1. Introduction
Molecular carbon formation, transportation and transformation is one of the great scientific challenges in astrochemistry. Complex carbon molecules have been proposed as primary carbon sources in astrochemical environments such as the interstellar medium (ISM) [
1], nebulae clouds [
2,
3] and supernovae remnants [
4,
5]. Carbon is expected to exist in a variety of forms including amorphous carbon and hydrocarbon dust [
1] as well as in molecular structures. Of these, three key families are carbon chains [
6], fullerenes [
1,
2,
3,
7], and polycyclic aromatic hydrocarbons (PAHs) [
3,
4]. UV-irradiation, hydrogen impacts and thermal cooling near supernovae present mechanisms for transformation between different species types. Growth and transformation models have predicted short chain presence during fullerene formation [
8,
9] while electron irradiation studies have demonstrated spontaneous transformation of planar aromatics into fullerenes [
10], but precise carbonaceous transformation processes and pathways remain a subject of debate.
Curvature should play a critical role in carbonaceous species transformation and stability. As an example, localized curvature has been shown to aid hydrogen trapping in interstellar graphenic dust grain models, catalysing subsequent pairwise release giving H
2 formation [
11]. Curvature typically destabilises aromaticity and localises reactive sites [
12]. It adds a driving mechanism for functionalisation and structural reorganisation in order to relieve curvature-induced strain, either by localising the curvature, or eliminating it through transformation from sp
2- to sp
3- bonded carbon. It is therefore interesting to examine the transformation behaviour under irradiation of curved aromatic hydrocarbons.
Despite the extensive available literature of damage-induced structural change in nanocarbons [
10,
13], it is very difficult to examine the damage breakdown process stepwise. This is because samples are typically non-uniform and data is either statistical across a range of related but different configurations, or based on observation of a single event (e.g., via high-resolution transmission electron microscopy, HRTEM). Here, we have chosen to study damage in carbon nanobelts [
14] (CNBs, [(CH)
2-C
6H
2]
2n), part of a family of cyclic aromatic species that began with cycloparaphenylenes first synthesised in 2006 [
15]. Nanobelts are of interest since they only contain hexagons and provide a continuous aromatic structure with uniform curvature. This is dependent purely on the ring circumference selected during synthesis. As such nanobelts can be considered archetypal curved polyaromatic hydrocarbons. At the same time, each molecule is identical to the others. This allows us to examine with statistical rigor the breakdown of identical, uniform nanorings, allowing us to identify the individual matter loss and transformation steps.
In this study we use laser-induced vaporisation to evaporate and ionise a source of carbon nanobelts. Collision impacts between species cause mass loss and the resultant ions are catalogued via mass-spectrometry. In order to understand the processes involved we perform a series of “in-silico”-simulated systematic hydrogen-loss studies using density functional theory (DFT) modelling. We sequentially remove hydrogen atoms using thermodynamic stability as a selection for subsequent dehydrogenation, allowing us to map and compare sequential hydrogen loss routes.
Our calculations show good agreement with experiment indicating initial pairwise hydrogen loss and local carbyne chain formation within the nanoring. The resultant species are a novel form of previously unreported circular strained dehydrobenzoannulenes, a new class of unsaturated chain-based hydrocarbons of great literature interest [
16,
17,
18]. Subsequent damage results in release of the chain ends, leading to subsequent C
2 and CH loss.
2. Method
Experimental gas phase studies of the carbon nanobelt C48H24 were performed with a laser cluster source using a 532 nm pulsed Nd:YAG laser system. The C48H24 sample is surface coated onto a quartz rod. A channel 2 mm in diameter and ~8.5 mm in length runs from a pulsed valve (10–70 psi backing pressure, 800 μs pulse width) into the region containing the rod to introduce helium. A second channel 2 mm in diameter was directed into the target area to admit the laser beam. Vaporization of the target rod is achieved by a single laser pulse fired from the Nd:YAG laser (5 ns, 5 mJ per pulse, ~1.5 mm beam diameter) in conjunction with the opening of the pulsed valve to admit He. A channel 4 mm in diameter and ~8.5 mm in length was located downstream from the target, aligned with the helium introduction channel to achieve confinement and clustering of the vapor produced. The gas then enters high vacuum and undergoes a free jet expansion.
