
The Kinetics of Aragonite Formation from Solution via Amorphous Calcium Carbonate


 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methodology
2.1. Sample Preparation
2.2. Analysis Techniques
3. Results
3.1. Characterisation of the Amorphous Phase
3.1.1. Thermal Gravimetric Analysis
3.1.2. X-ray Diffraction
3.1.3. Laser-Ablation Inductively Coupled Plasma Mass Spectrometry
3.1.4. Neutron Scattering
3.2. Characterisation of the Crystalline Phases
3.2.1. Raman Spectroscopy
3.2.2. X-ray Diffraction
3.2.3. Scanning Electron Microscopy
3.2.4. High-Precision X-ray Diffraction
3.3. Kinetics of Particle Growth and Crystallisation of Calcium Carbonate
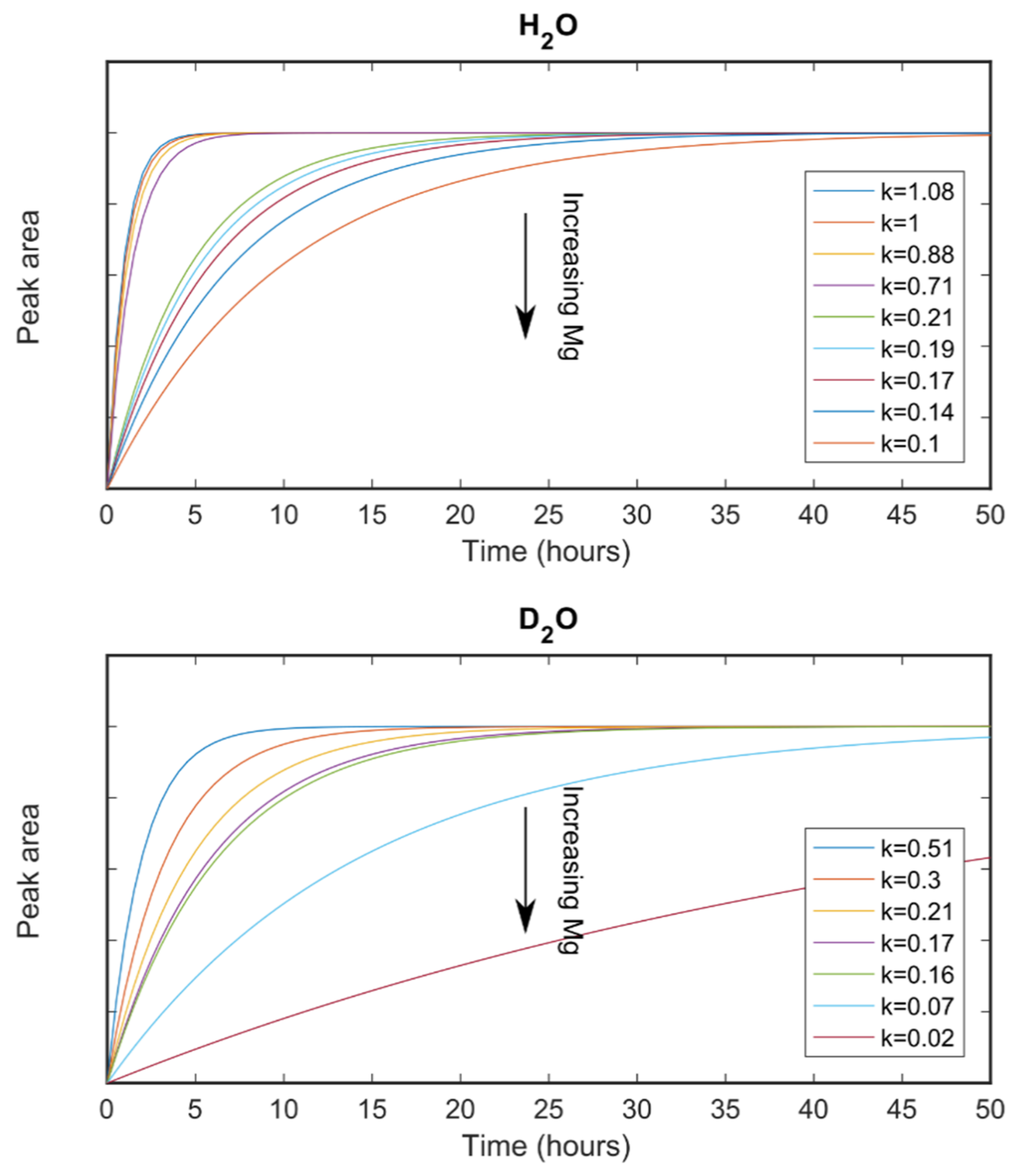
3.3.1. Small-Angle Neutron Scattering
3.3.2. X-ray Diffraction
4. Discussion and Conclusions
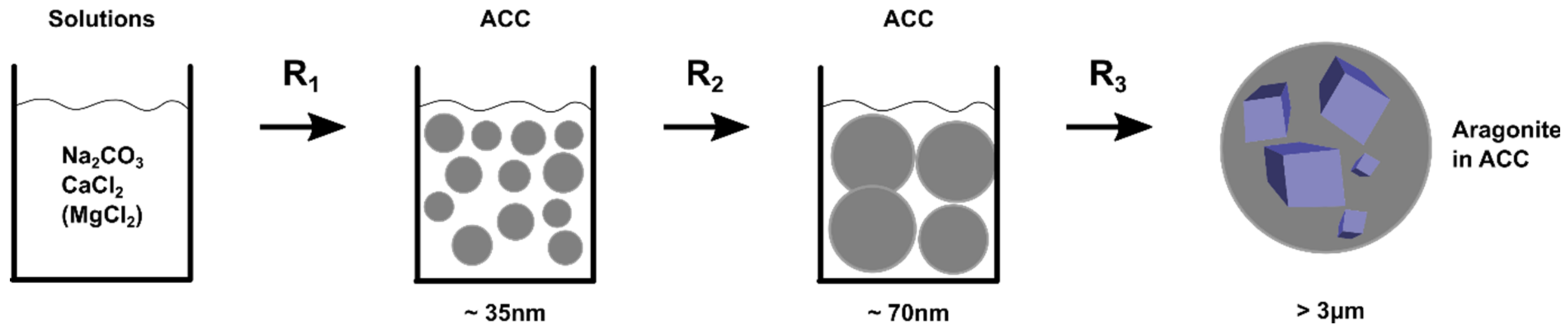
- Precipitation of ACC from solution forming 35 nm particles: reaction R1.
- The 35 nm particles grow into 70 nm particles: reaction R2.
- Formation of aragonite crystals by localised dissolution and reprecipitation: reaction R3.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Politi, Y.; Metzler, R.A.; Abrecht, M.; Gilbert, B.; Wilt, F.H.; Sagi, I.; Addadi, L.; Weiner, S.; Gilbert, P.U.P.A. Transformation mechanism of amorphous calcium carbonate into calcite in the sea urchin larval spicule. Proc. Natl. Acad. Sci. USA 2008, 105, 17362–17366. [Google Scholar] [CrossRef] [Green Version]
- Navrotsky, A. Energetic clues to pathways to biomineralization: Precursors, clusters, and nanoparticles. Proc. Natl. Acad. Sci. USA 2004, 101, 12096–12101. [Google Scholar] [CrossRef] [Green Version]
- De Yoreo, J.J.; Gilbert, P.U.P.A.; Sommerdijk, N.A.; Penn, R.L.; Whitelam, S.; Joester, D.; Zhang, H.; Rimer, J.D.; Navrotsky, A.; Banfield, J.F. Crystallisation by particle attachment in synthetic, biogenic, and geologic environments. Science 2015, 349, aaa6760. [Google Scholar] [CrossRef]
- Cartwright, J.H.E.; Checa, A.G.; Gale, J.D.; Gebauer, D.; Sainz-Días, C.I. Calcium carbonate polyamorphism and its role in biomineralisation: How many amorphous calcium carbonates are there? Angew. Chem. Int. Ed. 2012, 51, 11960–11970. [Google Scholar] [CrossRef] [PubMed]
- Bots, P.; Benning, L.G.; Rodriguez-Blanco, J.-D.; Roncal-Herrero, T.; Shaw, S. Mechanistic Insights into the Crystallization of Amorphous Calcium Carbonate (ACC). Cryst. Growth Des. 2012, 12, 3806–3814. [Google Scholar] [CrossRef]
