
The Cost of Improving the Precision of the Variational Quantum Eigensolver for Quantum Chemistry

 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
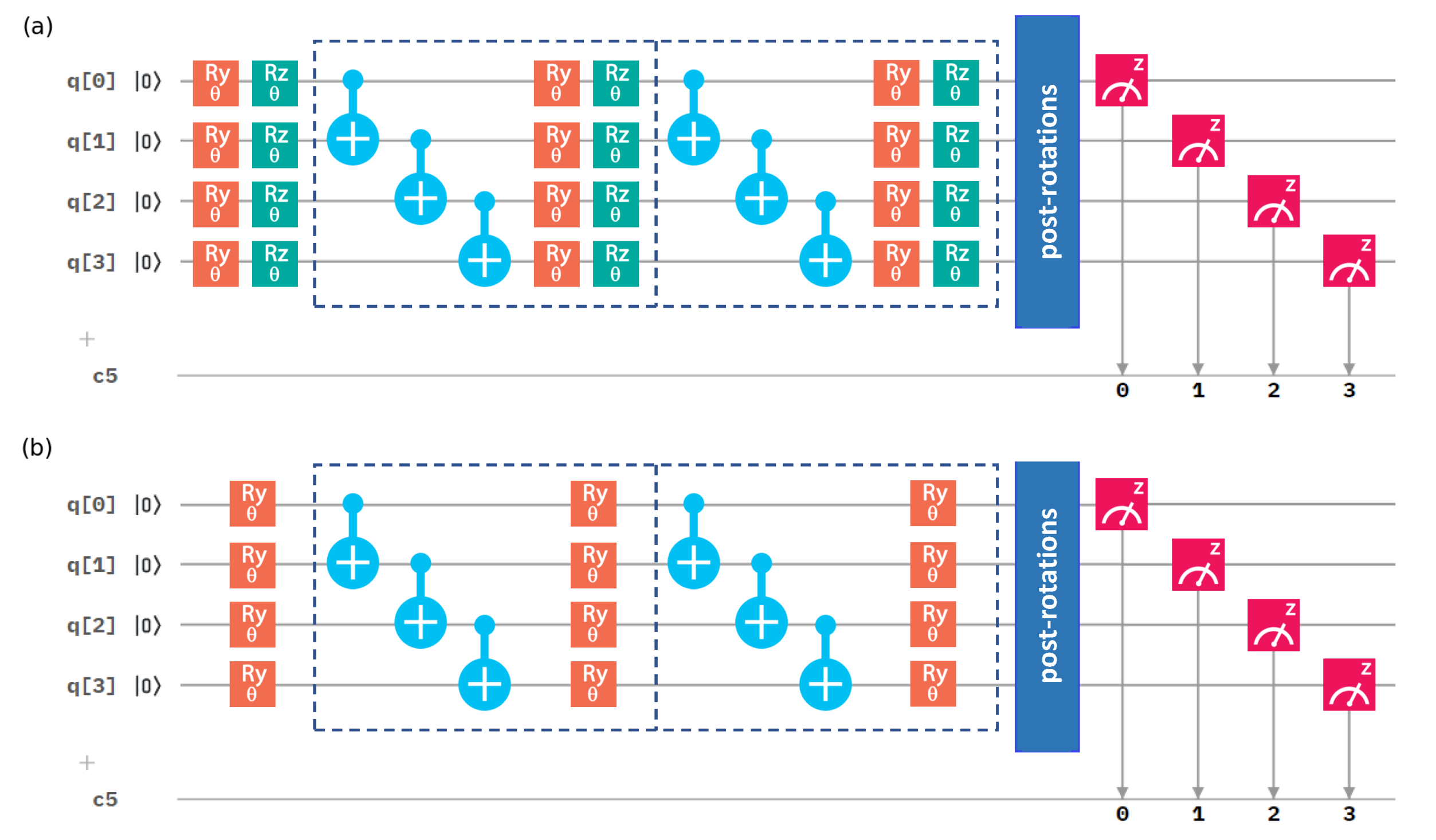
2. Methods
- Quantum: prepare a parametrized quantum state on a quantum device;
- Quantum: measure each Hamiltonian term (requires repetitions of step 1);
- Classical: sum the expectation values of the Hamiltonian terms to estimate the energy of the parametrized state;
- Classical: use the energy value to update the parameters of the trial quantum state.
3. Results and Discussion
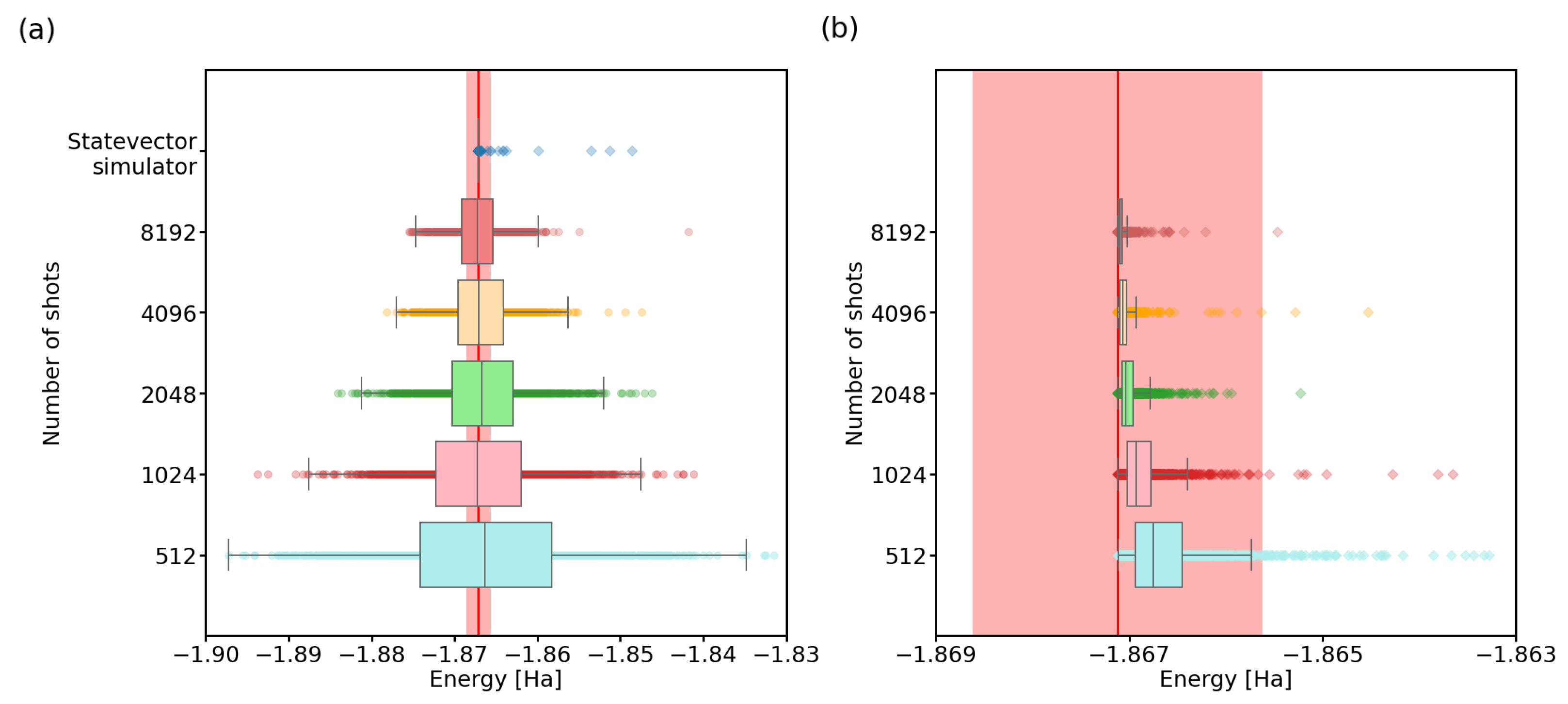
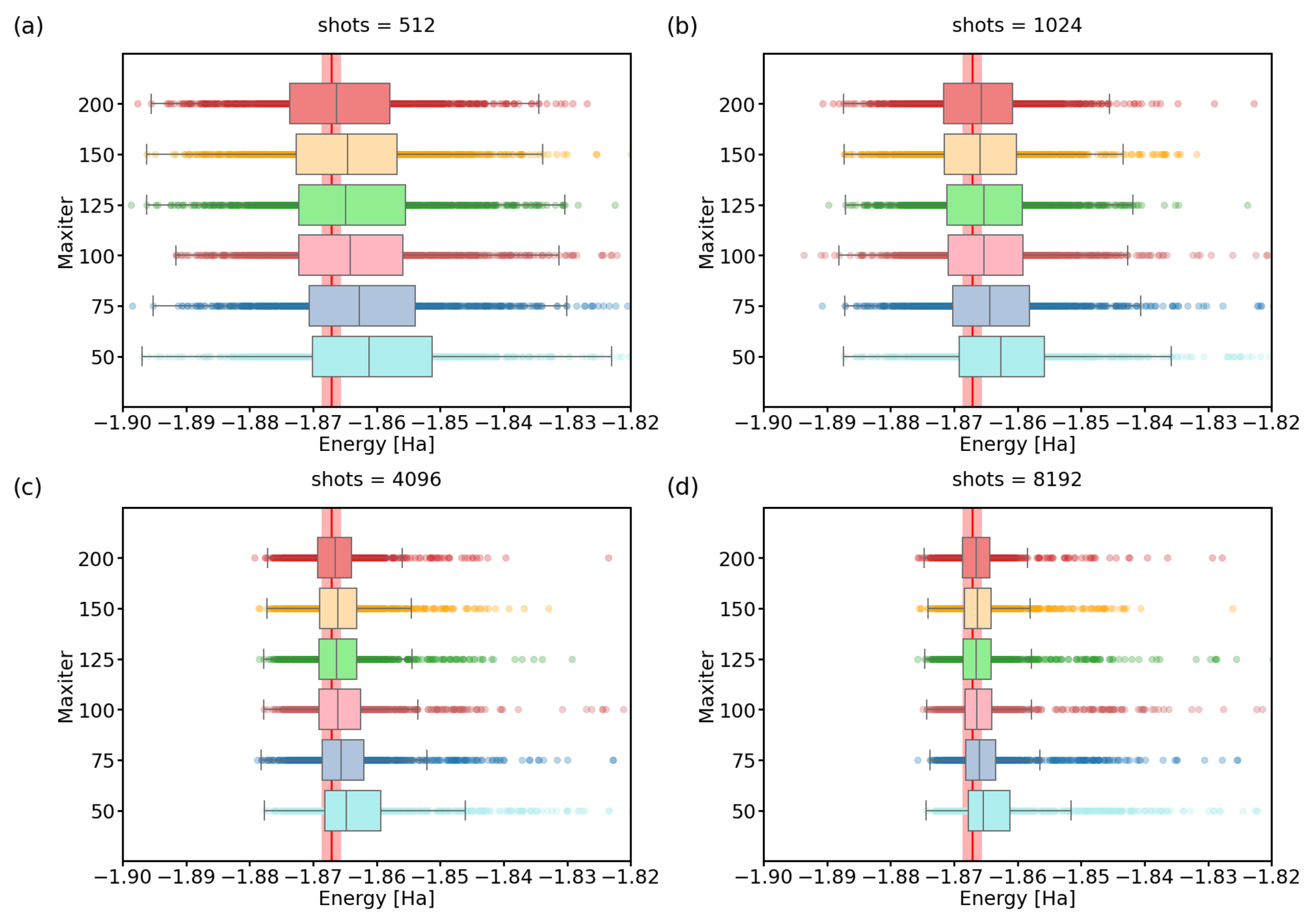
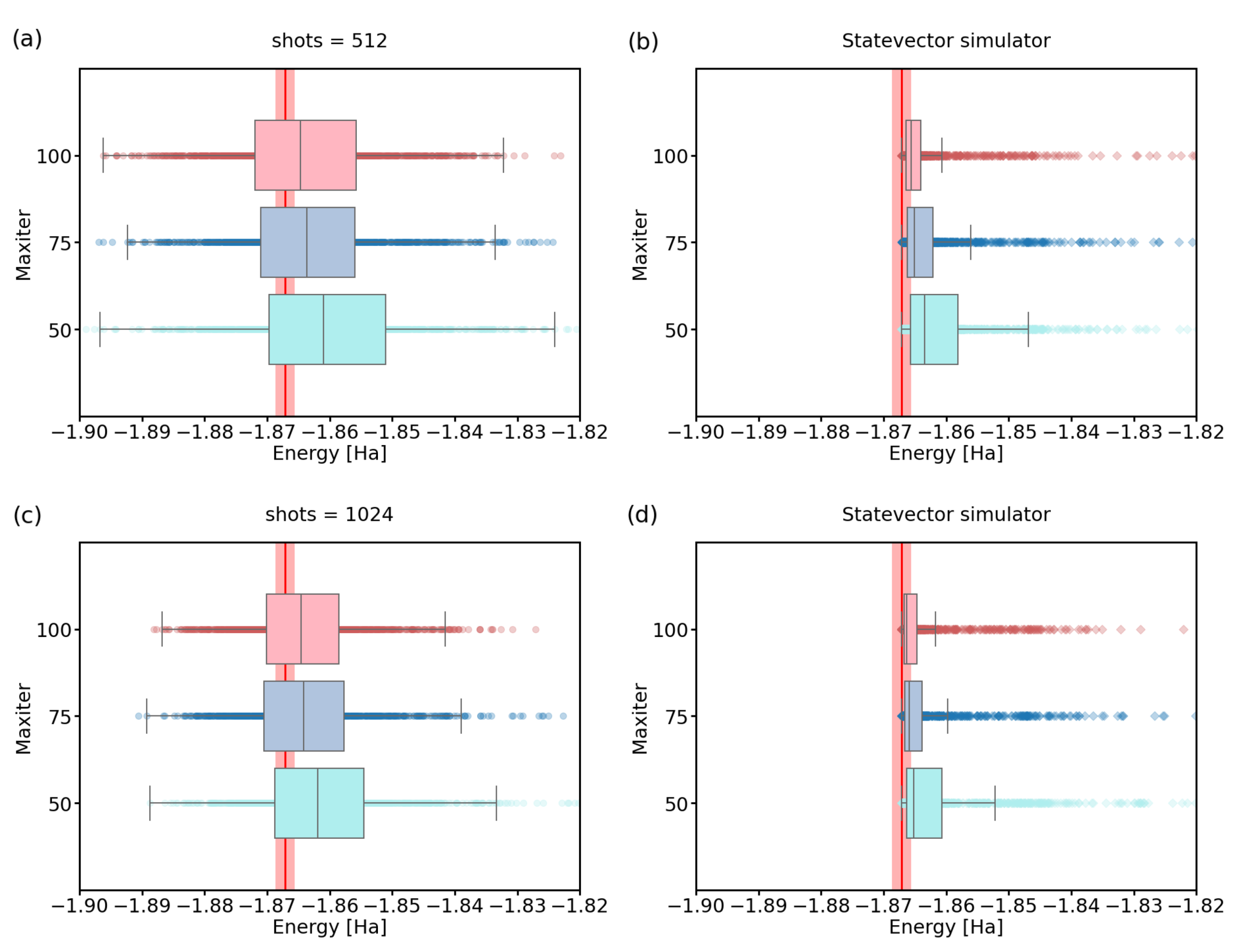
3.1. Number of Quantum Computer Calls
3.2. Choice of Hamiltonian and State-Preparation Ansatz
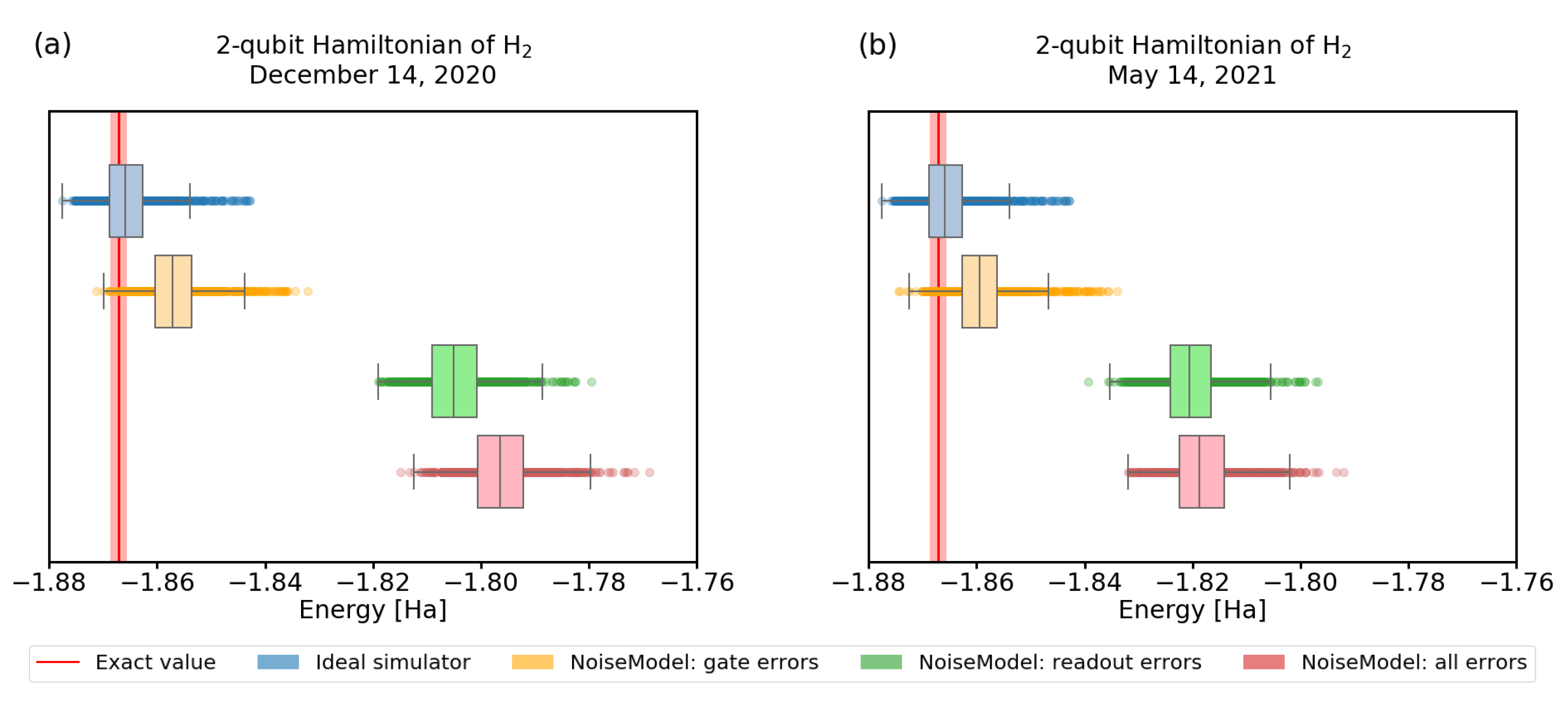
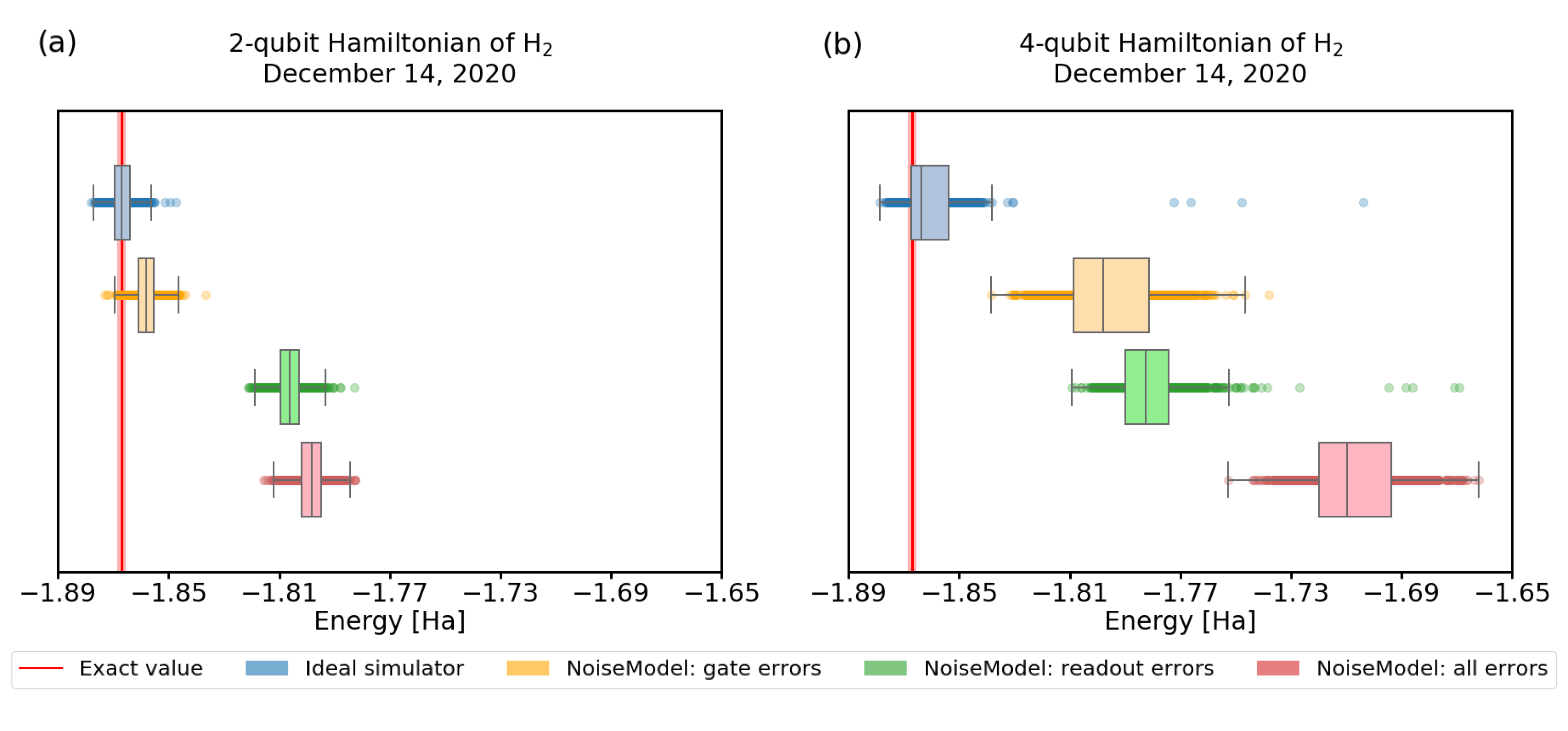
3.3. Imperfect Quantum Devices