The molecular species are isolated in the gas phase by use of the Sustained Off-Resonance Irradiation (SORI) technique [
19] and then probed by collision-induced dissociation (CID) [
20] to provide circumstantial structural information. This is achieved by accelerating the trapped ions in cyclotron motion, in the presence of Ar used as a collision gas. It is this step that causes molecular fracture as discussed further below. The cluster source is coupled directly to a Fourier Transform Ion Cyclotron (FT-ICR) Mass Spectrometer [
21] whose very high resolution enables unequivocal identification of the stoichiometry of each and every ionic species produced, as the resulting spectra are completely resolved. The FT-ICR mass spectrometer is custom-built with ultra-high resolution, based on a 9.4 T, 155 mm bore diameter actively shielded superconducting magnet [
22,
23,
24]. The cluster source was housed in a source chamber (1 × 10
−7 torr) evacuated by a large diffusion pump (3000 L/s). Ions produced in the cluster source were transported to the ICR cell via three stages of differential pumping, each supplied with a turbomolecular pump to achieve ultrahigh vacuum (10
−10 torr) in the ICR cell. After exiting the clustering region, the ions were skimmed into an octopole ion guide (175 V
p-p, 1.8 MHz, 570 mm length) and immediately transferred to a second octopole for ion accumulation. Ions were confined radially in the accumulation octopole (240 V
p-p, 2.8 MHz, 160 mm) by a time-varying electric field generated by a radiofrequency applied with 180° phase difference to adjacent rods and axially by the application of positive voltages to the end caps at the conductance limits at either side of the accumulation octopole. Further experimental details are available in Ref. [
21], and for a review of SORI-CID techniques as applied to the activation of large ions, the reader is referred to Ref. [
25].
We detect only the positive ion distribution created by the totality of all possible sequences of neutral–neutral, neutral–charged and charged–charged processes that result in positively charged species that emanate from the nozzle orifice. The experimental setup allows us to extract the resultant molecular fragments either before or after intermolecular collisions have been allowed to occur.
All calculations were carried out based on the density functional theory (DFT) calculations under the local density approximation (LDA) using the AIMPRO code [
26,
27,
28]. Pseudo potentials given by Hartwigsen, Goedecker, and Hutter (HGH) [
29] were used. Wavefunctions are handled using a basis set of Gaussian type orbitals [
30,
31], in which 38 (12) independent functions and used for carbon (hydrogen) atoms. Charge density is handled using plane waves with a cut-off of 300 Ha. Spin polarisation was used in systems with an odd number of electrons (µ
B = 1) and averaged for systems with an even number of electrons. Since only positively charged species are measured experimentally, all calculations performed here are in the +1 charge state. Such charging is handled by introducing a uniform homogenous compensating background charge to the cell. Electron temperature is handled using a Fermi function with temperature of 0.04 eV to aid convergence. All molecules are modelled in large hexagonal cells with over 20 Å until the next molecule to ensure no interaction and to minimize the density of the homogenous background charge. Vibrational modes are determined from numerical energy double derivatives (0.2 au atom shifts). Structures were geometrically optimised until system energy was converged to better than 10
−7 Ha, and positions to within 10
−7 atomic units. Enthalpies (in eV) are quoted for hydrogen removal via
where E(n) is the total energy of species n in isolation. For the +1 charge state it is assumed that H
2 always remains neutral, i.e., the positive charge is carried by the nanoring in each case. Note that this approach uses H
2 as a reference state and does not necessarily imply hydrogen extraction as H
2.
