
DFT Study on the CO2 Reduction to C2 Chemicals Catalyzed by Fe and Co Clusters Supported on N-Doped Carbon


 ,
, 
Abstract
:1. Introduction
2. Theoretical Method
3. Results and Discussion
3.1. The STABILITY Analysis of Fen-NC
3.2. Electrocatalytic CO2RR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Birdja, Y.Y.; Pérez-Gallent, E.; Figueiredo, M.C.; Göttle, A.J.; Calle-Vallejo, F.; Koper, M.T.M. Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy 2019, 4, 732–745. [Google Scholar] [CrossRef]
- Wei, X.; Xiao, S.; Wu, R.; Zhu, Z.; Zhao, L.; Li, Z.; Wang, J.; Chen, J.S.; Wei, Z. Activating COOH* intermediate by Ni/Ni3ZnC0.7 heterostructure in porous N-doped carbon nanofibers for boosting CO2 electroreduction. Appl. Catal. B Environ. 2022, 302, 120861. [Google Scholar] [CrossRef]
- Li, Z.; Wu, R.; Xiao, S.; Yang, Y.; Lai, L.; Chen, J.S.; Chen, Y. Axial chlorine coordinated iron-nitrogen-carbon single-atom catalysts for efficient electrochemical CO2 reduction. Chem. Eng. J. 2022, 430, 132882. [Google Scholar] [CrossRef]
- Lu, S.; Wang, Y.; Xiang, H.; Lei, H.; Xu, B.B.; Xing, L.; Yu, E.H.; Liu, T.X. Mass transfer effect to electrochemical reduction of CO2: Electrode, electrocatalyst and electrolyte. J. Energy Storage 2022, 52, 104764. [Google Scholar] [CrossRef]
- Wu, Y.; Jiang, Z.; Lu, X.; Liang, Y.; Wang, H. Domino electroreduction of CO2 to methanol on a molecular catalyst. Nature 2019, 575, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ngoc Pham, T.; Tian, Y.; Morikawa, Y.; Yan, L. Density functional theory study on a nitrogen-rich carbon nitride material C3N5 as photocatalyst for CO2 reduction to C1 and C2 products. J. Colloid Interface Sci. 2021, 585, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Zhang, J. Architectural Design for Enhanced C2 Product Selectivity in Electrochemical CO2 Reduction Using Cu-Based Catalysts: A Review. ACS Nano 2021, 15, 7975–8000. [Google Scholar] [CrossRef]
- Wang, Y.; Lei, H.; Lu, S.; Yang, Z.; Xu, B.B.; Xing, L.; Liu, T.X. Cu2O nano-flowers/graphene enabled scaffolding structure catalyst layer for enhanced CO2 electrochemical reduction. Appl. Catal. B Environ. 2022, 305, 121022. [Google Scholar] [CrossRef]