- Clark, S.M.; Colas, B.; Jacob, D.E.; Neuefeind, J.C.; Wang, H.-W.; Page, K.L.; Soper, A.K.; Schodder, P.I.; Duchstein, P.; Zubiri, B.A.; et al. The nano- and meso-scale structure of amorphous calcium carbonate indicate nanoscale assembly processes. Sci. Rep. 2022, 12, 6870. [Google Scholar] [CrossRef]
- Rodriguez-Blanco, J.D.; Shaw, S.; Benning, L.G. The kinetics and mechanisms of amorphous calcium carbonate (ACC) crystallization to calcite, via vaterite. Nanoscale 2011, 3, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Stephens, C.J.; Ladden, S.F.; Meldrum, F.C.; Christenson, H.K. Amorphous Calcium Carbonate is Stabilized in Confinement. Adv. Funct. Mater. 2010, 20, 2108–2115. [Google Scholar] [CrossRef]
- Addadi, L.; Raz, S.; Weiner, S. Taking Advantage of Disorder: Amorphous Calcium Carbonate and Its Roles in Biomineralization. Adv. Mater. 2003, 15, 959–970. [Google Scholar] [CrossRef]
- Ouhenia, S.; Chateigner, D.; Belkhir, M.; Guilmeau, E.; Krauss, C. Synthesis of calcium carbonate polymorphs in the presence of polyacrylic acid. J. Cryst. Growth 2008, 310, 2832–2841. [Google Scholar] [CrossRef]
- Huang, S.-C.; Naka, K.; Chujo, Y. A Carbonate Controlled-Addition Method for Amorphous Calcium Carbonate Spheres Stabilized by Poly(acrylic acid)s. Langmuir 2007, 23, 12086–12095. [Google Scholar] [CrossRef] [PubMed]
- Lam, R.S.K.; Charnock, J.M.; Lennie, A.; Meldrum, F.C. Synthesis-dependant structural variations in amorphous calcium carbonate. CrystEngComm 2007, 9, 1226–1236. [Google Scholar] [CrossRef]
- Loste, E.; Wilson, R.M.; Seshadri, R.; Meldrum, F.C. The role of magnesium in stabilising amorphous calcium carbonate and controlling calcite morphologies. J. Cryst. Growth 2003, 254, 206–218. [Google Scholar] [CrossRef]
- Rodriguez-Blanco, J.; Shaw, S.; Bots, P.; Roncal-Herrero, T.; Benning, L. The role of pH and Mg on the stability and crystallization of amorphous calcium carbonate. J. Alloys Compd. 2011, 536, S477–S479. [Google Scholar] [CrossRef]
- Foran, E.; Weiner, S.; Fine, M. Biogenic Fish-gut Calcium Carbonate is a Stable Amorphous Phase in the Gilt-head Seabream, Sparus aurata. Sci. Rep. 2013, 3, 1700–1705. [Google Scholar] [CrossRef] [Green Version]
- Pai, R.K.; Pillai, S. Nanoparticles of amorphous calcium carbonate by miniemulsion: Synthesis and mechanism. CrystEngComm 2008, 10, 865–872. [Google Scholar] [CrossRef]
- Sun, W.; Jayaraman, S.; Chen, W.; Persson, K.A.; Ceder, G. Nucleation of metastable aragonite CaCO3 in seawater. Proc. Natl. Acad. Sci. USA 2015, 112, 3199–3204. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Xie, Y.; Xu, X.; Pan, H.; Tang, R. Transformation of amorphous calcium carbonate into aragonite. J. Cryst. Growth 2012, 343, 62–67. [Google Scholar] [CrossRef]