3. Results
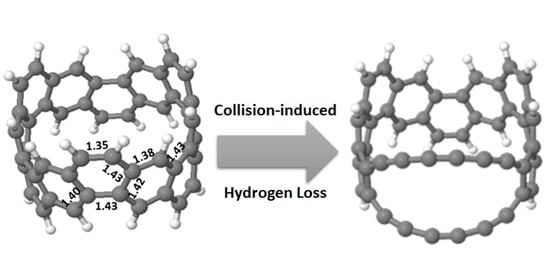
The insert of
Figure 1 shows the relaxed CNB (C
48H
24) structure. The ring has C
3 symmetry, with significant variation in C-C bond-length around the ring. In the +1 charge state the exterior C-C bonds have strong local double bond character (1.35 Å) while the interior bonds become increasing aromatic (1.38–1.43 Å); the neutral charge state is similar with slightly dilated interior bonds (up to 1.45 Å). There are two hydrogen types by symmetry alternating around the ring in pairs: “exterior” hydrogen atom pairs are on neighbouring sites of the same hexagon, and “interior” hydrogen atom pairs on opposing sites of a given hexagon (1.09 Å bond length).
Fragmentation of the C
48H
24 nanobelt after collision-induced dissociation is shown in detail in
Figure 1 (higher resolution details in
Supplementary Materials Figure S1), showing clearly that the primary loss species come from removal of nH
2, n = 1, …, 3. Similar intensity peaks correspond to removal of 3H
2 + n
1CH + n
2C
2, with (n
1 + n
2) ≤ 2, and increasingly weak signals for 2H
2 + CH and H
2 + CH loss. We are confident of the peak assignment since carbon isotopes can be readily distinguished due to the resolution of the spectrometer (
Figure S1). Molecular dissociation occurs following intermolecular collisions, since the two mass spectrometry figures in
Figure S2 show that exit species collected before collision do not show the presence of dissociated species in any notable amount.
We now turn to simulations to examine potential dehydrogenation routes.
Figure 2 shows three possible single dehydrogenation sites: exterior and “interior” hydrogenated sites (i.e., sites furthest and closest to the ring equator, respectively), and a third alternative where the structure in
Figure 2b undergoes a 90° C-C bond rotation about the bond centre (a Stone-Wales (SW) bond rotation), for the non-circumferential bond of the dehydrogenated carbon atom. The result is that two hexagons are replaced with a pentagon-heptagon pair, with the dehydrogenated carbon sitting in the heptagon. Single hydrogen atom removal is highly endothermic (+3.49 eV for the interior site, +3.67 eV exterior and +4.21 eV for the bond rotated structure) due to the creation of a dangling carbon bond.
We next remove a second hydrogen atom from all possible symmetrically distinct sites of these three structures, testing this way nearly 30 2H-loss structures. Second hydrogen removal is always much easier than the first due to the electronic closed shell produced from pairwise dehydrogenation (the energetic cost for the second dehydrogenation step is typically less than half that of the first). This is consistent with a picture of pairwise dehydrogenation as seen experimentally in
Figure 1.
Three structures show significantly lower energy than all others (
Figure 3). For the external dehydrogenation (
Figure 2a), the most stable C
48H
22 loses a neighbouring pair of external hydrogen atoms (
Figure 3a). The C-C bond between the dehydrogenated carbon neighbours stabilises the dangling bonds by rehybridising from a double (1.35 Å,
Figure 1) to a geometrically distorted triple bond (1.23 Å,
Figure 3a). This can be regarded as the shortest carbyne chain.
For interior dehydrogenation (
Figure 2b), the most stable structure has the two closest internal hydrogen atoms removed. In this case, the resultant dangling bonds point towards each other and are removed by forming a new single bond and creating a square (
Figure 3b). The three interior C-C bonds of the square remain at 1.41–1.42 Å, while the fourth bond, newly formed after dehydrogenation, is 1.52 Å, showing its single bond character. This rebonding creates a curved surface resembling locally the curved surface of a fullerene cage.
For the bond-rotated structure (
Figure 2c), the lowest energy dehydrogenation process is the mirror of the first, i.e., removal of the nearest internal hydrogen followed by another bond rotation (SW) on the other side of the heptagon. The result is a conversion from three hexagons in the initial nanoring structure, to pentagon-heptagon-hexagon for the first dehydrogenation step, to pentagon-octagon-pentagon for the second. The two dehydrogenated carbon atoms form the exposed side of the octagon, rehybridising to a carbyne chain with bonds of 1.24 and 1.34 Å (
Figure 3c).