- Lu, Q.; Rosen, J.; Jiao, F. Nanostructured Metallic Electrocatalysts for Carbon Dioxide Reduction. ChemCatChem 2015, 7, 38–47. [Google Scholar] [CrossRef]
- Mistry, H.; Choi, Y.W.; Bagger, A.; Scholten, F.; Bonifacio, C.S.; Sinev, I.; Divins, N.J.; Zegkinoglou, I.; Jeon, H.S.; Kisslinger, K.; et al. Enhanced Carbon Dioxide Electroreduction to Carbon Monoxide over Defect-Rich Plasma-Activated Silver Catalysts. Angew. Chem. Int. Ed. Engl. 2017, 56, 11394–11398. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Li, C.W.; Kanan, M.W. Aqueous CO2 reduction at very low overpotential on oxide-derived Au nanoparticles. J. Am. Chem. Soc. 2012, 134, 19969–19972. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Michalsky, R.; Metin, O.; Lv, H.; Guo, S.; Wright, C.J.; Sun, X.; Peterson, A.A.; Sun, S. Monodisperse Au nanoparticles for selective electrocatalytic reduction of CO2 to CO. J. Am. Chem. Soc. 2013, 135, 16833–16836. [Google Scholar] [CrossRef] [PubMed]
- Mistry, H.; Reske, R.; Zeng, Z.; Zhao, Z.J.; Greeley, J.; Strasser, P.; Cuenya, B.R. Exceptional size-dependent activity enhancement in the electroreduction of CO2 over Au nanoparticles. J. Am. Chem. Soc. 2014, 136, 16473–16476. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.; Bagger, A.; Hao, G.P.; Varela, A.S.; Sinev, I.; Bon, V.; Roldan Cuenya, B.; Kaskel, S.; Rossmeisl, J.; Strasser, P. Understanding activity and selectivity of metal-nitrogen-doped carbon catalysts for electrochemical reduction of CO2. Nat. Commun. 2017, 8, 944. [Google Scholar] [CrossRef]
- Bagger, A.; Ju, W.; Varela, A.S.; Strasser, P.; Rossmeisl, J. Single site porphyrine-like structures advantages over metals for selective electrochemical CO2 reduction. Catal. Today 2017, 288, 74–78. [Google Scholar] [CrossRef]
- Zu, X.; Li, X.; Liu, W.; Sun, Y.; Xu, J.; Yao, T.; Yan, W.; Gao, S.; Wang, C.; Wei, S.; et al. Efficient and Robust Carbon Dioxide Electroreduction Enabled by Atomically Dispersed Snδ+ Sites. Adv. Mater. 2019, 31, e1808135. [Google Scholar] [CrossRef]
- Gao, D.; Arán-Ais, R.M.; Jeon, H.S.; Roldan Cuenya, B. Rational catalyst and electrolyte design for CO2 electroreduction towards multicarbon products. Nat. Catal. 2019, 2, 198–210. [Google Scholar] [CrossRef]
