; Hsu, S.-C.; Matyjaszewski, K. Synthesis of Poly(N-vinyl carbazole)-based Block Copolymers by Sequential Polymerizations of RAFT-ATRP. Polymer 2014, 55, 6051–6057. [Google Scholar] [CrossRef]
- Wayland, B.B.; Poszmik, G.; Mukerjee, S.L.; Fryd, M. Living radical polymerization of acrylates by organocobalt porphyrin complexes. J. Am. Chem. Soc. 1994, 116, 7943–7944. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Wei, M.L.; Xia, J.H.; McDermott, N.E. Controlled/“living” radical polymerization of styrene and methyl methacrylate catalyzed by iron complexes. Macromolecules 1997, 30, 8161–8164. [Google Scholar] [CrossRef]
- Braunecker, W.A.; Itami, Y.; Matyjaszewski, K. Osmium-mediated radical polymerization. Macromolecules 2005, 38, 9402–9404. [Google Scholar] [CrossRef]
- Braunecker, W.A.; Brown, W.C.; Morelli, B.C.; Tang, W.; Poli, R.; Matyjaszewski, K. Origin of activity in Cu-, Ru-, and Os-mediated radical polymerization. Macromolecules 2007, 40, 8576–8585. [Google Scholar] [CrossRef]
- Nakamura, Y.; Ebeling, B.; Wolpers, A.; Monteil, V.; D’Agosto, F.; Yamago, S. Controlled Radical Polymerization of Ethylene Using Organotellurium Compounds. Angew. Chem. Int. Ed. 2018, 57, 305–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawker, C.J.; Frechet, J.M.J.; Grubbs, R.B.; Dao, J. Preparation of hyperbranched and star polymers by a living, self-condensing free-radical polymerization. J. Am. Chem. Soc. 1995, 117, 10763–10764. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Gaynor, S.G.; Kulfan, A.; Podwika, M. Preparation of hyperbranched polyacrylates by atom transfer radical polymerization. 1. Acrylic AB* monomers in “living” radical polymerizations. Macromolecules 1997, 30, 5192–5194. [Google Scholar] [CrossRef]
- Hsu, C.-J.; Tu, C.-W.; Huang, Y.-W.; Kuo, S.-W.; Lee, R.-H.; Liu, Y.-T.; Hsueh, H.-Y.; Aimi, J.; Huang, C.-F. Synthesis of Poly(styrene)-b-Poly(2-vinyl pyridine) Four-arm Star Block Copolymers via ATRP and Their Self-assembly Behaviors. Polymer 2021, 213, 123212. [Google Scholar] [CrossRef]
- Huang, C.F.; Huang, Y.S.; Lai, K.Y. Synthesis and Self-Assembly of Poly(N-octyl benzamide)-μ-Poly(ε-caprolactone) Miktoarm Star Copolymers Displaying Uniform Nanofibril Morphology. Polymer 2019, 178, 121582. [Google Scholar] [CrossRef]
- Aimi, J.; Wang, P.-H.; Shih, C.-C.; Huang, C.-F.; Nakanishi, T.; Takeuchi, M.; Hsueh, H.-Y.; Chen, W.-C. A Star Polymer with a Metallo-Phthalocyanine Core as a Tunable Charge Storage Material for Nonvolatile Transistor Memory Devices. J. Mater. Chem. C 2018, 6, 2724–2732. [Google Scholar] [CrossRef]
- Huang, C.F.; Aimi, J.; Lai, K.Y. Synthesis of Novel mu-Star Copolymers with Poly(N-octyl benzamide) and Poly(epsilon-caprolactone) Miktoarms through Chain-Growth Condensation Polymerization, Styrenics-Assisted Atom Transfer Radical Coupling, and Ring-Opening Polymerization. Macromol. Rapid Commun. 2017, 38, 1600607. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.-Y.; Huang, Y.-S.; Chu, C.-Y.; Huang, C.-F. Synthesis of Poly(N-H benzamide)-b-Poly(lauryl methacrylate)-b-Poly(N-H benzamide) Symmetrical Triblock Copolymers by Combinations of CGCP, SARA ATRP, and SA ATRC. Polymer 2018, 137, 385–394. [Google Scholar] [CrossRef]