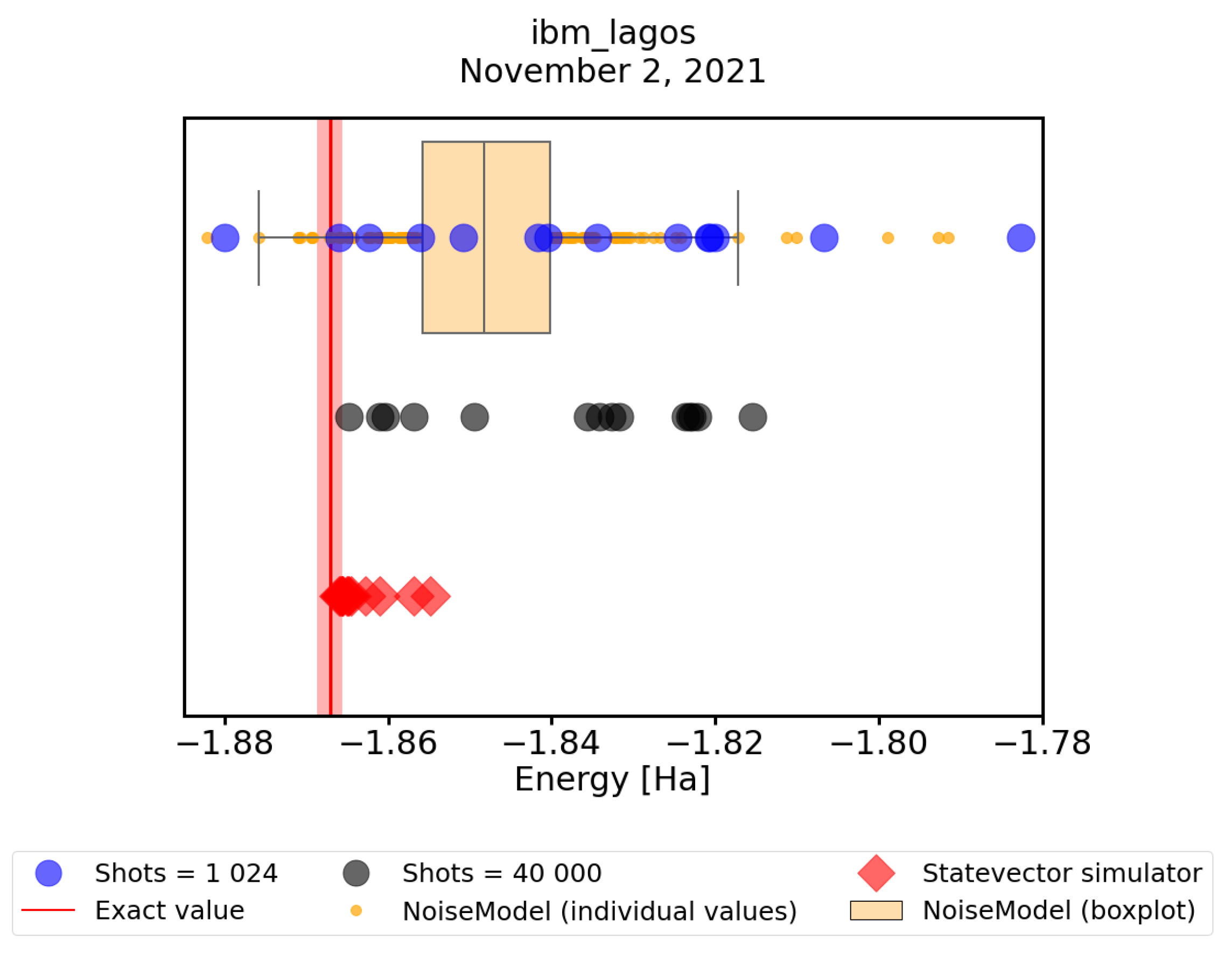
3.4. Convergence of VQE on Real Quantum Hardware
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| VQE | Variational Quantum Eigensolver |
| STO-3G | Slater-type Orbital basis set with each orbital expanded into 3 Gaussian functions. |
| 6-31G | A double-zeta basis set, which uses six primitive Gaussians to describe each core |
| atomic orbital. Further, each valence orbital is described by two basis functions. | |
| The first one is composed of a linear combination of 3 primitive Gaussian functions | |
| and the other one is composed of a single primitive Gaussian function. | |
| SPSA | Simultaneous Perturbation Stochastic Approximation |
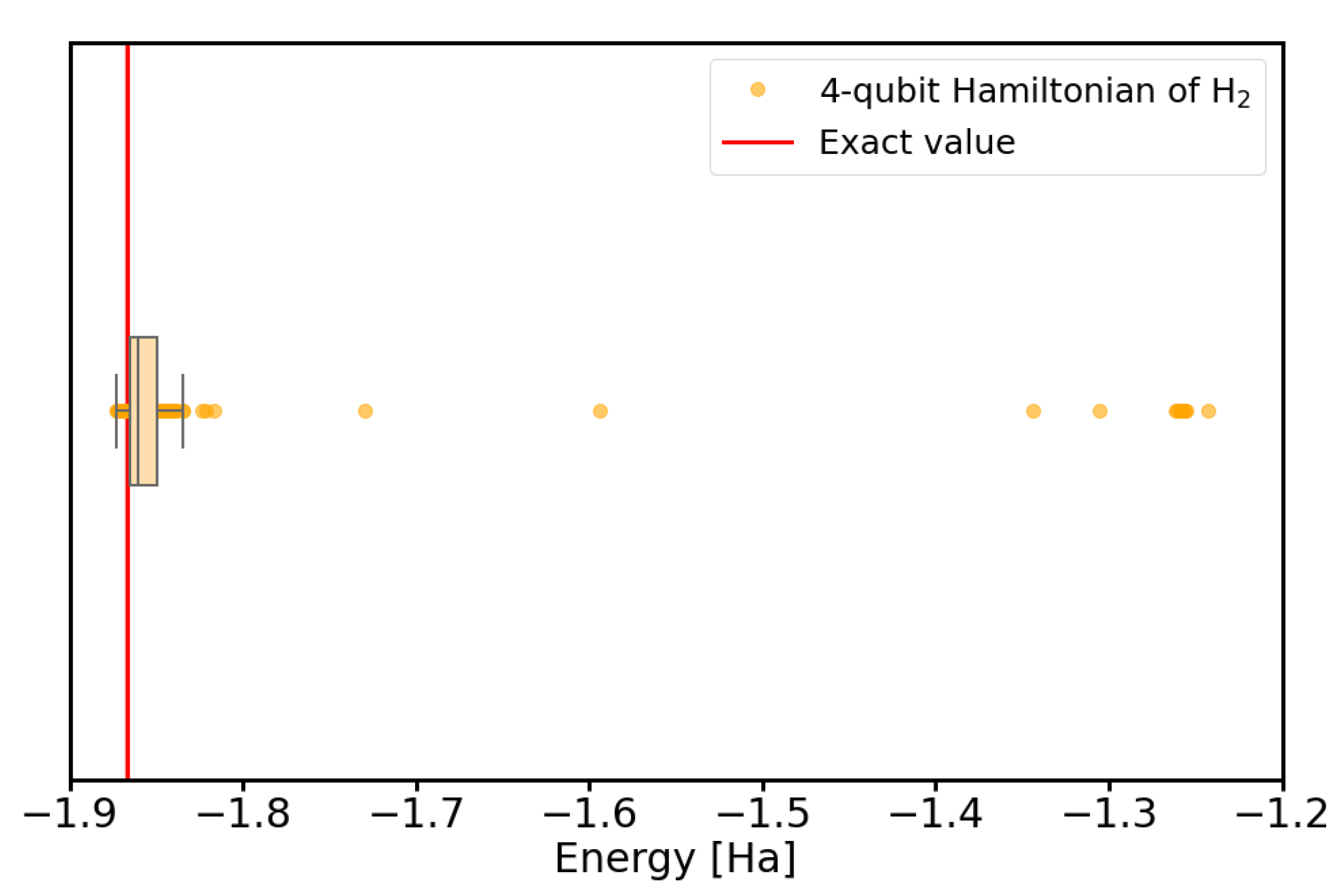
Appendix A. Convergence to Local Minima


Appendix B. Data Tables

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
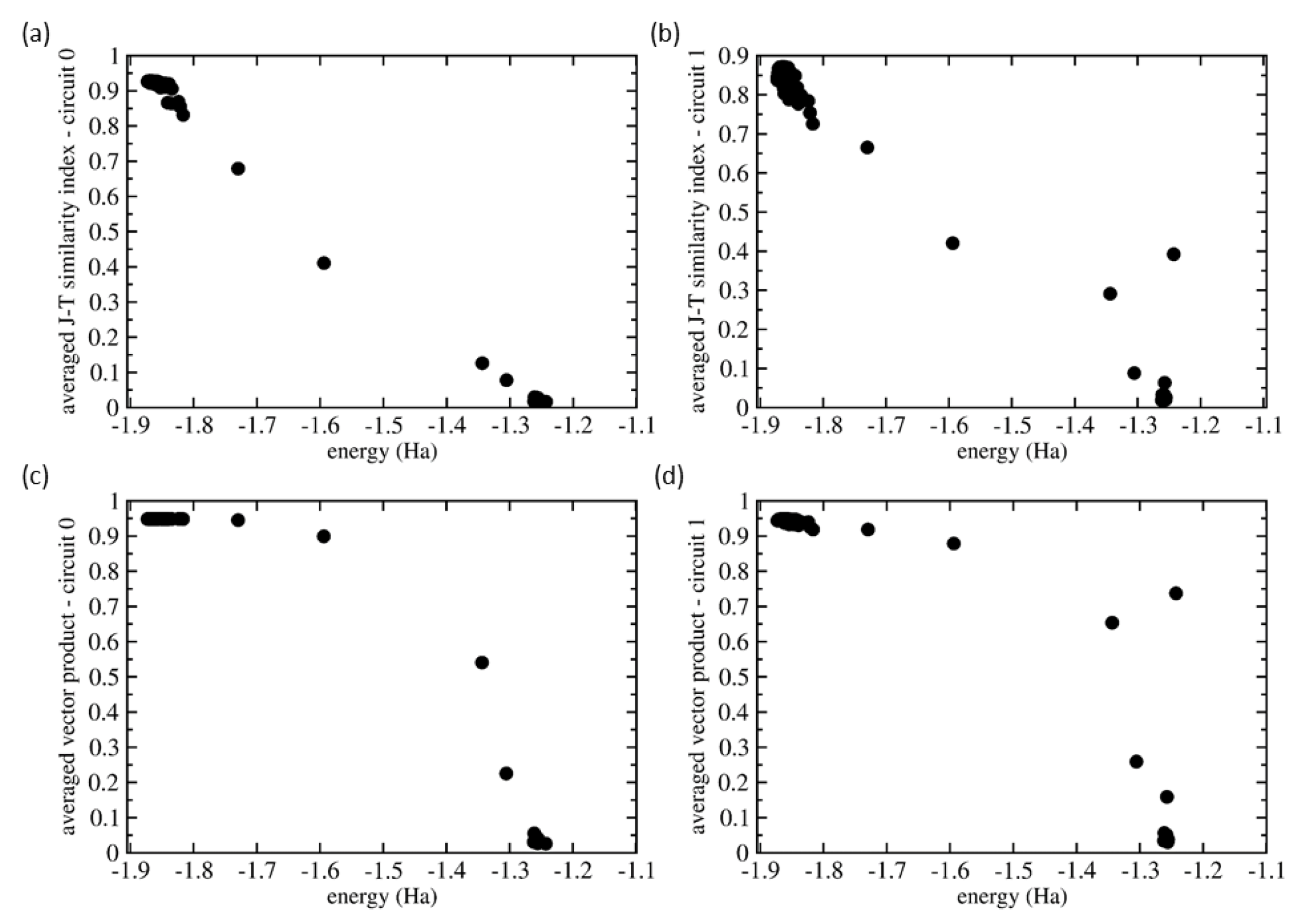{kind=link}