- Blue, C.; Dove, P. Chemical controls on the magnesium content of amorphous calcium carbonate. Geochim. Cosmochim. Acta 2015, 148, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Gal, A.; Habraken, W.; Gur, D.; Fratzl, P.; Weiner, S.; Addadi, L. Calcite Crystal Growth by a Solid-State Transformation of Stabilized Amorphous Calcium Carbonate Nanospheres in a Hydrogel. Angew. Chem. Int. Ed. 2013, 52, 4867–4870. [Google Scholar] [CrossRef]
- Ihli, J.; Kim, Y.-Y.; Noel, E.H.; Meldrum, F.C. The Effect of Additives on Amorphous Calcium Carbonate (ACC): Janus Behavior in Solution and the Solid State. Adv. Funct. Mater. 2012, 23, 1575–1585. [Google Scholar] [CrossRef]
- Pontoni, D.; Bolze, J.; Dingenouts, N.; Narayanan, T.; Ballauff, M. Crystallization of Calcium Carbonate Observed In-situ by Combined Small- and Wide-angle X-ray Scattering. J. Phys. Chem. B 2003, 107, 5123–5125. [Google Scholar] [CrossRef]
- Giuffre, A.J.; Gagnon, A.C.; De Yoreo, J.J.; Dove, P.M. Isotopic tracer evidence for the amorphous calcium carbonate to calcite transformation by dissolution–reprecipitation. Geochim. Cosmochim. Acta 2015, 165, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhang, Z.; Wang, Z.; Jin, B.; Li, D.; Tao, J.; Tang, R.; De Yoreo, J.J. Shape-preserving amorphous-to-crystalline transformation of CaCO3 revealed by in situ TEM. Proc. Natl. Acad. Sci. USA 2020, 117, 3397–3404. [Google Scholar] [CrossRef] [PubMed]
- Jacob, D.E.; Wirth, R.; Agbaje, O.B.A.; Branson, O.; Eggins, S.M. Planktic foraminifera form their shells via metastable carbonate phases. Nat. Commun. 2017, 8, 1265. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.H.; Aloni, S.; De Yoreo, J.J. In situ TEM imaging of CaCO3 nucleation reveals coexistence of direct and indirect pathways. Science 2014, 345, 1158–1162. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Martinez, A.; Kalkan, B.; Clark, S.M.; Waychunas, G.A. Pressure-Induced Polyamorphism and Formation of ‘Aragonitic’ Amorphous Calcium Carbonate. Angew. Chem. Int. Ed. 2013, 52, 8354–8357. [Google Scholar] [CrossRef]
- Jochum, K.P.; Nohl, U.; Herwig, K.; Lammel, E.; Stoll, B.; Hofmann, A.W. GeoReM: A new geochemical database for reference materials and standards. Geostand. Geoanal. Res. 2006, 29, 333–338. [Google Scholar] [CrossRef]
- Doebelin, N.; Kleeberg, R. Profex: A graphical user interface for the Rietveld refinement program BGMN. J. Appl. Crystallogr. 2015, 48, 1573–1580. [Google Scholar] [CrossRef] [Green Version]