- Huang, C.-F.; Chen, W.-H.; Aimi, J.; Huang, Y.-S.; Venkatesan, S.; Chiang, Y.-W.; Huang, S.-H.; Kuo, S.-W.; Chen, T. Synthesis of Well-defined PCL-b-PnBA-b-PMMA ABC-type Triblock Copolymers: Toward the Construction of Nanostructures in Epoxy Thermosets. Polym. Chem. 2018, 9, 5644–5654. [Google Scholar] [CrossRef]
- Gao, Y.S.; Zhou, D.Z.; Lyu, J.; Sigen, A.; Xu, Q.; Newland, B.; Matyjaszewski, K.; Tai, H.Y.; Wang, W.X. Complex polymer architectures through free-radical polymerization of multivinyl monomers. Nat. Rev. Chem. 2020, 4, 194–212. [Google Scholar] [CrossRef]
- Huang, C.-F.; Kuo, S.-W.; Chen, J.-K.; Chang, F.-C. Synthesis and Characterization of Polystyrene-b-Poly(4-vinyl pyridine) Block Copolymers by Atom Transfer Radical Polymerization. J. Polym. Res. 2005, 12, 449–456. [Google Scholar] [CrossRef]
- Huang, Y.S.; Chen, J.K.; Kuo, S.W.; Hsieh, Y.A.; Yamamoto, S.; Nakanishi, J.; Huang, C.F. Synthesis of Poly(N-vinylpyrrolidone)-Based Polymer Bottlebrushes by ATRPA and RAFT Polymerization: Toward Drug Delivery Application. Polymers 2019, 11, 1079. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.C.; Chou, L.C.; Huang, C.F. Iron-Catalysed Atom Transfer Radical Polyaddition for the Synthesis and Modification of Novel Aliphatic Polyesters Displaying Lower Critical Solution Temperature and pH-Dependent Release Behaviors. Polym. Chem. 2019, 10, 3912–3921. [Google Scholar] [CrossRef]
- Di Sacco, F.; Pucci, A.; Raffa, P. Versatile Multi-Functional Block Copolymers Made by Atom Transfer Radical Polymerization and Post-Synthetic Modification: Switching from Volatile Organic Compound Sensors to Polymeric Surfactants for Water Rheology Control via Hydrolysis. Nanomaterials 2019, 9, 458. [Google Scholar] [CrossRef] [Green Version]
- Sciannamea, V.; Guerrero-Sanchez, A.; Schubert, U.S.; Catala, J.M.; Jerome, R.; Detrembleur, C. Ability of nitrones of various structures to control the radical polymerization of styrene mediated by in situ formed nitroxides. Polymer 2005, 46, 9632–9641. [Google Scholar] [CrossRef] [Green Version]
- Sciannamea, V.; Jerome, R.; Detrembleur, C. In-situ nitroxide-mediated radical polymerization (NMP) processes: Their understanding and optimization. Chem. Rev. 2008, 108, 1104–1126. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.H.H.; Junkers, T.; Barner-Kowollik, C. Enhanced Spin Capturing Polymerization: An Efficient and Versatile Protocol for Controlling Molecular Weight Distributions. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 7273–7279. [Google Scholar] [CrossRef]
- Junkers, T.; Wong, E.H.H.; Stenzel, M.H.; Barner-Kowollik, C. Formation Efficiency of ABA Blockcopolymers via Enhanced Spin Capturing Polymerization (ESCP): Locating the Alkoxyamine Function. Macromolecules 2009, 42, 5027–5035. [Google Scholar] [CrossRef]