| Simulator | Shots | Median [Ha] | % | Recalculation | |
|---|---|---|---|---|---|
| Median [Ha] | % | ||||
| qasm_simulator | 512 | ||||
| 1024 | |||||
| 2048 | |||||
| 4096 | |||||
| 8192 | |||||
| statevector_simulator | − | − | − | ||
| Settings | shots | Median [Ha] | % |
|---|---|---|---|
| SPSA(maxiter = 50) | 512 | ||
| 1024 | |||
| 4096 | |||
| 8192 | |||
| SPSA(maxiter = 75) | 512 | ||
| 1024 | |||
| 4096 | |||
| 8192 | |||
| SPSA(maxiter = 100) | 512 | ||
| 1024 | |||
| 4096 | |||
| 8192 | |||
| SPSA(maxiter = 125) | 512 | ||
| 1024 | |||
| 4096 | |||
| 8192 | |||
| SPSA(maxiter = 150) | 512 | ||
| 1024 | |||
| 4096 | |||
| 8192 | |||
| SPSA(maxiter = 200) | 512 | ||
| 1024 | |||
| 4096 | |||
| 8192 |
| Settings | Shots | Median [Ha] | % | Recalculation | |
|---|---|---|---|---|---|
| Median [Ha] | % | ||||
| SPSA(maxiter = 50) | 512 | ||||
| 1024 | |||||
| SPSA(maxiter = 75) | 512 | ||||