- Cheng, M.J.; Clark, E.L.; Pham, H.H.; Bell, A.T.; Head-Gordon, M. Quantum Mechanical Screening of Single-Atom Bimetallic Alloys for the Selective Reduction of CO2 to C1 Hydrocarbons. ACS Catal. 2016, 6, 7769–7777. [Google Scholar] [CrossRef] [Green Version]
- Prieto, G. Carbon Dioxide Hydrogenation into Higher Hydrocarbons and Oxygenates: Thermodynamic and Kinetic Bounds and Progress with Heterogeneous and Homogeneous Catalysis. ChemSusChem 2017, 10, 1056–1070. [Google Scholar] [CrossRef]
- Alvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef]
- Jiang, K.; Sandberg, R.B.; Akey, A.J.; Liu, X.; Bell, D.C.; Nørskov, J.K.; Chan, K.; Wang, H. Metal ion cycling of Cu foil for selective C–C coupling in electrochemical CO2 reduction. Nat. Catal. 2018, 1, 111–119. [Google Scholar] [CrossRef]
- Li, X.; Yu, J.; Jaroniec, M.; Chen, X. Cocatalysts for Selective Photoreduction of CO2 into Solar Fuels. Chem. Rev. 2019, 119, 3962–4179. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.B.; De Luna, P.; Li, Y.F.; Dinh, C.-T.; Kim, D.; Yang, P.; Sargent, E.H. Designing materials for electrochemical carbon dioxide recycling. Nat. Catal. 2019, 2, 648–658. [Google Scholar] [CrossRef] [Green Version]
- Karapinar, D.; Huan, N.T.; Ranjbar Sahraie, N.; Li, J.; Wakerley, D.; Touati, N.; Zanna, S.; Taverna, D.; Galvao Tizei, L.H.; Zitolo, A.; et al. Electroreduction of CO2 on Single-Site Copper-Nitrogen-Doped Carbon Material: Selective Formation of Ethanol and Reversible Restructuration of the Metal Sites. Angew. Chem. Int. Ed. Engl. 2019, 58, 15098–15103. [Google Scholar] [CrossRef]
- Zhu, Y.; Yang, X.; Peng, C.; Priest, C.; Mei, Y.; Wu, G. Carbon-Supported Single Metal Site Catalysts for Electrochemical CO2 Reduction to CO and Beyond. Small 2021, 17, e2005148. [Google Scholar] [CrossRef]
- Imaoka, T.; Akanuma, Y.; Haruta, N.; Tsuchiya, S.; Ishihara, K.; Okayasu, T.; Chun, W.J.; Takahashi, M.; Yamamoto, K. Platinum clusters with precise numbers of atoms for preparative-scale catalysis. Nat. Commun. 2017, 8, 688. [Google Scholar] [CrossRef]
- Xu, J.; Li, R.; Zeng, R.; Li, Y.; Pan, Q.; Zhong, H.; Chen, J. Underpotential deposition of Cu clusters on sulfur and nitrogen Co-doped graphite foam for the oxygen reduction reaction. ChemElectroChem 2019, 6, 5682–5687. [Google Scholar] [CrossRef]