- Radha, A.; Fernandez-Martinez, A.; Hu, Y.; Jun, Y.-S.; Waychunas, G.A.; Navrotsky, A. Energetic and structural studies of amorphous Ca1−xMgxCO3·nH2O (0⩽x⩽1). Geochim. Cosmochim. Acta 2012, 90, 83–95. [Google Scholar] [CrossRef]
- Wood, K.; Mata, J.; Garvey, C.J.; Wu, C.-M.; Hamilton, W.A.; Abbeywick, P.; Bartlett, D.; Bartsch, F.; Baxter, P.; Booth, N.; et al. QUOKKA, the pinhole small-angle neutron scattering instrument at the OPAL Research Reactor, Australia: Design, performance, operation and scientific highlights. J. Appl. Crystallogr. 2018, 51, 294–314. [Google Scholar] [CrossRef]
- Wang, D.; Hamm, L.M.; Giuffre, A.J.; Echigo, T.; Rimstidt, J.D.; De Yoreo, J.J.; Grotzinger, J.; Dove, P.M. Revisiting geochemical controls on patterns of carbonate deposition through the lens of multiple pathways to mineralization. Faraday Discuss. 2012, 159, 371–386. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Yan, C.; Zhang, F.; Konishi, H.; Xu, H.; Teng, H.H. Testing the cation-hydration effect on the crystallization of Ca–Mg–CO3 systems. Proc. Natl. Acad. Sci. USA 2013, 110, 17750–17755. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-W.; Kim, Y.-Y.; Stephens, C.J.; Meldrum, F.C.; Christenson, H.K. In Situ Study of the Precipitation and Crystallization of Amorphous Calcium Carbonate (ACC). Cryst. Growth Des. 2012, 12, 1212–1217. [Google Scholar] [CrossRef]
- Parker, J.E.; Thompson, S.P.; Lennie, A.R.; Potter, J.; Tang, C.C. A study of the aragonite-calcite transformation using Raman spectroscopy, synchrotron powder diffraction and scanning electron microscopy. CrystEngComm 2010, 12, 1590–1599. [Google Scholar] [CrossRef]
- Wehrmeister, U.; Jacob, D.E.; Soldati, A.L.; Loges, N.; Häger, T.; Hofmeister, W. Amorphous, nanocrystalline and crystalline calcium carbonates in biological materials. J. Raman Spectrosc. 2010, 42, 926–935. [Google Scholar] [CrossRef]
- Lafuente, B.; Downs, R.T.; Yang, H.; Stone, N. 1. The Power of Databases: The RRUFF Project. In Highlights in Mineralogical Crystallography; Armbruster, T., Danisi, R.M., Eds.; De Gruyter: Berlin, Germany, 2016; pp. 1–30. ISBN 9783110417104. [Google Scholar]
- Nudelman, F. Nacre biomineralisation: A review on the mechanisms of crystal nucleation. Semin. Cell Dev. Biol. 2015, 46, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Dudnikova, V.B.; Eremin, N.N. Simulation of the local structure, properties of mixing, and stability of solid solutions Ba x Sr1–x CO3 by the interatomic potential method. Phys. Solid State 2016, 58, 1101–1107. [Google Scholar] [CrossRef]