| 1024 | |||||
| SPSA(maxiter = 100) | 512 | ||||
| 1024 | |||||
| 2-Qubit Hamiltonian H | |||
|---|---|---|---|
| Form | Depth (Parameters) | Median [Ha] | % |
| 3 | |||
| 2 | |||
| 2 | |||
| 1 | |||
| 1 | |||
| 4-qubit Hamiltonian H | |||
| Form | Depth (Parameters) | Median [Ha] | % |
| 5 | |||
| 2 | |||
| 2 | |||
| 1 | |||
| 1 | |||
| Simulator NoiseModel | Qubits | Date | R Form | Form | ||
|---|---|---|---|---|---|---|
| Median [Ha] | % | Median [Ha] | % | |||
| gate errors | 2 | 14 December 2020 | ||||
| 14 May 2021 | ||||||
| 4 | 14 December 2020 | |||||
| readout errors | 2 | 14 December 2020 | 0.0 | |||
| 14 May 2021 | ||||||
| 4 | 14 December 2020 | |||||
| all errors | 2 | 14 December 2020 | ||||
| 14 May 2021 | ||||||
| 4 | 14 December 2020 | |||||
| ibm_lagos | Statevector Simulator | |
|---|---|---|
| shots = 1024 | shots = 40,000 | |
| Ha | Ha | Ha |
| Ha | Ha | Ha |
| Ha | Ha | Ha |