Thus, we have three candidate structural routes for dehydrogenation resulting in either carbyne chains (
Figure 3a), cage closing towards fullerene structures (
Figure 3b) or carbyne chain formation linked with end pentagons (
Figure 3c). For 2H-loss, this latter SW-carbyne route (c) is the least stable and the fullerene route (b) the most stable, although the energetic difference is very small (+0.03 eV/H atom).
We now continue sequentially removing hydrogen atoms from each structure, at each step selecting the most stable structure for subsequent hydrogen removal. The full sequences are given in
Figure S3, a summary with key steps in
Figure 4.
For the “fullerene” closure route (
Figure 2b), the next step involves the formation of a second square on the opposing side of the ring (
Figure 4b). This localizes curvature at the two “ends” with squares, allowing the rest of the ring to flatten and increase aromaticity. However, thereafter there is no symmetrically obvious location for a third subsequent H
2 removal via this route, and instead we see a diversity of possible structures. Notably external hydrogen removal (as via the orange route) is energetically almost identical to the formation of further squares along the sides of the ring. Since this route is energetically much less stable than the alternatives, we do not pursue it further.
For the “carbyne” route starting with removal of two exterior hydrogen atoms (
Figure 2a), for 4H it becomes more stable to break a cross-linking C-C bond, allowing the structure to rehybridise and open out into two carbyne chains (
Figure 5a). While this process also results in strain relaxation in the ring with an increase in overall ring circumference, the two chains formed are not of equal length (containing two and four carbon atoms, respectively), which creates strong compression and tension, destabilising the structures. Thus, this route is also energetically relatively unfavourable. Bond length distribution shows a clear polyyne (single-triple) bonding in the shorter chain, with more cumulene-type bonding in the centre of the longer chain.
The third, SW-carbyne route (
Figure 2c,
Figure 3c and
Figure 4c) results in significantly more stable structures than the other routes. Although these are also carbyne chains (
Figure 4c), in this case the pentagon cross-bonding is maintained, the two carbyne chains are symmetric and contain only one C-C triply bound pair each (
Figure 5b). In particular, this stabilises removal of the third hydrogen atom (
Figure 4d), and as a result these structures are 0.7–0.8 eV more stable than the alternatives. The equivalent chain lengths avoid inter-chain strain while still allowing an increase in diameter, relieving curvature-induced strain in the rest of the ring. These structures form part of the family of strained dehydrobenzoannulenes [
32], organic species with cross-linking triple-bonded chains.
We note furthermore that these routes are not mutually exclusive. As marked in
Figure 4a, by rotating a C-C bond it is possible to shift from the “interior” fullerene route onto the SW-carbyne route. Thus, although the fullerene structures are energetically favoured for the initial hydrogen removal steps, they can easily divert to the more stable SW-carbyne structures upon further dehydrogenation.
As dehydrogenation continues (
Figure 4), this route always results in the most stable structures, extending the two carbyne chains symmetrically. While this process can in principle continue around the ring, after removal of six hydrogen atoms, the carbyne chains are already very long (4 carbon atoms each). From this point on, the system becomes increasingly fragile and susceptible to breakage during intermolecular collisions. When the ends of the chains break away this allows the nanorings to open out and will not be reversible. Thereafter the unbonded ends of the carbyne chains will easily fragment during collision, losing C
2 and CH species as seen experimentally (
Figure 1).
