
Simultaneous Influence of Gradients in Natural Organic Matter and Abiotic Parameters on the Behavior of Silver Nanoparticles in the Transition Zone from Freshwater to Saltwater Environments



Abstract
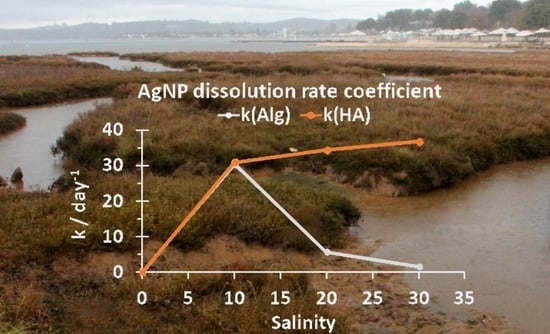
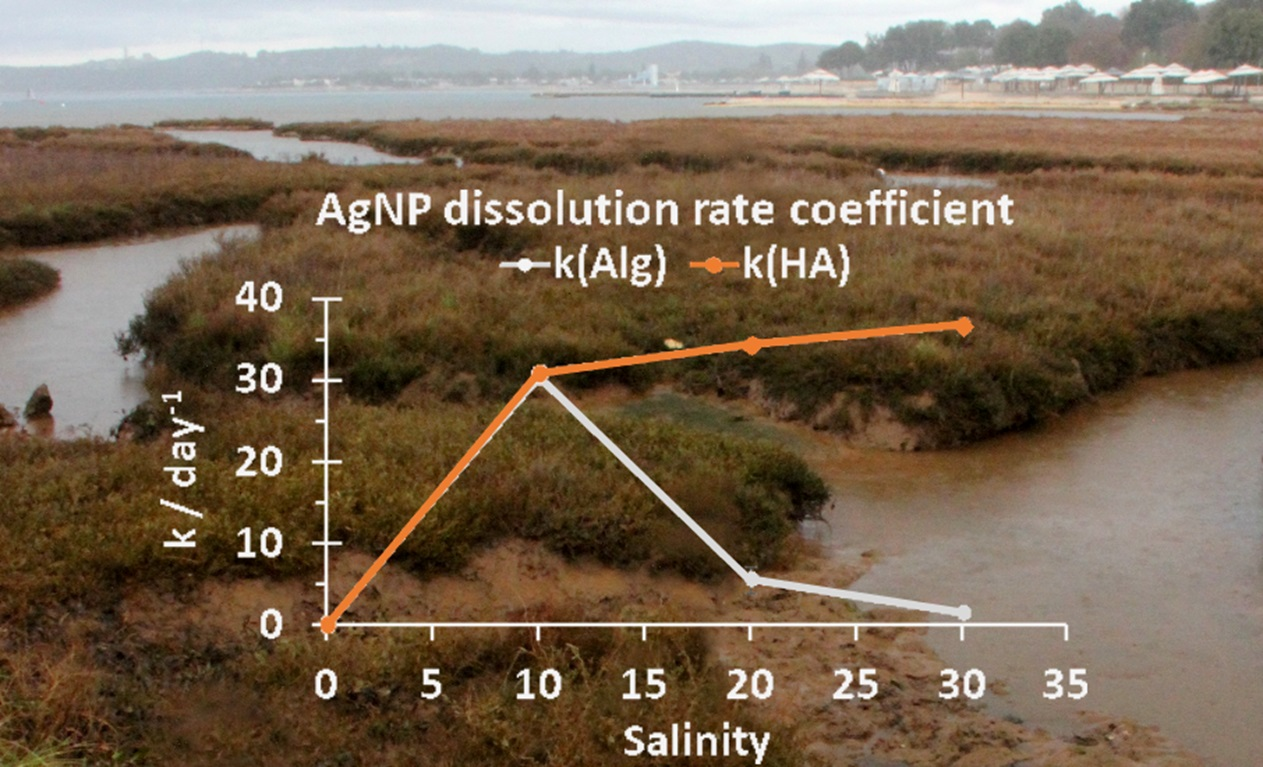
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
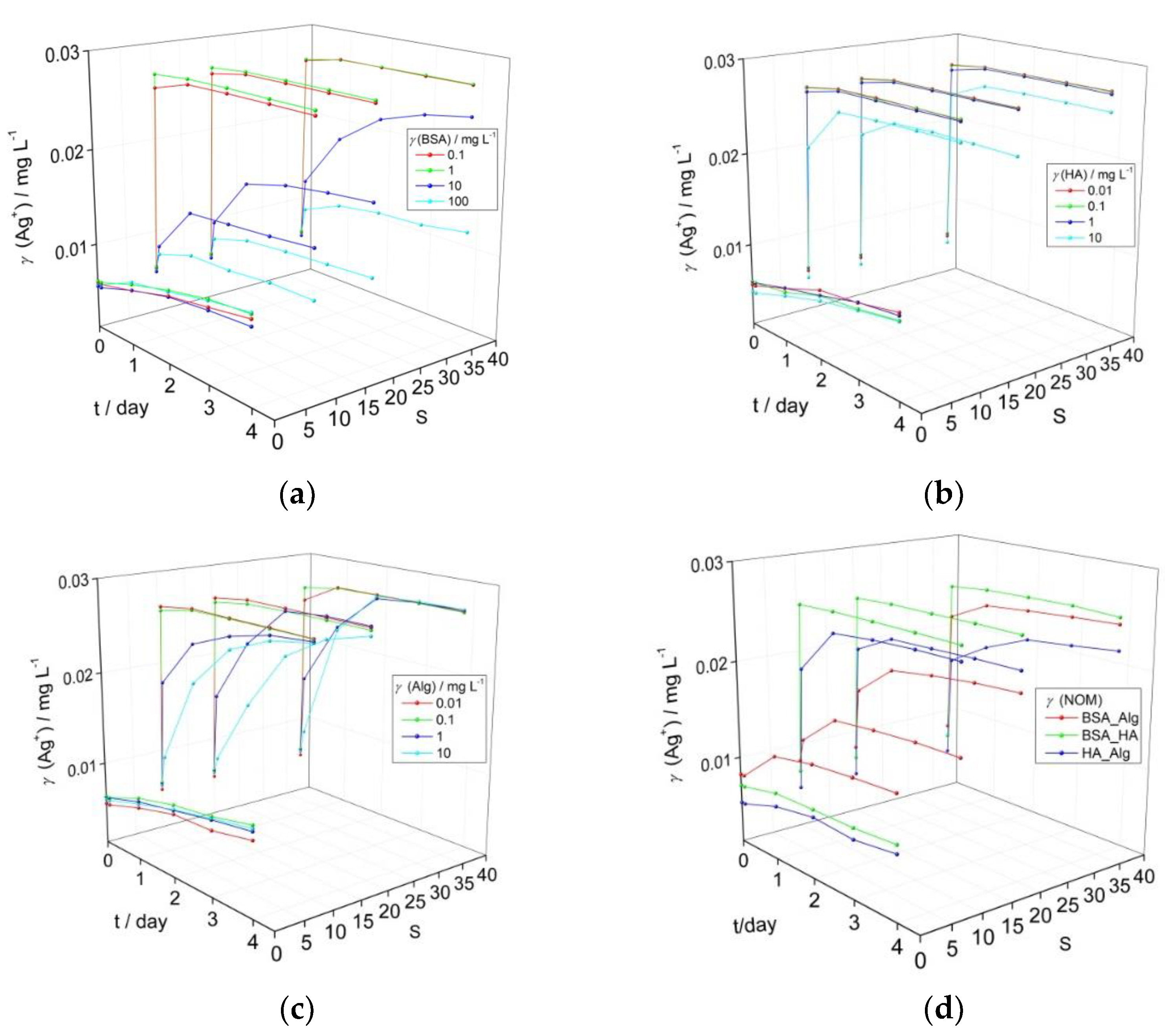
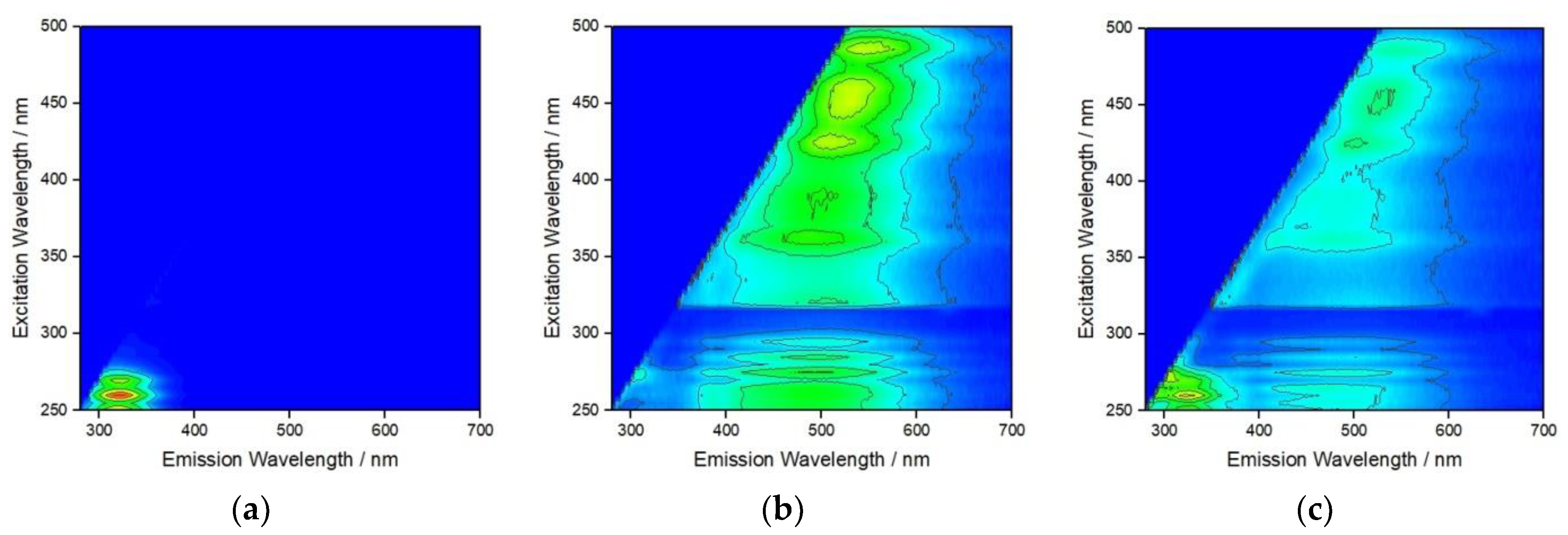{kind=link}
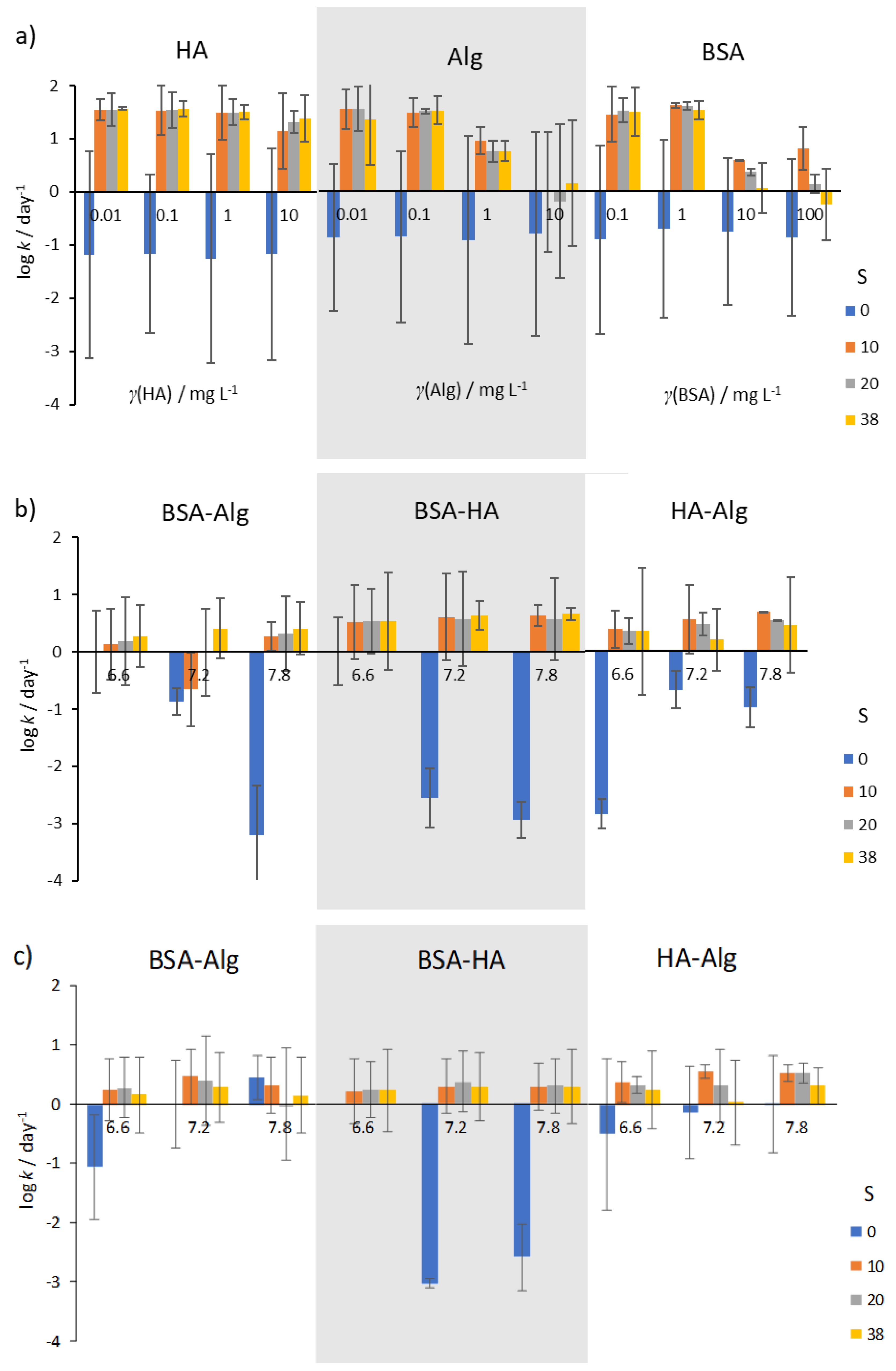{kind=link}
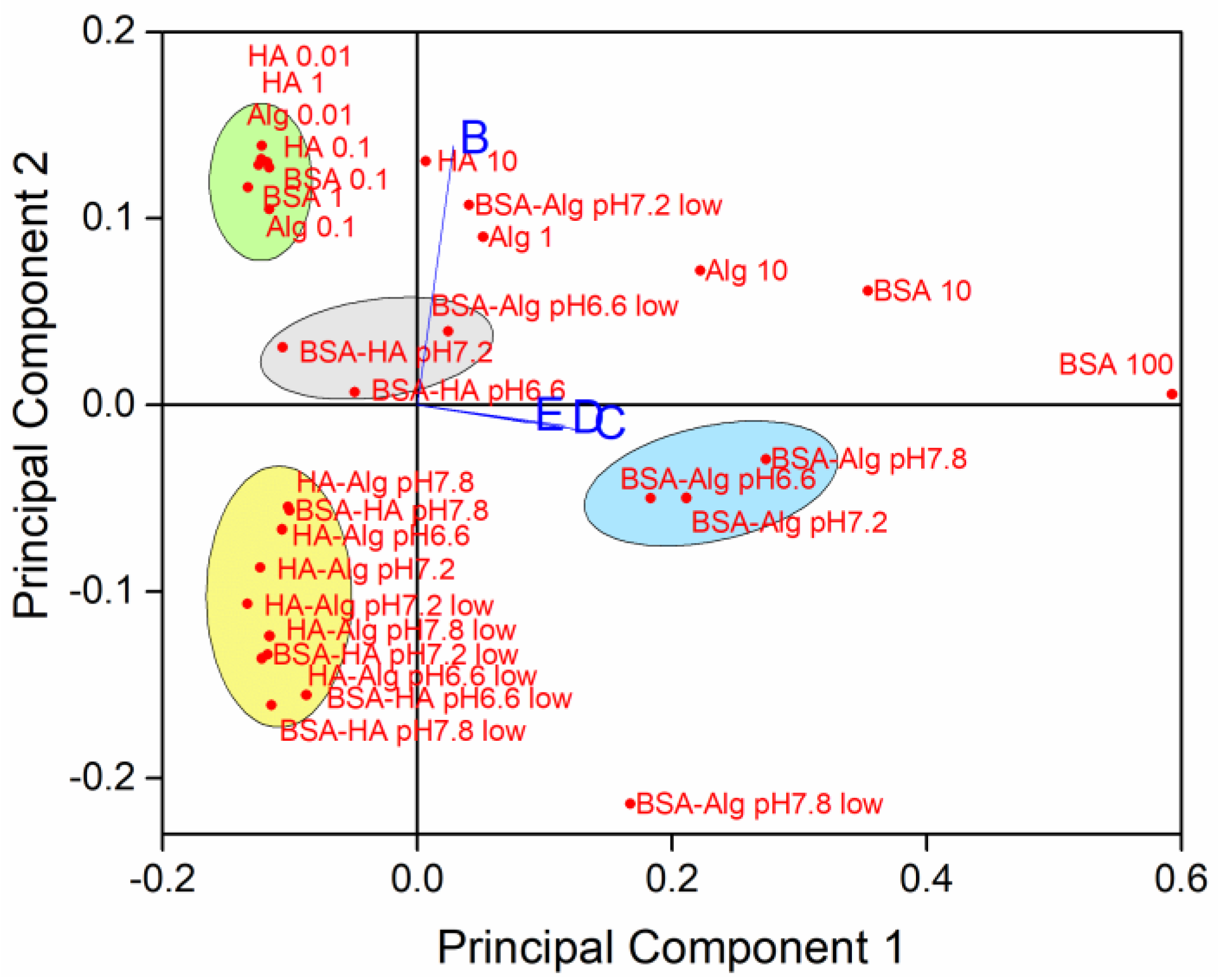{kind=link}
1. Introduction
2. Experimental
3. Results and Discussion
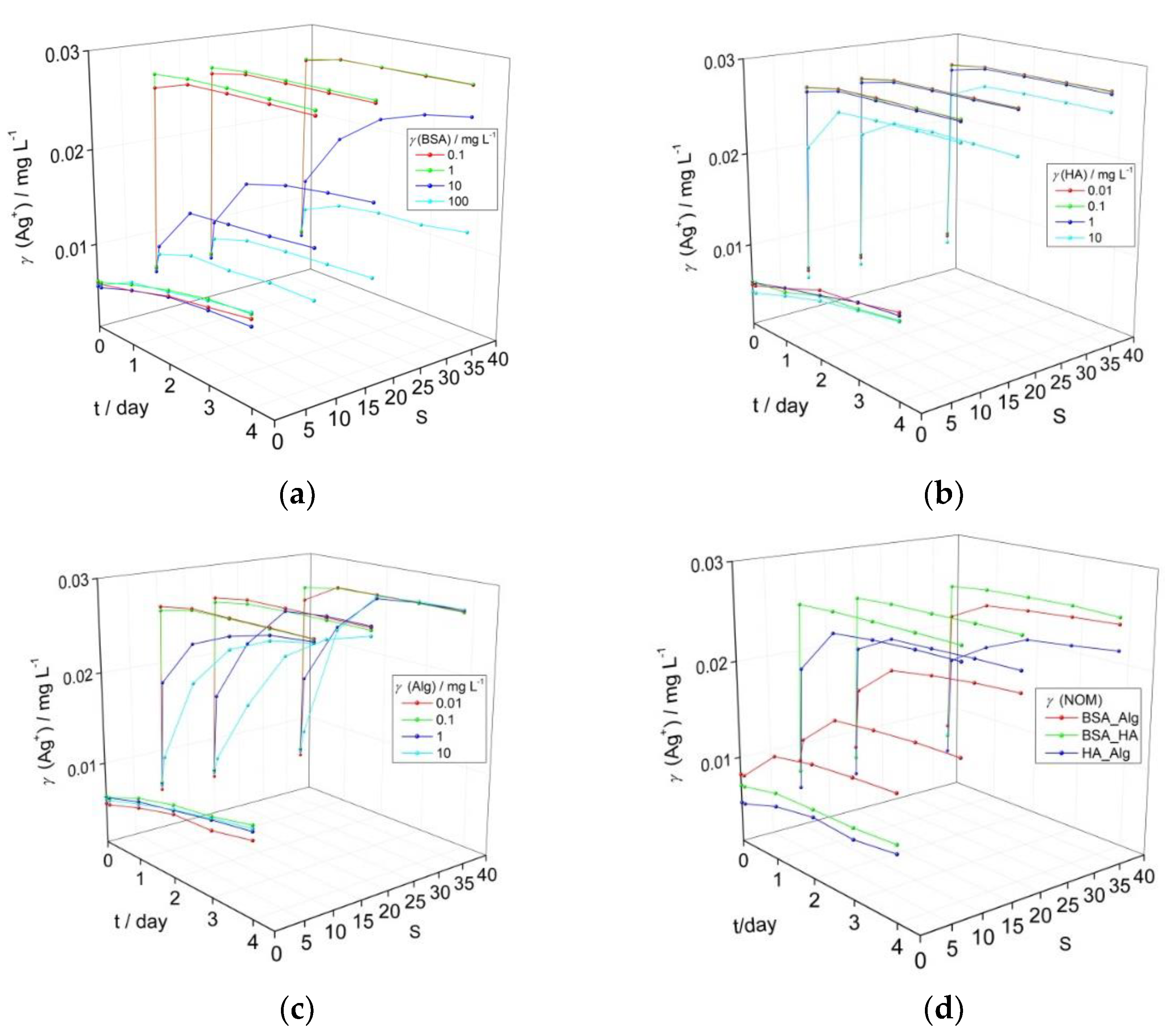
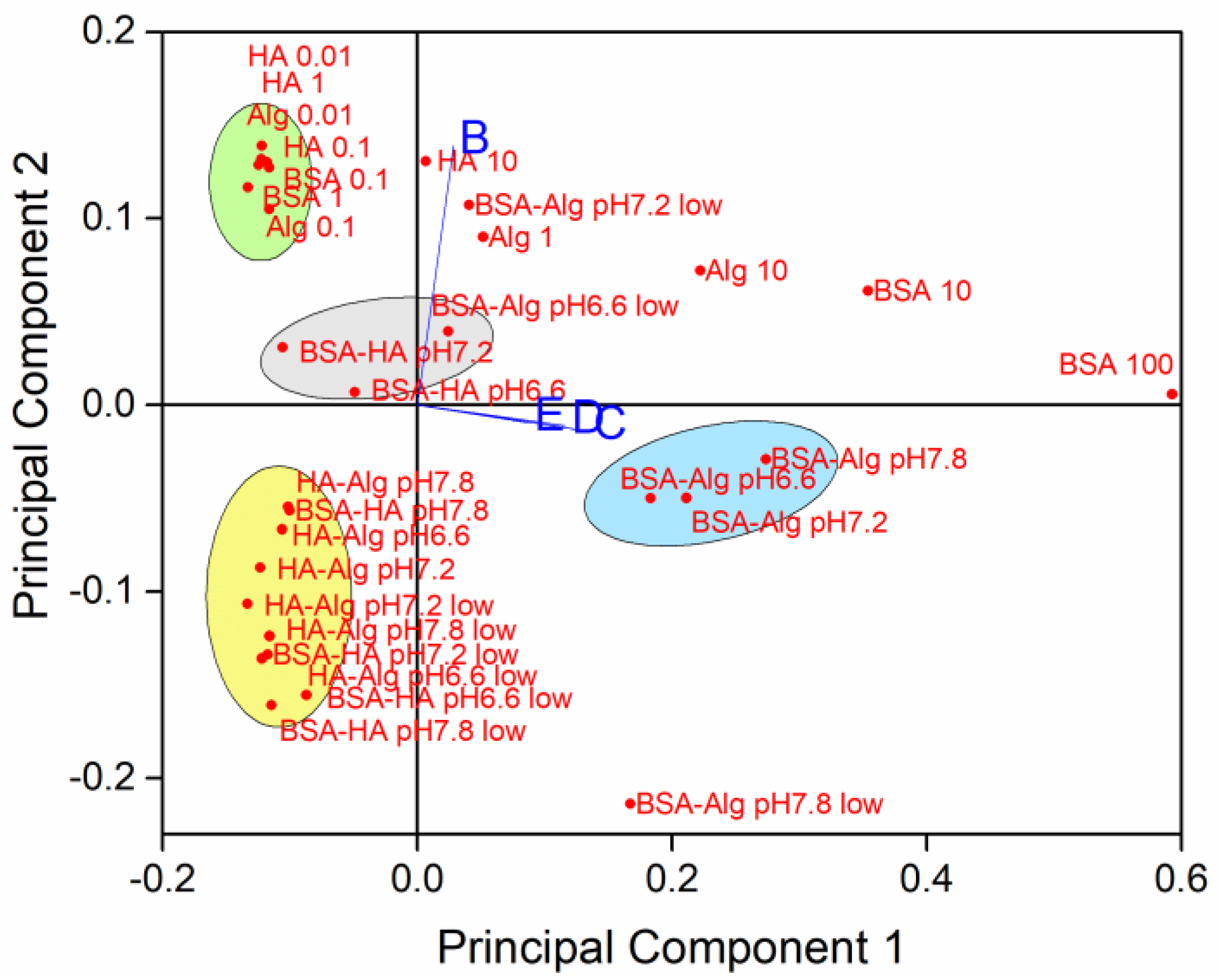
3.1. Behavior of AgNPs as a Function of NOM and Salinity
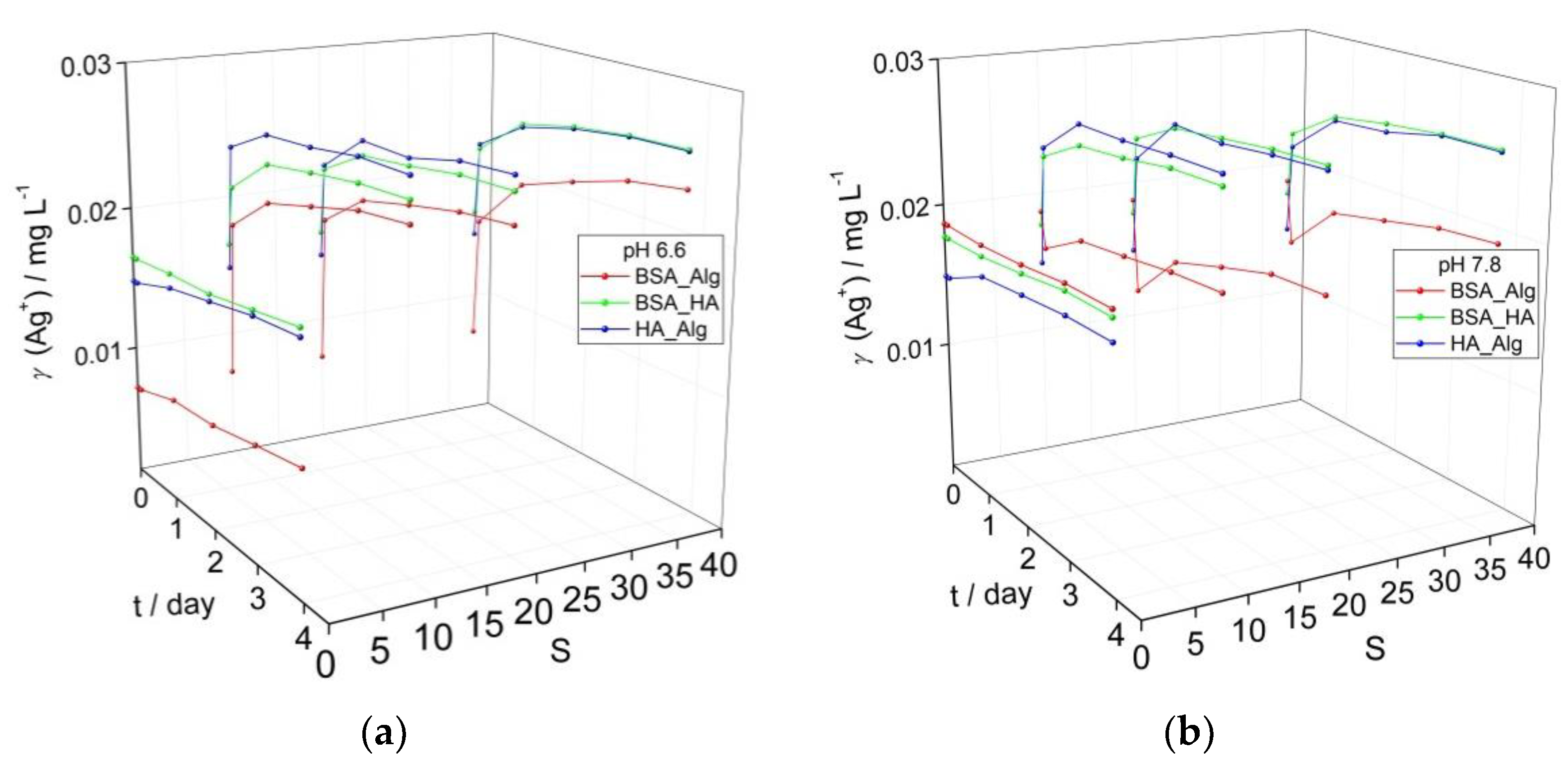
3.2. Behavior of AgNPs as a Function of NOM Combinations, Salinity, and pH
3.3. Behavior of AgNPs as a Function of NOM Combinations, Salinity, pH and Oxygen Saturation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, S.H.; Jun, B.H. Silver Nanoparticles: Synthesis and Application for Nanomedicine. Int. J. Mol. Sci. 2019, 20, 865. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, Q.; Scotter, M.; Blackburn, J.; Ross, B.; Boxall, A.; Castle, L.; Aitken, R.; Watkins, R. Applications and implications of nanotechnologies for the food sector. Food Addit. Contam. 2008, 25, 241–258. [Google Scholar] [CrossRef]
- Burdușel, A.-C.; Gherasim, O.; Grumezescu, A.; Mogoantă, L.; Ficai, A.; Andronescu, E. Biomedical Applications of Silver Nanoparticles: An Up-to-Date Overview. Nanomaterials 2018, 8, 681. [Google Scholar] [CrossRef] [Green Version]
- Dale, A.L.; Casman, E.A.; Lowry, G.V.; Lead, J.R.; Viparelli, E.; Baalousha, M. Modeling Nanomaterial Environmental Fate in Aquatic Systems. Environ. Sci. Technol. 2015, 49, 2587–2593. [Google Scholar] [CrossRef] [PubMed]
- Lead, J.R.; Batley, G.E.; Alvarez, P.J.; Croteau, M.N.; Handy, R.D.; McLaughlin, M.J.; Judy, J.D.; Schirmer, K. Nanomaterials in the environment: Behavior, fate, bioavailability, and effects-An updated review. Environ. Toxicol. Chem. 2018, 37, 2029–2063. [Google Scholar] [CrossRef]