| Ha | Ha | Ha |
| Ha | Ha | Ha |
| Ha | Ha | Ha |
| Ha | Ha | Ha |
| Ha | Ha | Ha |
| Ha | Ha | Ha |
| Ha | Ha | Ha |
| Ha | Ha | Ha |
| Ha | Ha | Ha |
| Ha | Ha | Ha |
| Ha | Ha | Ha |
References
- Baiardi, A.; Reiher, M. The density matrix renormalization group in chemistry and molecular physics: Recent developments and new challenges. J. Chem. Phys. 2020, 152, 040903. [Google Scholar] [CrossRef] [Green Version]
- Bowler, D.R.; Miyazaki, T. O(N) methods in electronic structure calculations. Rep. Prog. Phys. 2012, 75, 036503. [Google Scholar] [CrossRef]
- Montanaro, A. Quantum speedup of Monte Carlo methods: Table 1. Proc. R. Soc. A Math. Phys. Eng. Sci. 2015, 471. [Google Scholar] [CrossRef] [Green Version]
- Friesner, R.A. Ab initio quantum chemistry: Methodology and applications. Proc. Natl. Acad. Sci. USA 2005, 102, 6648–6653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helgaker, T.; Klopper, W.; Tew, D.P. Quantitative quantum chemistry. Mol. Phys. 2008, 106, 2107–2143. [Google Scholar] [CrossRef]
- Cremer, D. Møller–Plesset perturbation theory: From small molecule methods to methods for thousands of atoms. WIREs Comput. Mol. Sci. 2011, 1, 509–530. [Google Scholar] [CrossRef]
- Lyakh, D.I.; Musiał, M.; Lotrich, V.F.; Bartlett, R.J. Multireference Nature of Chemistry: The Coupled-Cluster View. Chem. Rev. 2012, 112, 182–243. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.S.; Li, S.L.; Truhlar, D.G. Perspective: Kohn-Sham density functional theory descending a staircase. J. Chem. Phys. 2016, 145, 130901. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Feynman, R. Simulating physics with computers. Int. J. Theor. Phys. 1982, 21, 467–488. [Google Scholar] [CrossRef]
- Jordan, S. Quantum Algorithm ZOO. Available online: https://quantumalgorithmzoo.org/ (accessed on 11 November 2021).