- Stuhrmann, H. Small-angle scattering of X-rays. Prog. Cryst. Growth Charact. 1989, 18, 1–19. [Google Scholar] [CrossRef]
- Jacob, D.; Wirth, R.; Soldati, A.; Wehrmeister, U.; Schreiber, A. Amorphous calcium carbonate in the shells of adult Unionoida. J. Struct. Biol. 2011, 173, 241–249. [Google Scholar] [CrossRef]
- Wolf, S.E.; Böhm, C.; Harris, J.; Hajir, M.; Mondeshki, M.; Marin, F. Single Nanogranules Preserve Intracrystalline Amorphicity in Biominerals. Key Eng. Mater. 2016, 672, 47–59. [Google Scholar] [CrossRef]
- Nassif, N.; Pinna, N.; Gehrke, N.; Antonietti, M.; Jäger, C.; Cölfen, H. Amorphous layer around aragonite platelets in nacre. Proc. Natl. Acad. Sci. USA 2005, 102, 12653–12655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macías-Sánchez, E.; Willinger, M.G.; Pina, C.M.; Checa, A.G. Transformation of ACC into aragonite and the origin of the nanogranular structure of nacre. Sci. Rep. 2017, 7, 12728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, D.; Soldati, A.; Wirth, R.; Huth, J.; Wehrmeister, U.; Hofmeister, W. Nanostructure, composition and mechanisms of bivalve shell growth. Geochim. Cosmochim. Acta 2008, 72, 5401–5415. [Google Scholar] [CrossRef]
- Hovden, R.; Wolf, S.E.; Holtz, M.E.; Marin, F.; Muller, D.A.; Estroff, L.A. Nanoscale assembly processes revealed in the nacroprismatic transition zone of Pinna nobilis mollusc shells. Nat. Commun. 2015, 6, 10097. [Google Scholar] [CrossRef] [Green Version]
- Ihli, J.; Wong, W.C.; Noel, E.H.; Kim, Y.-Y.; Kulak, A.N.; Christenson, H.K.; Duer, M.J.; Meldrum, F.C. Dehydration and crystallization of amorphous calcium carbonate in solution and in air. Nat. Commun. 2014, 5, 3169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeets, P.J.M.; Cho, K.R.; Kempen, R.G.E.; Sommerdijk, N.A.J.M.; De Yoreo, J.J. Calcium carbonate nucleation driven by ion binding in a biomimetic matrix revealed by in situ electron microscopy. Nat. Mater. 2015, 14, 394–399. [Google Scholar] [CrossRef]
- Pouget, E.M.; Bomans, P.H.H.; Goos, J.A.C.M.; Frederik, P.M.; de With, G.; Sommerdijk, N.A.J.M. The Initial Stages of Template-Controlled CaCO3 Formation Revealed by Cryo-TEM. Science 2009, 323, 1455–1458. [Google Scholar] [CrossRef]
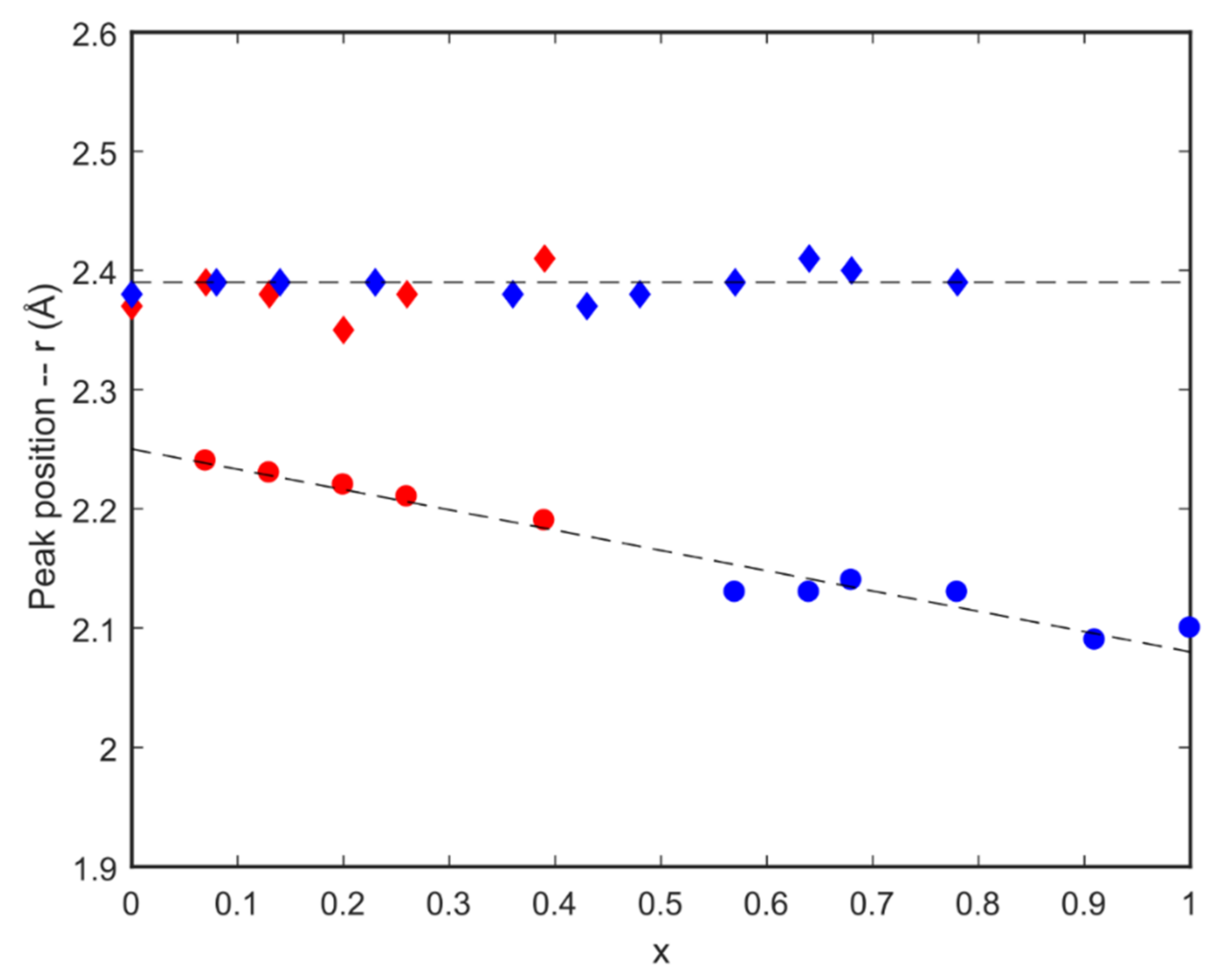
 from this study,
from this study,  mixed with Formamide [33],
mixed with Formamide [33],  [31],
[31],  [32],
[32],  [23],
[23],  with water [33]; the dashed lines are linear fits to the data points of corresponding colour.
with water [33]; the dashed lines are linear fits to the data points of corresponding colour.