- Saleem, H.; Zaidi, S.J. Sustainable Use of Nanomaterials in Textiles and Their Environmental Impact. Materials 2020, 13, 5134. [Google Scholar] [CrossRef] [PubMed]
- Nowack, B.; Bucheli, T.D. Occurrence, behavior and effects of nanoparticles in the environment. Environ. Pollut. 2007, 150, 5–22. [Google Scholar] [CrossRef]
- Kaegi, R.; Voegelin, A.; Sinnet, B.; Zuleeg, S.; Hagendorfer, H.; Burkhardt, M.; Siegrist, H. Behavior of Metallic Silver Nanoparticles in a Pilot Wastewater Treatment Plant. Environ. Sci. Technol. 2011, 45, 3902–3908. [Google Scholar] [CrossRef] [PubMed]
- Kent, R.D.; Oser, J.G.; Vikesland, P.J. Controlled Evaluation of Silver Nanoparticle Sulfidation in a Full-Scale Wastewater Treatment Plant. Environ. Sci. Technol. 2014, 48, 8564–8572. [Google Scholar] [CrossRef] [PubMed]
- Baker, T.J.; Tyler, C.R.; Galloway, T.S. Impacts of metal and metal oxide nanoparticles on marine organisms. Environ. Pollut. 2014, 186, 257–271. [Google Scholar] [CrossRef]
- Levard, C.; Hotze, E.M.; Lowry, G.V.; Brown, G.E. Environmental Transformations of Silver Nanoparticles: Impact on Stability and Toxicity. Environ. Sci. Technol. 2012, 46, 6900–6914. [Google Scholar] [CrossRef]
- Lodeiro, P.; Achterberg, E.P.; Pampín, J.; Affatati, A.; El-Shahawi, M.S. Silver nanoparticles coated with natural polysaccharides as models to study AgNP aggregation kinetics using UV-Visible spectrophotometry upon discharge in complex environments. Sci. Total Environ. 2016, 539, 7–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akaighe, N.; MacCuspie, R.I.; Navarro, D.A.; Aga, D.S.; Banerjee, S.; Sohn, M.; Sharma, V.K. Humic Acid-Induced Silver Nanoparticle Formation Under Environmentally Relevant Conditions. Environ. Sci. Technol. 2011, 45, 3895–3901. [Google Scholar] [CrossRef]