- Abhijith, J.; Adedoyin, A.; Ambrosiano, J.; Anisimov, P.; Bärtschi, A.; Casper, W.; Chennupati, G.; Coffrin, C.; Djidjev, H.; Gunter, D.; et al. Quantum Algorithm Implementations for Beginners. arXiv 2018, arXiv:1804.03719. [Google Scholar]
- Nielsen, M.A.; Chuang, I.L. Quantum Computation and Quantum Information: 10th Anniversary Edition, 10th ed.; Cambridge University Press: Cambridge, CA, USA, 2011. [Google Scholar]
- Degen, C.L.; Reinhard, F.; Cappellaro, P. Quantum sensing. Rev. Mod. Phys. 2017, 89, 035002. [Google Scholar] [CrossRef] [Green Version]
- Gisin, N.; Ribordy, G.; Tittel, W.; Zbinden, H. Quantum cryptography. Rev. Mod. Phys. 2002, 74, 145–195. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Ma, X.; Zhang, Q.; Lo, H.K.; Pan, J.W. Secure quantum key distribution with realistic devices. Rev. Mod. Phys. 2020, 92, 025002. [Google Scholar] [CrossRef]
- Herrero-Collantes, M.; Garcia-Escartin, J.C. Quantum random number generators. Rev. Mod. Phys. 2017, 89, 015004. [Google Scholar] [CrossRef] [Green Version]
- Arute, F.; Arya, K.; Babbush, R.; Bacon, D.; Bardin, J.C.; Barends, R.; Biswas, R.; Boixo, S.; Brandao, F.G.S.L.; Buell, D.A.; et al. Quantum supremacy using a programmable superconducting processor. Nature 2019, 574, 505–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Bao, W.S.; Cao, S.; Chen, F.; Chen, M.C.; Chen, X.; Chung, T.H.; Deng, H.; Du, Y.; Fan, D.; et al. Strong Quantum Computational Advantage Using a Superconducting Quantum Processor. Phys. Rev. Lett. 2021, 127, 180501. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.S.; Deng, Y.H.; Qin, J.; Wang, H.; Chen, M.C.; Peng, L.C.; Luo, Y.H.; Wu, D.; Gong, S.Q.; Su, H.; et al. Phase-Programmable Gaussian Boson Sampling Using Stimulated Squeezed Light. Phys. Rev. Lett. 2021, 127, 180502. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Romero, J.; Olson, J.P.; Degroote, M.; Johnson, P.D.; Kieferová, M.; Kivlichan, I.D.; Menke, T.; Peropadre, B.; Sawaya, N.P.D.; et al. Quantum Chemistry in the Age of Quantum Computing. Chem. Rev. 2019, 119, 10856–10915. [Google Scholar] [CrossRef] [Green Version]
- Peruzzo, A.; McClean, J.; Shadbolt, P.; Yung, M.H.; Zhou, X.Q.; Love, P.; Aspuru-Guzik, A.; O’Brien, J. A variational eigenvalue solver on a photonic quantum processor. Nat. Commun. 2014, 5, 4213. [Google Scholar] [CrossRef] [Green Version]
- O’Malley, P.J.J.; Babbush, R.; Kivlichan, I.D.; Romero, J.; McClean, J.R.; Barends, R.; Kelly, J.; Roushan, P.; Tranter, A.; Ding, N.; et al. Scalable Quantum Simulation of Molecular Energies. Phys. Rev. X 2016, 6, 031007. [Google Scholar] [CrossRef]
- Bloch, F. Generalized theory of relaxation. Phys. Rev. 1957, 105, 1206–1222. [Google Scholar] [CrossRef]
- Krantz, P.; Kjaergaard, M.; Yan, F.; Orlando, T.P.; Gustavsson, S.; Oliver, W.D. A quantum engineer’s guide to superconducting qubits. Appl. Phys. Rev. 2019, 6, 021318. [Google Scholar] [CrossRef]
- Preskill, J. Quantum Computing in the NISQ era and beyond. Quantum 2018, 2, 79. [Google Scholar] [CrossRef]
- Kandala, A.; Mezzacapo, A.; Temme, K.; Takita, M.; Brink, M.; Chow, J.M.; Gambetta, J.M. Hardware-efficient variational quantum eigensolver for small molecules and quantum magnets. Nature 2017, 549, 242–246. [Google Scholar] [CrossRef]
- Kandala, A.; Temme, K.; Córcoles, A.D.; Mezzacapo, A.; Chow, J.M.; Gambetta, J.M. Error mitigation extends the computational reach of a noisy quantum processor. Nature 2019, 567, 491–495. [Google Scholar] [CrossRef] [Green Version]
- Temme, K.; Bravyi, S.; Gambetta, J.M. Error Mitigation for Short-Depth Quantum Circuits. Phys. Rev. Lett. 2017, 119, 180509. [Google Scholar] [CrossRef] [Green Version]
- Endo, S.; Benjamin, S.C.; Li, Y. Practical Quantum Error Mitigation for Near-Future Applications. Phys. Rev. X 2018, 8, 031027. [Google Scholar] [CrossRef] [Green Version]
- Geller, M.R.; Sun, M. Toward efficient correction of multiqubit measurement errors: Pair correlation method. Quantum Sci. Technol. 2021, 6, 025009. [Google Scholar] [CrossRef]
- Bravyi, S.; Sheldon, S.; Kandala, A.; Mckay, D.C.; Gambetta, J.M. Mitigating measurement errors in multiqubit experiments. Phys. Rev. A 2021, 103. [Google Scholar] [CrossRef]
- Nachman, B.; Urbanek, M.; de Jong, W.A.; Bauer, C.W. Unfolding quantum computer readout noise. NPJ Quantum Inf. 2020, 6, Art. [Google Scholar] [CrossRef]
- Maciejewski, F.B.; Zimborás, Z.; Oszmaniec, M. Mitigation of readout noise in near-term quantum devices by classical post-processing based on detector tomography. Quantum 2020, 4, 257. [Google Scholar] [CrossRef]
- Cai, Z. Quantum Error Mitigation using Symmetry Expansion. Quantum 2021, 5, 548. [Google Scholar] [CrossRef]
- Suchsland, P.; Tacchino, F.; Fischer, M.H.; Neupert, T.; Barkoutsos, P.K.; Tavernelli, I. Algorithmic Error Mitigation Scheme for Current Quantum Processors. Quantum 2021, 5, 492. [Google Scholar] [CrossRef]
- McArdle, S.; Endo, S.; Aspuru-Guzik, A.; Benjamin, S.; Yuan, X. Quantum computational chemistry. Rev. Mod. Phys. 2020, 92, 15003. [Google Scholar] [CrossRef] [Green Version]
- Bravyi, S.; Gambetta, J.M.; Mezzacapo, A.; Temme, K. Tapering off qubits to simulate fermionic Hamiltonians. arXiv 2017, arXiv:1701.08213. [Google Scholar]
- Bravyi, S.; Kitaev, A. Fermionic quantum computation. Ann. Phys. 2002, 298, 210–226. [Google Scholar] [CrossRef] [Green Version]
- Jordan, P.; Wigner, E. Über das Paulische Äquivalenzverbot. Z. Phys. 1928, 47, 631–651. [Google Scholar] [CrossRef]
- Qiskit: An Open-Source Framework for Quantum Computing. Available online: https://qiskit.org/ (accessed on 4 January 2021).