- Pei, W.; Zhou, S.; Zhao, J.; Xu, X.; Du, Y.; Dou, S.X. Immobilized trimeric metal clusters: A family of the smallest catalysts for selective CO2 reduction toward multi-carbon products. Nano Energy 2020, 76, 105049. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmiiller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B Condens. Matter. 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmiiller, J. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B Condens. Matter. 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Logadottir, A.; Nørskov, J.K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 2005, 319, 178–184. [Google Scholar] [CrossRef]
- Peterson, A.A.; Abild-Pedersen, F.; Felix Studt, F.; Rossmeisl, J.; Nørskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Wang, Y.; Yuan, H.; Li, Y.; Chen, Z. Two-dimensional iron-phthalocyanine (Fe-Pc) monolayer as a promising single-atom-catalyst for oxygen reduction reaction: A computational study. Nanoscale 2015, 7, 11633–11641. [Google Scholar] [CrossRef]
- Lim, D.H.; Wilcox, J. Mechanisms of the Oxygen Reduction Reaction on Defective Graphene-Supported Pt Nanoparticles from First-Principles. J. Phys. Chem. C 2012, 116, 3653–3660. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.; Zhang, H.; Luo, Z. Nitrogen reduction reaction on small iron clusters supported by N-doped graphene: A theoretical study of the atomically precise active-site mechanism. Nano Res. 2020, 13, 2280–2288. [Google Scholar] [CrossRef]
- Li, X.F.; Li, Q.K.; Cheng, J.; Liu, L.; Yan, Q.; Wu, Y.; Zhang, X.H.; Wang, Z.Y.; Qiu, Q.; Luo, Y. Conversion of Dinitrogen to Ammonia by FeN3-Embedded Graphene. J. Am. Chem. Soc. 2016, 138, 8706–8709. [Google Scholar] [CrossRef]
- Xiao, H.; Goddard, W.A., 3rd; Cheng, T.; Liu, Y. Cu metal embedded in oxidized matrix catalyst to promote CO2 activation and CO dimerization for electrochemical reduction of CO2. Proc. Natl. Acad. Sci. USA 2017, 114, 6685–6688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Che, F.; Liu, M.; Zou, C.; Liang, Z.; De Luna, P.; Yuan, H.; Li, J.; Wang, Z.; Xie, H.; et al. Dopant-induced electron localization drives CO2 reduction to C2 hydrocarbons. Nat. Chem. 2018, 10, 974–980. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, B.; Zhu, J.; Wang, L.; Ou, H.; Zhang, Z.; Liang, X.; Zheng, L.; Zhou, L.; Su, Y.Q.; et al. Lewis Acid Site-Promoted Single-Atomic Cu Catalyzes Electrochemical CO2 Methanation. Nano Lett. 2021, 21, 7325–7331. [Google Scholar] [CrossRef] [PubMed]