- Yin, Y.; Liu, J.; Jiang, G. Sunlight-Induced Reduction of Ionic Ag and Au to Metallic Nanoparticles by Dissolved Organic Matter. ACS Nano 2012, 6, 7910–7919. [Google Scholar] [CrossRef] [PubMed]
- Adegboyega, N.F.; Sharma, V.; Siskova, K.; Zbořil, R.; Sohn, M.; Schultz, B.J.; Banerjee, S. Interactions of Aqueous Ag+ with Fulvic Acids: Mechanisms of Silver Nanoparticle Formation and Investigation of Stability. Environ. Sci. Technol. 2012, 47, 757–764. [Google Scholar] [CrossRef]
- Hou, W.C.; Stuart, B.; Howes, R.; Zepp, R.G. Sunlight-Driven Reduction of Silver Ions by Natural Organic Matter: Formation and Transformation of Silver Nanoparticles. Environ. Sci. Technol. 2013, 47, 7713–7721. [Google Scholar] [CrossRef]
- Sánchez-Cortés, S.; Francioso, O.; Ciavatta, C.; Garcia-Ramos, J.; Gessa, C. pH-Dependent Adsorption of Fractionated Peat Humic Substances on Different Silver Colloids Studied by Surface-Enhanced Raman Spectroscopy. J. Colloid Interface Sci. 1998, 198, 308–318. [Google Scholar] [CrossRef]
- Huangfu, X.; Jiang, J.; Ma, J.; Liu, Y.; Yang, J. Aggregation Kinetics of Manganese Dioxide Colloids in Aqueous Solution: Influence of Humic Substances and Biomacromolecules. Environ. Sci. Technol. 2013, 47, 10285–10292. [Google Scholar] [CrossRef]
- Levak, M.; Burić, P.; Dutour Sikirić, M.; Domazet Jurašin, D.; Mikac, N.; Bačić, N.; Drexel, R.; Meier, F.; Jakšić, Ž.; Lyons, D.M. Effect of Protein Corona on Silver Nanoparticle Stabilization and Ion Release Kinetics in Artificial Seawater. Environ. Sci. Technol. 2017, 51, 1259–1266. [Google Scholar] [CrossRef]
- Chen, S.-F.; Zhang, H. Aggregation kinetics of nanosilver in different water conditions. Adv. Nat. Sci. Nanosci. Nanotechnol. 2012, 3, 035006. [Google Scholar] [CrossRef]
- Delay, M.; Dolt, T.; Woellhaf, A.; Sembritzki, R.; Frimmel, F.H. Interactions and stability of silver nanoparticles in the aqueous phase: Influence of natural organic matter (NOM) and ionic strength. J. Chromatogr. A 2011, 1218, 4206–4212. [Google Scholar] [CrossRef]