- Miháliková, I. Implementation of the Variational Quantum Eigensolver. 2021. Available online: https://github.com/imihalik/VQE_H2 (accessed on 11 November 2021).
- VQE Tutorial. Available online: https://pennylane.ai/qml/demos/tutorial_vqe.html (accessed on 1 November 2021).
- Qiskit’s Chemistry Module. Available online: https://qiskit.org/documentation/apidoc/qiskit_chemistry.html (accessed on 4 January 2021).
- Spall, J.C. Multivariate Stochastic Approximation Using a Simultaneous Perturbation Gradient Approximation. IEEE Trans. Autom. Control 1992, 37, 332–341. [Google Scholar] [CrossRef] [Green Version]
- Spall, J. Implementation of the simultaneous perturbation algorithm for stochastic optimization. IEEE Trans. Aerosp. Electron. Syst. 1998, 34, 817–823. [Google Scholar] [CrossRef]
- Spall, J.C. An Overview of the Simultaneous Perturbation Method for Efficient Optimization. Johns Hopkins Apl Tech. Dig. 1998, 19, 482–492. [Google Scholar]
- Havlíček, V.; Córcoles, A.; Temme, K.; Harrow, A.; Kandala, A.; Chow, J.; Gambetta, J. Supervised learning with quantum-enhanced feature spaces. Nature 2019, 567, 209–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IBM Quantum. Available online: https://quantum-computing.ibm.com/ (accessed on 4 November 2021).
- Least Square Fitting Error Mitigation. Available online: https://qiskit.org/textbook/ch-quantum-hardware/measurement-error-mitigation.html (accessed on 2 November 2021).
- Amazon Braket Pricing. Available online: https://aws.amazon.com/braket/pricing/ (accessed on 1 November 2021).
- Tukey, J.W. Exploratory Data Analysis; Addison-Wesley: Boston, MA, USA, 1977. [Google Scholar]
- Letham, B.; Karrer, B.; Ottoni, G.; Bakshy, E. Constrained Bayesian Optimization with Noisy Experiments. arXiv 2017, arXiv:1706.07094. [Google Scholar] [CrossRef]
- Frazier, P. A Tutorial on Bayesian Optimization. arXiv 2018, arXiv:1807.02811. [Google Scholar]
- Powell, M.J.D. The NEWUOA software for unconstrained optimization without derivatives. In Large-Scale Nonlinear Optimization; Di Pillo, G., Roma, M., Eds.; Springer: Boston, MA, USA, 2006; pp. 255–297. [Google Scholar] [CrossRef]
- Powell, M.J.D. A Direct Search Optimization Method That Models the Objective and Constraint Functions by Linear Interpolation. In Advances in Optimization and Numerical Analysis; Gomez, S., Hennart, J.P., Eds.; Springer: Dordrecht, The Netherlands, 1994; pp. 51–67. [Google Scholar] [CrossRef]
- Derby, C.; Klassen, J.; Bausch, J.; Cubitt, T. Compact fermion to qubit mappings. Phys. Rev. B 2021, 104, 035118. [Google Scholar] [CrossRef]
- Lee, J.; Berry, D.W.; Gidney, C.; Huggins, W.J.; McClean, J.R.; Wiebe, N.; Babbush, R. Even More Efficient Quantum Computations of Chemistry Through Tensor Hypercontraction. PRX Quantum 2021, 2, 030305. [Google Scholar] [CrossRef]
- Cerezo, M.; Sone, A.; Volkoff, T.; Cincio, L.; Coles, P.J. Cost function dependent barren plateaus in shallow parametrized quantum circuits. Nat. Commun. 2021, 12, 1791. [Google Scholar] [CrossRef] [PubMed]
- Funcke, L.; Hartung, T.; Jansen, K.; Kühn, S.; Stornati, P. Dimensional Expressivity Analysis of Parametric Quantum Circuits. Quantum 2021, 5, 422. [Google Scholar] [CrossRef]
- Nakaji, K.; Yamamoto, N. Expressibility of the alternating layered ansatz for quantum computation. Quantum 2021, 5, 434. [Google Scholar] [CrossRef]
- Jaccard, P. The distribution of the flora IN the alpine ZONE.1. New Phytol. 1912, 11, 37–50. [Google Scholar] [CrossRef]
- Tanimoto, T.T. An Elementary Mathematical theory of Classification and Prediction; Internal IBM Technical Report; International Business Machines Corporation: New York, NY, USA, 1958; p. 8. [Google Scholar]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miháliková, I.; Pivoluska, M.; Plesch, M.; Friák, M.; Nagaj, D.; Šob, M. The Cost of Improving the Precision of the Variational Quantum Eigensolver for Quantum Chemistry. Nanomaterials 2022, 12, 243. https://doi.org/10.3390/nano12020243
Miháliková I, Pivoluska M, Plesch M, Friák M, Nagaj D, Šob M. The Cost of Improving the Precision of the Variational Quantum Eigensolver for Quantum Chemistry. Nanomaterials. 2022; 12(2):243. https://doi.org/10.3390/nano12020243
Chicago/Turabian StyleMiháliková, Ivana, Matej Pivoluska, Martin Plesch, Martin Friák, Daniel Nagaj, and Mojmír Šob. 2022. "The Cost of Improving the Precision of the Variational Quantum Eigensolver for Quantum Chemistry" Nanomaterials 12, no. 2: 243. https://doi.org/10.3390/nano12020243
APA StyleMiháliková, I., Pivoluska, M., Plesch, M., Friák, M., Nagaj, D., & Šob, M. (2022). The Cost of Improving the Precision of the Variational Quantum Eigensolver for Quantum Chemistry. Nanomaterials, 12(2), 243. https://doi.org/10.3390/nano12020243