- Varela, A.S.; Ranjbar Sahraie, N.; Steinberg, J.; Ju, W.; Oh, H.S.; Strasser, P. Metal-Doped Nitrogenated Carbon as an Efficient Catalyst for Direct CO2 Electroreduction to CO and Hydrocarbons. Angew. Chem Int. Ed. Engl. 2015, 54, 10758–10762. [Google Scholar] [CrossRef] [PubMed]

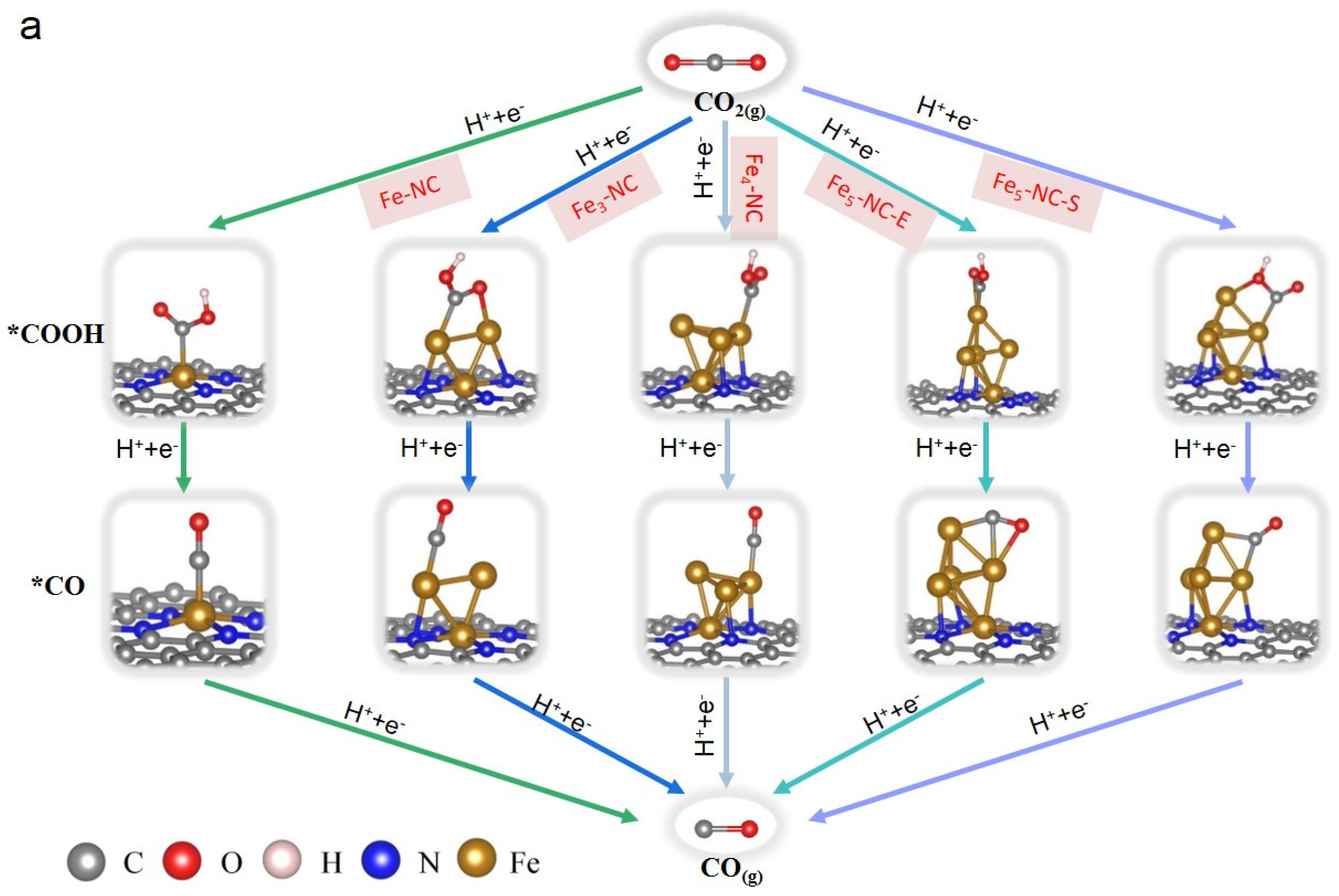



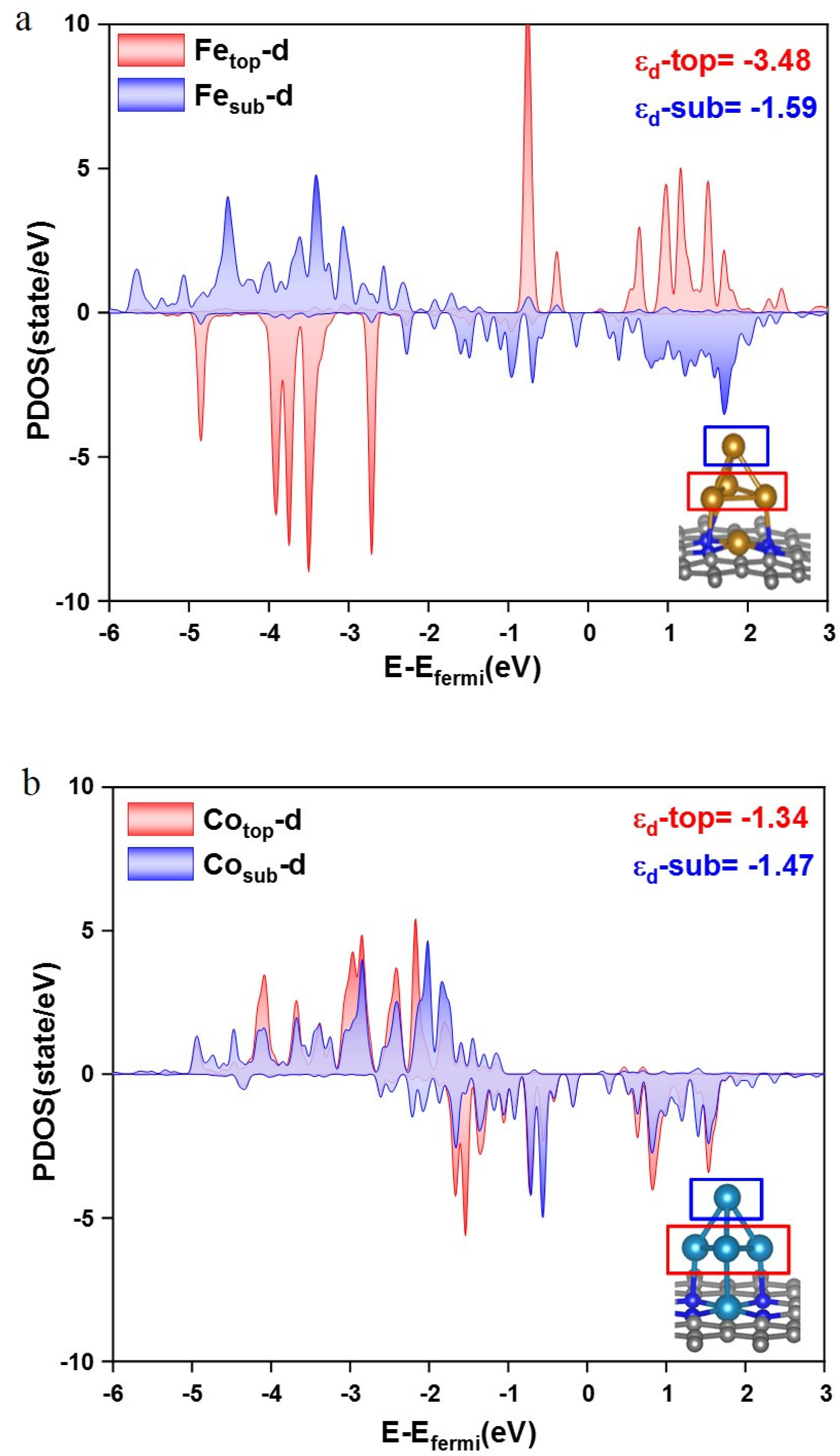

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | * Bader Charge (e) | Eb (eV) | Ec (eV) | ||||
|---|---|---|---|---|---|---|---|
| Fe 1site | Fe 2site | Fe 3site | Fe 4site | Fe 5site | |||
| Fe1-NC | −1.07 | - | - | - | - | −9.01 | 0 |
| Fe3-NC | −0.80 | −0.32 | −0.31 | - | - | −9.73 | −0.35 |
| Fe4-NC | −0.81 | −0.34 | −0.33 | −0.19 | - | −9.79 | −0.62 |
| Fe5-NC | −0.81 | −0.36 | −0.35 | −0.28 | −0.01 | −8.34 | −1.01 |
| Catalyst | Bader Charge (e) | Eb (eV) | Ec (eV) | ||||
|---|---|---|---|---|---|---|---|
| Co 1site | Co 2site | Co 3site | Co 4site | Co 5site | |||
| Co5-NC | −0.71 | −0.34 | −0.31 | −0.24 | 0.06 | −10.00 | −1.39 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, Q.; Qi, X.; Yang, T.; Jiang, J.; Zhou, Q.; Fu, C.; Yang, N. DFT Study on the CO2 Reduction to C2 Chemicals Catalyzed by Fe and Co Clusters Supported on N-Doped Carbon. Nanomaterials 2022, 12, 2239. https://doi.org/10.3390/nano12132239
Xue Q, Qi X, Yang T, Jiang J, Zhou Q, Fu C, Yang N. DFT Study on the CO2 Reduction to C2 Chemicals Catalyzed by Fe and Co Clusters Supported on N-Doped Carbon. Nanomaterials. 2022; 12(13):2239. https://doi.org/10.3390/nano12132239
Chicago/Turabian StyleXue, Qian, Xueqiang Qi, Tingting Yang, Jinxia Jiang, Qi Zhou, Chuang Fu, and Na Yang. 2022. "DFT Study on the CO2 Reduction to C2 Chemicals Catalyzed by Fe and Co Clusters Supported on N-Doped Carbon" Nanomaterials 12, no. 13: 2239. https://doi.org/10.3390/nano12132239
APA StyleXue, Q., Qi, X., Yang, T., Jiang, J., Zhou, Q., Fu, C., & Yang, N. (2022). DFT Study on the CO2 Reduction to C2 Chemicals Catalyzed by Fe and Co Clusters Supported on N-Doped Carbon. Nanomaterials, 12(13), 2239. https://doi.org/10.3390/nano12132239