- Baalousha, M.; Nur, Y.; Romer, I.; Tejamaya, M.; Lead, J.R. Effect of monovalent and divalent cations, anions and fulvic acid on aggregation of citrate-coated silver nanoparticles. Sci. Total Environ. 2013, 454, 119–131. [Google Scholar] [CrossRef]
- González-Márquez, L.C.; Hansen, A.M.; González-Farias, F.A. Effect of mono and divalent salts on the conformation and composition of a humic acid and on atrazine adsorption. Environ. Sci. Pollut. Res. 2018, 25, 17509–17518. [Google Scholar] [CrossRef] [PubMed]
- Salas, P.; Odzak, N.; Echegoyen, Y.; Kägi, R.; Sancho, M.C.; Navarro, E. The role of size and protein shells in the toxicity to algal photosynthesis induced by ionic silver delivered from silver nanoparticles. Sci. Total Environ. 2019, 692, 233–239. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, J.; Li, X.; Shao, J.; Peijnenburg, W.J.G.M. Aquatic toxicity of nanosilver colloids to different trophic organisms: Contributions of particles and free silver ion. Environ. Toxicol. Chem. 2012, 31, 2408–2413. [Google Scholar] [CrossRef]
- Gambardella, C.; Morgana, S.; Di Bari, G.; Ramoino, P.; Bramini, M.; Diaspro, A.; Falugi, C.; Faimali, M. Multidisciplinary screening of toxicity induced by silica nanoparticles during sea urchin development. Chemosphere 2015, 139, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Reinsch, B.C.; Levard, C.; Li, Z.; Ma, R.; Wise, A.; Gregory, K.B.; Brown, G.E., Jr.; Lowry, G.V. Sulfidation of Silver Nanoparticles DecreasesEscherichia coliGrowth Inhibition. Environ. Sci. Technol. 2012, 46, 6992–7000. [Google Scholar] [CrossRef]
- Liu, J.; Hurt, R.H. Ion Release Kinetics and Particle Persistence in Aqueous Nano-Silver Colloids. Environ. Sci. Technol. 2010, 44, 2169–2175. [Google Scholar] [CrossRef]
- El Badawy, A.M.; Luxton, T.P.; Silva, R.G.; Scheckel, K.G.; Suidan, M.T.; Tolaymat, T.M. Impact of Environmental Conditions (pH, Ionic Strength, and Electrolyte Type) on the Surface Charge and Aggregation of Silver Nanoparticles Suspensions. Environ. Sci. Technol. 2010, 44, 1260–1266. [Google Scholar] [CrossRef] [PubMed]