We have calculated the vibrational modes associated with the SW-Carbyne chain structures (
Figure 4c). With 2H removal (
Figure 3c) a triple bond stretch mode appears at 2060.4 cm
−1. This is slightly lower than the 2150 cm
−1 mode associated with triple bonds in ion-implanted graphite [
33] and is due to the local curvature and the +1 charge state in the current case, which will weaken the carbyne bonding and lead to frequency downshift. Long chain coupling, which can also downshift this mode, does not apply to the short carbyne chains in the current case [
34]. In the 4H-loss case, this mode splits into two at 2068.3 and 2033.7 cm
−1 due to the symmetry difference between the upper and lower chain, respectively (the lower lying more axially), showing weak vibronic coupling between the chains. In the 6H-loss case the chains are longer, the ring-induced curvature is less dominating and the +1 charge more distributed, and as a result the frequencies rise towards the conventional triple bond values, reaching 2080.8–2051.6 cm
−1. For the simple 2H-loss structure in
Figure 3a where no bond rotation has occurred, the triple bond is less linear, and its stretch frequency is only 2046 cm
−1. These frequencies are all higher than those seen during damage-induced merging of double walled nanotube bundles at 1855 cm
−1 ascribed to short linear carbon chains (3 to 7 atoms long) forming as bridging precursors to the merging of the tubes [
35]. This is likely because short chain bonding will be highly sensitive to the nature of the end connections to the host nanocarbon.
There are no strongly localized characteristic modes on the pentagons at the chain ends, their modes instead strongly coupled to the rest of the structure. Two peaks at 1283/1302 cm−1 show relatively strong pentagon character, but as these lie very close to conventional damage peaks in carbon materials such as the D-peak in graphite they cannot serve as indicators for the presence of local hydrogenated pentagons.
4. Discussion and Conclusions
Experimental collision-induced mass loss of laser-ablated gas-phase C48H24 nanorings occurs through the loss of nH2, n = 1–3, followed by subsequent loss of nC2 and/or nCH, n = 0–2. Theoretical modelling of dehydrogenation routes, selecting favoured structures based on their relative enthalpic stability, provides a complete picture that is fully consistent with the experimental collision-induced dissociation mass spectrometry data.
Initial pairwise hydrogen removal of up to six hydrogen atoms occurs through the formation of two parallel carbyne chains. In order to obtain parallel symmetric carbynes it is necessary to rotate carbon-carbon bonds at their ends, creating local pentagons that connect the chain pair to the rest of the nanoring. The carbyne chains stabilize the dangling bonds produced through dehydrogenation by rehybridising to triple bonds, with the added energetic advantage that the ring diameter can increase, relieving the internal curvature induced strain of the ring.
As dehydrogenation progresses and the chains become longer, intermolecular collisions break open the rings at the carbyne ends, and subsequent mass loss occurs through removal of C2 and CH from the chain ends. Alternative mass-loss routes, notably through introduction of further local curvature and closure towards fullerene-type structures, are energetically unfavourable beyond the first dehydrogenation step.
There are many literature examples of damage giving rise to both carbyne [
36], and fullerene-like structures. Electron irradiation of small carbon flakes on graphene produces vacancies and results in closure to fullerenes [
10], and metal templating also appears to catalyse fullerene formation during both growth and irradiation [
8,
21]. Carbyne formation is seen during irradiation damage of double-walled nanotube bundles [
35] and graphite [
33], and spontaneous short-lived chains are predicted to form during fullerene growth from small amorphous carbon clusters [
9]. A common argument for carbyne production during irradiation is constrained thinning, where carbon is removed at a junction between nanoobjects, resulting in an increasingly thin connecting region which stabilizes as a carbyne chain before eventually breaking [
37,
38]. The current study is a limiting example of this; rather than thinning the connection between two carbon nanoobjects, there is instead thinning of the connection between two ends of the same curved nanoobject. Curvature in the aromatic network induced by the ring structure is thus a critical factor, applying tension to the damaged zone and thereby favouring carbyne formation. Additionally, the two hydrogenated nanoring edges in close proximity provide an easy route to rehybridisation into dual carbyne chains once dehydrogenated. Wider rings with more hexagons along their axis may become harder to rehybridise, although we note that field emission experiments from carbon nanotube tips suggested indirect evidence of carbon chain formation [
39,
40].
Finally, we note that these structures with parallel attached carbyne chains form part of the family of strained dehydrobenzoannulenes [
32]. These molecules, of interest for their optoelectronic behaviour, are extremely difficult to synthesise through bottom-up organic chemistry routes, and the current “top-down” route of collision-induced dehydrogenation may be of interest as an alternative route to their production.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