 Data from this study with the linear fit of the data points;
Data from this study with the linear fit of the data points;  Strontianite (SrCO3)-aragonite (CaCO3) solid solutions from [39] with the linear fit of the data points;
Strontianite (SrCO3)-aragonite (CaCO3) solid solutions from [39] with the linear fit of the data points;  Estimate for Mg substituting Ca in the aragonite.
Data from this study with the linear fit of the data points; Strontianite (SrCO3)-aragonite (CaCO3) solid solutions from [39] with the linear fit of the data points; Estimate for Mg substituting Ca in the aragonite.
Estimate for Mg substituting Ca in the aragonite.
Data from this study with the linear fit of the data points; Strontianite (SrCO3)-aragonite (CaCO3) solid solutions from [39] with the linear fit of the data points; Estimate for Mg substituting Ca in the aragonite.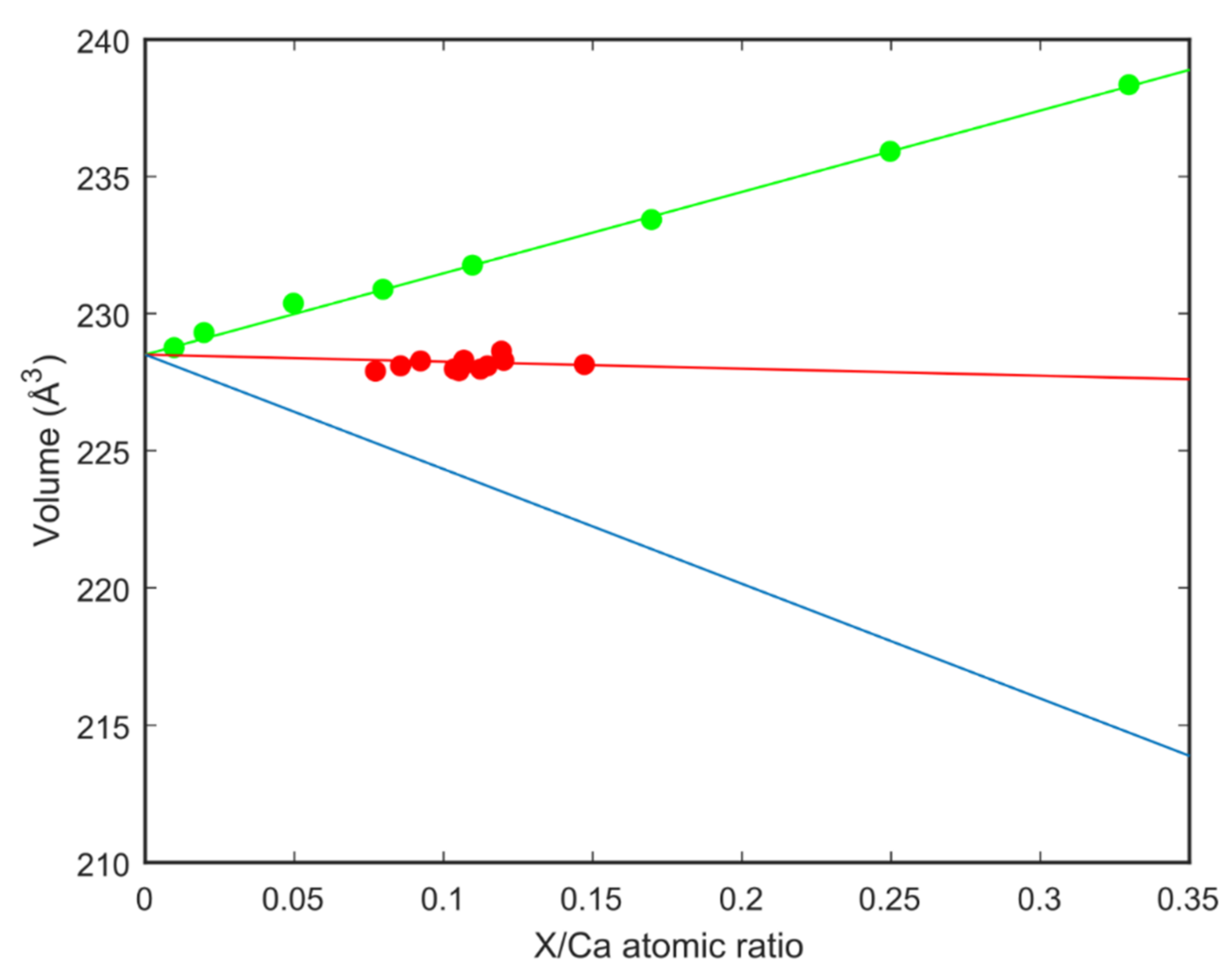