- Axson, J.L.; Stark, D.I.; Bondy, A.L.; Capracotta, S.S.; Maynard, A.D.; Philbert, M.A.; Bergin, I.L.; Ault, A.P. Rapid Kinetics of Size and pH-Dependent Dissolution and Aggregation of Silver Nanoparticles in Simulated Gastric Fluid. J. Phys. Chem. C 2015, 119, 20632–20641. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Liu, Y.L.; Stallworth, A.M.; Ye, C.; Lenhart, J.J. Effects of pH, Electrolyte, Humic Acid, and Light Exposure on the Long-Term Fate of Silver Nanoparticles. Environ. Sci. Technol. 2016, 50, 12214–12224. [Google Scholar] [CrossRef]
- Adegboyega, N.F.; Sharma, V.K.; Siskova, K.M.; Vecerova, R.; Kolar, M.; Zbořil, R.; Gardea-Torresdey, J.L. Enhanced Formation of Silver Nanoparticles in Ag+-NOM-Iron (II, III) Systems and Antibacterial Activity Studies. Environ. Sci. Technol. 2014, 48, 3228–3235. [Google Scholar] [CrossRef]
- Mittelman, A.M.; Fortner, J.D.; Pennell, K.D. Effects of ultraviolet light on silver nanoparticle mobility and dissolution. Environ. Sci. Nano 2015, 2, 683–691. [Google Scholar] [CrossRef]
- Chambers, B.A.; Afrooz, A.R.M.N.; Bae, S.; Aich, N.; Katz, L.; Saleh, N.B.; Kirisits, M.J. Effects of Chloride and Ionic Strength on Physical Morphology, Dissolution, and Bacterial Toxicity of Silver Nanoparticles. Environ. Sci. Technol. 2014, 48, 761–769. [Google Scholar] [CrossRef]
- Valenti, L.E.; Giacomelli, C.E. Stability of silver nanoparticles: Agglomeration and oxidation in biological relevant conditions. J. Nanoparticle Res. 2017, 19, 156. [Google Scholar] [CrossRef]
- Zhang, W.; Yao, Y.; Li, K.; Huang, Y.; Chen, Y. Influence of dissolved oxygen on aggregation kinetics of citrate-coated silver nanoparticles. Environ. Pollut. 2011, 159, 3757–3762. [Google Scholar] [CrossRef]
- Zou, X.; Li, P.; Lou, J.; Fu, X.; Zhang, H. Stability of single dispersed silver nanoparticles in natural and synthetic freshwaters: Effects of dissolved oxygen. Environ. Pollut. 2017, 230, 674–682. [Google Scholar] [CrossRef]