- Huang, C.-F.; Ohta, Y.; Yokoyama, A.; Yokozawa, T. Efficient low-temperature atom transfer radical coupling and its application to synthesis of well-defined symmetrical polybenzamides. Macromolecules 2011, 44, 4140–4148. [Google Scholar] [CrossRef]
- Yokozawa, T.; Ogawa, M.; Sekino, A.; Sugi, R.; Yokoyama, A. Chain-growth polycondensation for well-defined aramide. Synthesis of unprecedented block copolymer containing aramide with low polydispersity. J. Am. Chem. Soc. 2002, 124, 15158–15159. [Google Scholar] [CrossRef]
- Kamber, N.E.; Jeong, W.; Waymouth, R.M.; Pratt, R.C.; Lohmeijer, B.G.G.; Hedrick, J.L. Organocatalytic ring-opening polymerization. Chem. Rev. 2007, 107, 5813–5840. [Google Scholar] [CrossRef]
- Bielawski, C.W.; Grubbs, R.H. Living ring-opening metathesis polymerization. Prog. Polym. Sci. 2007, 32, 1–29. [Google Scholar] [CrossRef]
- Coulembier, O.; Degee, P.; Hedrick, J.L.; Dubois, P. From controlled ring-opening polymerization to biodegradable aliphatic polyester: Especially poly(beta-malic acid) derivatives. Prog. Polym. Sci. 2006, 31, 723–747. [Google Scholar] [CrossRef]
- Huang, C.-F.; Yokoyama, A.; Yokozawa, T. Synthesis of Polybenzamide-b-Polystyrene Block Copolymer via Combination of Chain-Growth Condensation Polymerization and Atom Transfer Radical Polymerization. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 2948–2954. [Google Scholar] [CrossRef]
- Huang, C.-F.; Kuo, S.-W.; Lee, H.-F.; Chang, F.-C. A New Strategy for the One-step Synthesis of Block Copolymers through Simultaneous Free Radical and Ring Opening Polymerizations Using A Dual-functional Initiator. Polymer 2005, 46, 1561–1565. [Google Scholar] [CrossRef]
- Yagci, Y.; Tasdelen, M.A. Mechanistic transformations involving living and controlled/living polymerization methods. Prog. Polym. Sci. 2006, 31, 1133–1170. [Google Scholar] [CrossRef]
- Sun, Z.W.; Chen, Z.B.; Zhang, W.X.; Choi, J.; Huang, C.L.; Jeong, G.J.; Coughlin, E.B.; Hsu, Y.T.; Yang, X.M.; Lee, K.Y.; et al. Directed Self-Assembly of Poly(2-vinylpyridine)-b-polystyrene-b-poly(2-vinylpyridine) Triblock Copolymer with Sub-15 nm Spacing Line Patterns Using a Nanoimprinted Photoresist Template. Adv. Mater. 2015, 27, 4364–4370. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.W.; Zhang, W.X.; Hong, S.; Chen, Z.B.; Liu, X.H.; Xiao, S.G.; Coughlin, E.B.; Russell, T.P. Using block copolymer architecture to achieve sub-10 nm periods. Polymer 2017, 121, 297–303. [Google Scholar] [CrossRef]
- Kim, H.; Kang, B.G.; Choi, J.W.; Sun, Z.W.; Yu, D.M.; Mays, J.; Russell, T.P. Morphological Behavior of A(2)B Block Copolymers in Thin Films. Macromolecules 2018, 51, 1181–1188. [Google Scholar] [CrossRef]
- Kim, H.; Arras, M.M.L.; Mahalik, J.P.; Wang, W.Y.; Yu, D.M.; Chernyy, S.; Goswami, M.; Kumar, R.; Sumpter, B.G.; Hong, K.L.; et al. Studies on the 3-Lamellar Morphology of Miktoarm Terpolymers. Macromolecules 2018, 51, 7491–7499. [Google Scholar] [CrossRef]
- Ren, J.M.; McKenzie, T.G.; Fu, Q.; Wong, E.H.H.; Xu, J.T.; An, Z.S.; Shanmugam, S.; Davis, T.P.; Boyer, C.; Qiao, G.G. Star Polymers. Chem. Rev. 2016, 116, 6743–6836. [Google Scholar] [CrossRef] [PubMed]