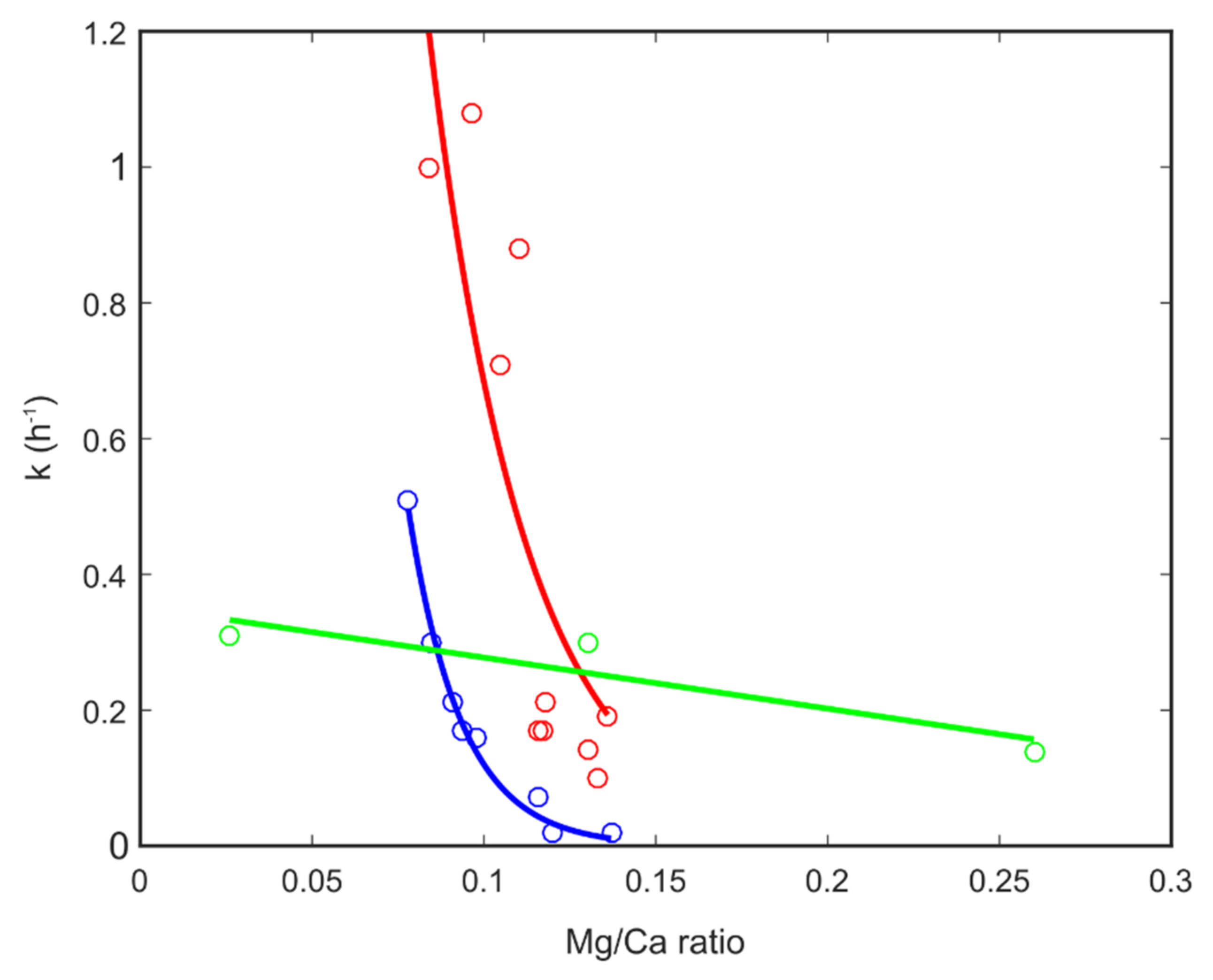
 is for the sample with Mg/Ca = 0.26;
is for the sample with Mg/Ca = 0.26;  is for the sample with Mg/Ca = 0.20; and
is for the sample with Mg/Ca = 0.20; and  is for the sample with Mg/Ca = 0.03. The corresponding lines are guides for the eye. The table contains estimates of the rate of transformation between the 35 nm and nm particles for three Mg/Ca ratios.
is for the sample with Mg/Ca = 0.26; is for the sample with Mg/Ca = 0.20; and is for the sample with Mg/Ca = 0.03. The corresponding lines are guides for the eye. The table contains estimates of the rate of transformation between the 35 nm and nm particles for three Mg/Ca ratios.
is for the sample with Mg/Ca = 0.03. The corresponding lines are guides for the eye. The table contains estimates of the rate of transformation between the 35 nm and nm particles for three Mg/Ca ratios.