- Zhang, W.; Yao, Y.; Sullivan, N.; Chen, Y. Modeling the Primary Size Effects of Citrate-Coated Silver Nanoparticles on Their Ion Release Kinetics. Environ. Sci. Technol. 2011, 45, 4422–4428. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ge, M.; Zhang, Z.; Wang, W.; Wu, G. Spectroscopic studies on the interaction between riboflavin and albumins. Spectrochim. Acta A 2006, 65, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Sikder, M.; Lead, J.R.; Chandler, G.T.; Baalousha, M. A rapid approach for measuring silver nanoparticle concentration and dissolution in seawater by UV–Vis. Sci. Total Environ. 2018, 618, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Paramelle, D.; Sadovoy, A.; Gorelik, S.; Free, P.; Hobley, J.; Fernig, D.G. A rapid method to estimate the concentration of citrate capped silver nanoparticles from UV-visible light spectra. Analyst 2014, 139, 4855–4861. [Google Scholar] [CrossRef]
- Kittler, S.; Greulich, C.; Diendorf, J.; Köller, M.; Epple, M. Toxicity of Silver Nanoparticles Increases during Storage Because of Slow Dissolution under Release of Silver Ions. Chem. Mater. 2010, 22, 4548–4554. [Google Scholar] [CrossRef]
- Lodeiro, P.; Browning, T.J.; Achterberg, E.P.; Guillou, A.; El-Shahawi, M.S. Mechanisms of silver nanoparticle toxicity to the coastal marine diatom Chaetoceros curvisetus. Sci. Rep. 2017, 7, 10777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baalousha, M.; Afshinnia, K.; Guo, L. Natural organic matter composition determines the molecular nature of silver nanomaterial-NOM corona. Environ. Sci. Nano 2018, 5, 868–881. [Google Scholar] [CrossRef]
- Grillo, R.; Rosa, A.H.; Fraceto, L.F. Engineered nanoparticles and organic matter: A review of the state-of-the-art. Chemosphere 2015, 119, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Erhayem, M.; Sohn, M. Stability studies for titanium dioxide nanoparticles upon adsorption of Suwannee River humic and fulvic acids and natural organic matter. Sci. Total Environ. 2014, 468-469, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Loosli, F.; Le Coustumer, P.; Stoll, S. TiO2 nanoparticles aggregation and disaggregation in presence of alginate and Suwannee River humic acids. pH and concentration effects on nanoparticle stability. Water Res. 2013, 47, 6052–6063. [Google Scholar] [CrossRef] [PubMed]