- Goseki, R.; Hirao, A.; Kakimoto, M.; Hayakawa, T. Cylindrical Nanostructure of Rigid-Rod POSS-Containing Polymethacrylate from a Star-Branched Block Copolymer. ACS Macro Lett. 2013, 2, 625–629. [Google Scholar] [CrossRef]
- Lo, T.Y.; Dehghan, A.; Georgopanos, P.; Avgeropoulos, A.; Shi, A.C.; Ho, R.M. Orienting Block Copolymer Thin Films via Entropy. Macromolecules 2016, 49, 624–633. [Google Scholar] [CrossRef]
- Jang, S.; Lee, K.; Moon, H.C.; Kwak, J.; Park, J.; Jeon, G.; Lee, W.B.; Kim, J.K. Vertical Orientation of Nanodomains on Versatile Substrates through Self-Neutralization Induced by Star-Shaped Block Copolymers. Adv. Funct. Mater. 2015, 25, 5414–5419. [Google Scholar] [CrossRef]
- Sathesh, V.; Chen, J.K.; Chang, C.J.; Aimi, J.; Chen, Z.C.; Hsu, Y.C.; Huang, Y.S.; Huang, C.F. Synthesis of Poly(epsilon-caprolactone)-Based Miktoarm Star Copolymers through ROP, SA ATRC, and ATRP. Polymers 2018, 10, 858. [Google Scholar] [CrossRef] [Green Version]
- Gryn’ova, G.; Lin, C.Y.; Coote, M.L. Which side-reactions compromise nitroxide mediated polymerization? Polym. Chem. 2013, 4, 3744–3754. [Google Scholar] [CrossRef]
- Huang, C.-F.; Lee, H.-F.; Kuo, S.-W.; Xu, H.; Chang, F.-C. Star Polymers via Atom Transfer Radical Polymerization from Adamantane-based Cores. Polymer 2004, 45, 2261–2269. [Google Scholar] [CrossRef]
- Huang, C.-F.; Chen, J.-K.; Tsai, T.-Y.; Hsieh, Y.-A.; Lin, K.-Y.A. Dual-functionalized Cellulose Nanofibrils Prepared through TEMPO-mediated Oxidation and Surface-initiated ATRP. Polymer 2015, 72, 395–405. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, T.-Y.; Tu, C.-W.; Aimi, J.; Huang, Y.-W.; Jamnongkan, T.; Hsueh, H.-Y.; Lin, K.-Y.A.; Huang, C.-F. Miktoarm Star Copolymers Prepared by Transformation from Enhanced Spin Capturing Polymerization to Nitroxide-Mediated Polymerization (ESCP-Ŧ-NMP) toward Nanomaterials. Nanomaterials 2021, 11, 2392. https://doi.org/10.3390/nano11092392
Lin T-Y, Tu C-W, Aimi J, Huang Y-W, Jamnongkan T, Hsueh H-Y, Lin K-YA, Huang C-F. Miktoarm Star Copolymers Prepared by Transformation from Enhanced Spin Capturing Polymerization to Nitroxide-Mediated Polymerization (ESCP-Ŧ-NMP) toward Nanomaterials. Nanomaterials. 2021; 11(9):2392. https://doi.org/10.3390/nano11092392
Chicago/Turabian StyleLin, Tzu-Yao, Cheng-Wei Tu, Junko Aimi, Yu-Wen Huang, Tongsai Jamnongkan, Han-Yu Hsueh, Kun-Yi Andrew Lin, and Chih-Feng Huang. 2021. "Miktoarm Star Copolymers Prepared by Transformation from Enhanced Spin Capturing Polymerization to Nitroxide-Mediated Polymerization (ESCP-Ŧ-NMP) toward Nanomaterials" Nanomaterials 11, no. 9: 2392. https://doi.org/10.3390/nano11092392
APA StyleLin, T.-Y., Tu, C.-W., Aimi, J., Huang, Y.-W., Jamnongkan, T., Hsueh, H.-Y., Lin, K.-Y. A., & Huang, C.-F. (2021). Miktoarm Star Copolymers Prepared by Transformation from Enhanced Spin Capturing Polymerization to Nitroxide-Mediated Polymerization (ESCP-Ŧ-NMP) toward Nanomaterials. Nanomaterials, 11(9), 2392. https://doi.org/10.3390/nano11092392