is for the sample with Mg/Ca = 0.26; is for the sample with Mg/Ca = 0.20; and is for the sample with Mg/Ca = 0.03. The corresponding lines are guides for the eye. The table contains estimates of the rate of transformation between the 35 nm and nm particles for three Mg/Ca ratios.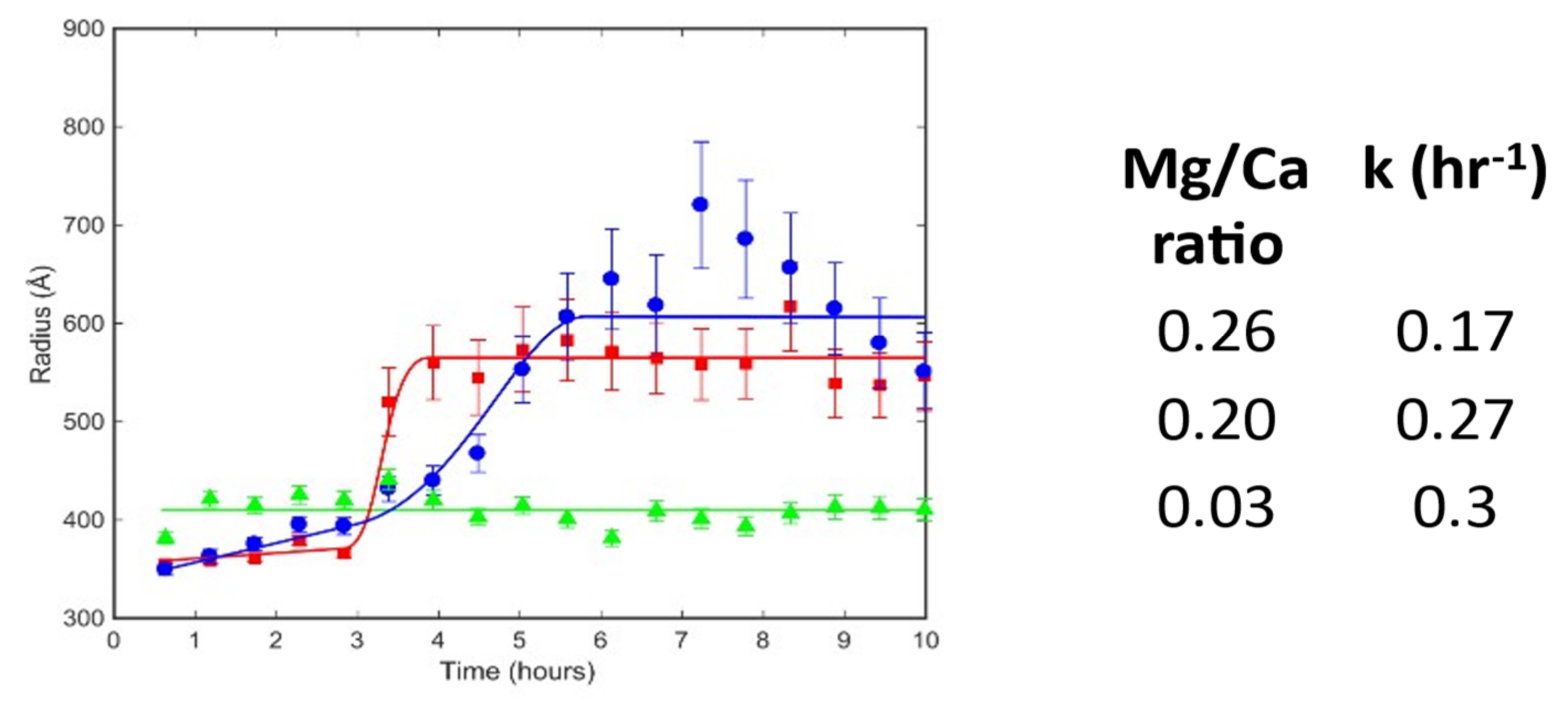
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clark, S.M.; Grigorova, V.; Colas, B.; Darwish, T.A.; Wood, K.; Neuefeind, J.; Jacob, D.E. The Kinetics of Aragonite Formation from Solution via Amorphous Calcium Carbonate. Nanomaterials 2022, 12, 4151. https://doi.org/10.3390/nano12234151
Clark SM, Grigorova V, Colas B, Darwish TA, Wood K, Neuefeind J, Jacob DE. The Kinetics of Aragonite Formation from Solution via Amorphous Calcium Carbonate. Nanomaterials. 2022; 12(23):4151. https://doi.org/10.3390/nano12234151
Chicago/Turabian StyleClark, Simon M., Vili Grigorova, Bruno Colas, Tamim A. Darwish, Kathleen Wood, Joerg Neuefeind, and Dorrit E. Jacob. 2022. "The Kinetics of Aragonite Formation from Solution via Amorphous Calcium Carbonate" Nanomaterials 12, no. 23: 4151. https://doi.org/10.3390/nano12234151
APA StyleClark, S. M., Grigorova, V., Colas, B., Darwish, T. A., Wood, K., Neuefeind, J., & Jacob, D. E. (2022). The Kinetics of Aragonite Formation from Solution via Amorphous Calcium Carbonate. Nanomaterials, 12(23), 4151. https://doi.org/10.3390/nano12234151