- Thalmann, B.; Voegelin, A.; Morgenroth, E.; Kaegi, R. Effect of humic acid on the kinetics of silver nanoparticle sulfidation. Environ. Sci. Nano 2016, 3, 203–212. [Google Scholar] [CrossRef]
- António, D.C.; Cascio, C.; Jakšić, Ž.; Jurašin, D.; Lyons, D.M.; Nogueira, A.J.A.; Rossi, F.; Calzolai, L. Assessing silver nanoparticles behaviour in artificial seawater by mean of AF4 and spICP-MS. Mar. Environ. Res. 2015, 111, 162–169. [Google Scholar] [CrossRef]
- Bian, S.-W.; Mudunkotuwa, I.A.; Rupasinghe, T.; Grassian, V.H. Aggregation and Dissolution of 4 nm ZnO Nanoparticles in Aqueous Environments: Influence of pH, Ionic Strength, Size, and Adsorption of Humic Acid. Langmuir 2011, 27, 6059–6068. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Westerhoff, P.; Crittenden, J. Impact of natural organic matter and divalent cations on the stability of aqueous nanoparticles. Water Res. 2009, 43, 4249–4257. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Medina, S.; Blankenburg, J.; Olson, J.; Landes, C.F.; Link, S. Adsorption of a Protein Monolayer via Hydrophobic Interactions Prevents Nanoparticle Aggregation under Harsh Environmental Conditions. ACS Sustain. Chem. Eng. 2013, 1, 833–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiu, Z.-M.; Zhang, Q.-B.; Puppala, H.L.; Colvin, V.L.; Alvarez, P.J.J. Negligible Particle-Specific Antibacterial Activity of Silver Nanoparticles. Nano Lett. 2012, 12, 4271–4275. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, B.; Engstrom, A.M.; Harper, B.J.; Harper, S.L.; Mackiewicz, M.R. Silver Nanoparticles Stable to Oxidation and Silver Ion Release Show Size-Dependent Toxicity In Vivo. Nanomaterials 2021, 11, 1516. [Google Scholar] [CrossRef] [PubMed]
- Burić, P.; Jakšić, Ž.; Štajner, L.; Dutour Sikirić, M.; Jurašin, D.; Cascio, C.; Calzolai, L.; Lyons, D.M. Effect of silver nanoparti-cles on Mediterranean Sea urchin embryonal development is species specific and depends on moment of first exposure. Mar. Environ. Res. 2015, 111, 50–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biba, R.; Košpić, K.; Komazec, B.; Markulin, D.; Cvjetko, P.; Pavoković, D.; Peharec Štefanić, P.; Tkalec, M.; Balen, B. Surface Coating-Modulated Phytotoxic Responses of Silver Nanoparticles in Plants and Freshwater Green Algae. Nanomaterials 2022, 12, 24. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Čarapar, I.; Jurković, L.; Pavičić-Hamer, D.; Hamer, B.; Lyons, D.M. Simultaneous Influence of Gradients in Natural Organic Matter and Abiotic Parameters on the Behavior of Silver Nanoparticles in the Transition Zone from Freshwater to Saltwater Environments. Nanomaterials 2022, 12, 296. https://doi.org/10.3390/nano12020296
Čarapar I, Jurković L, Pavičić-Hamer D, Hamer B, Lyons DM. Simultaneous Influence of Gradients in Natural Organic Matter and Abiotic Parameters on the Behavior of Silver Nanoparticles in the Transition Zone from Freshwater to Saltwater Environments. Nanomaterials. 2022; 12(2):296. https://doi.org/10.3390/nano12020296
Chicago/Turabian StyleČarapar, Ivana, Lara Jurković, Dijana Pavičić-Hamer, Bojan Hamer, and Daniel Mark Lyons. 2022. "Simultaneous Influence of Gradients in Natural Organic Matter and Abiotic Parameters on the Behavior of Silver Nanoparticles in the Transition Zone from Freshwater to Saltwater Environments" Nanomaterials 12, no. 2: 296. https://doi.org/10.3390/nano12020296
APA StyleČarapar, I., Jurković, L., Pavičić-Hamer, D., Hamer, B., & Lyons, D. M. (2022). Simultaneous Influence of Gradients in Natural Organic Matter and Abiotic Parameters on the Behavior of Silver Nanoparticles in the Transition Zone from Freshwater to Saltwater Environments. Nanomaterials, 12(2), 296. https://doi.org/10.3390/nano12020296