
Orbital Modulation with P Doping Improves Acid and Alkaline Hydrogen Evolution Reaction of MoS2



Abstract
:1. Introduction
2. Computational Model and Methods
3. Results and Discussion
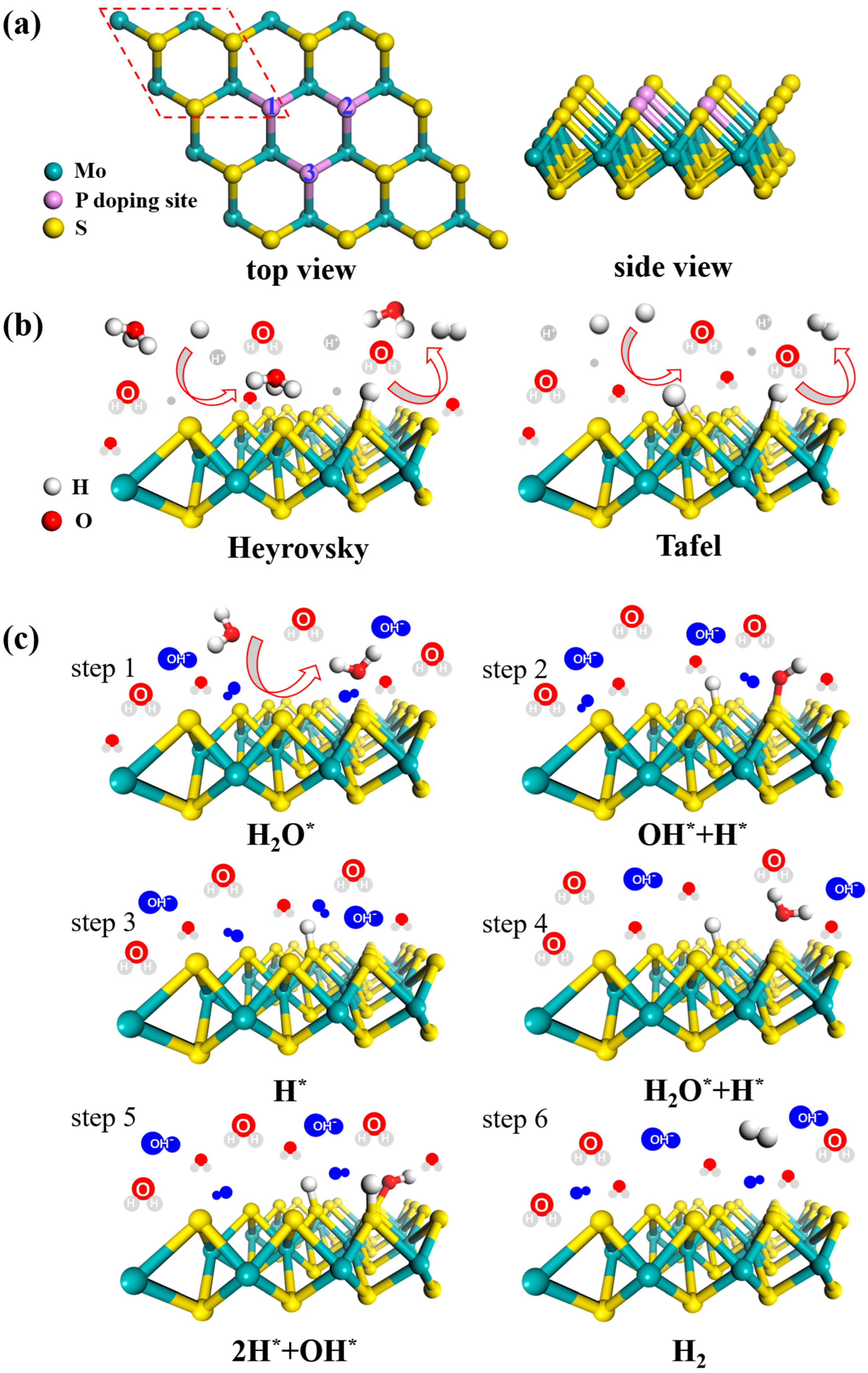
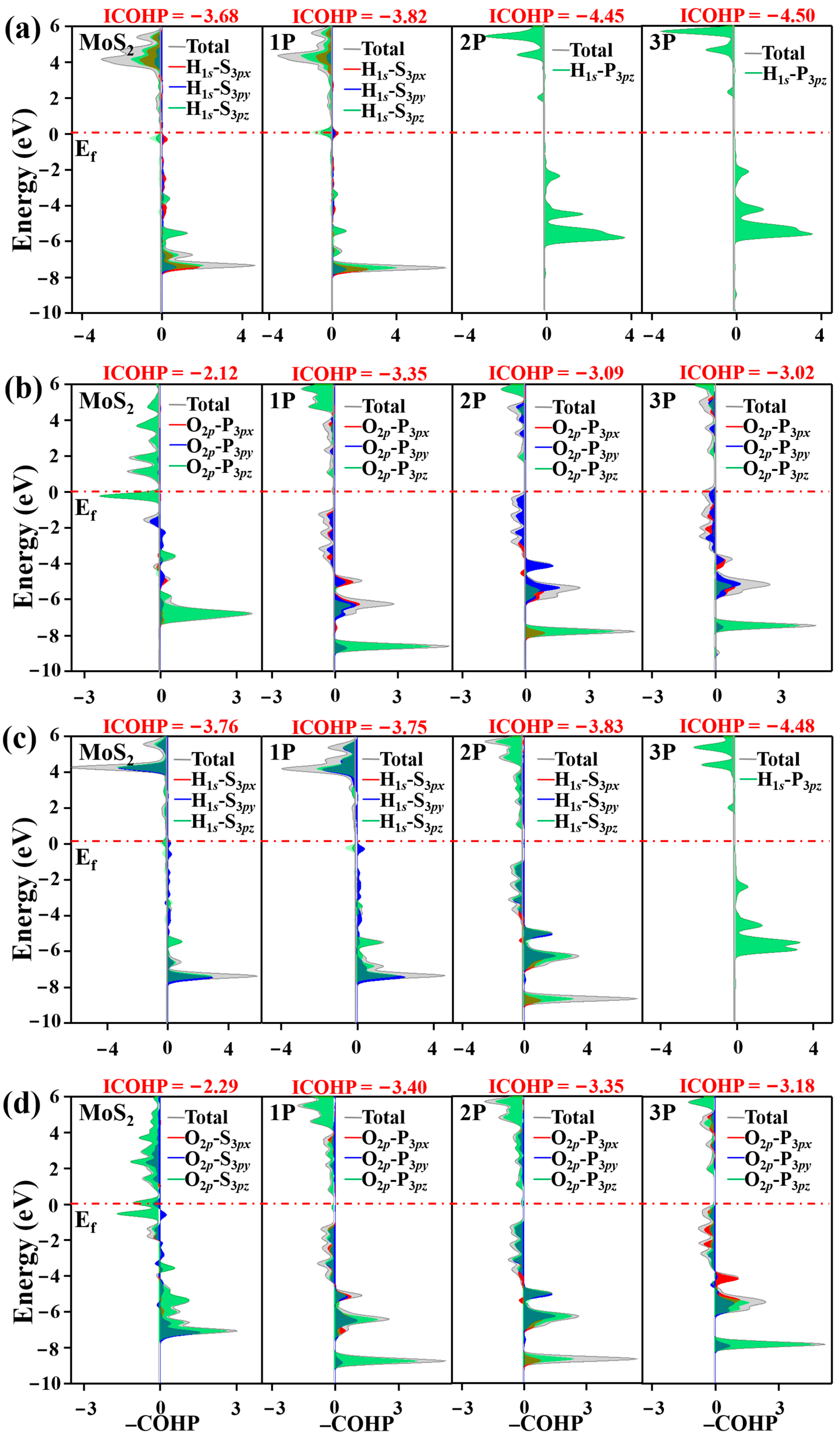
3.1. Electric Structure and Stability of P-doped MoS2
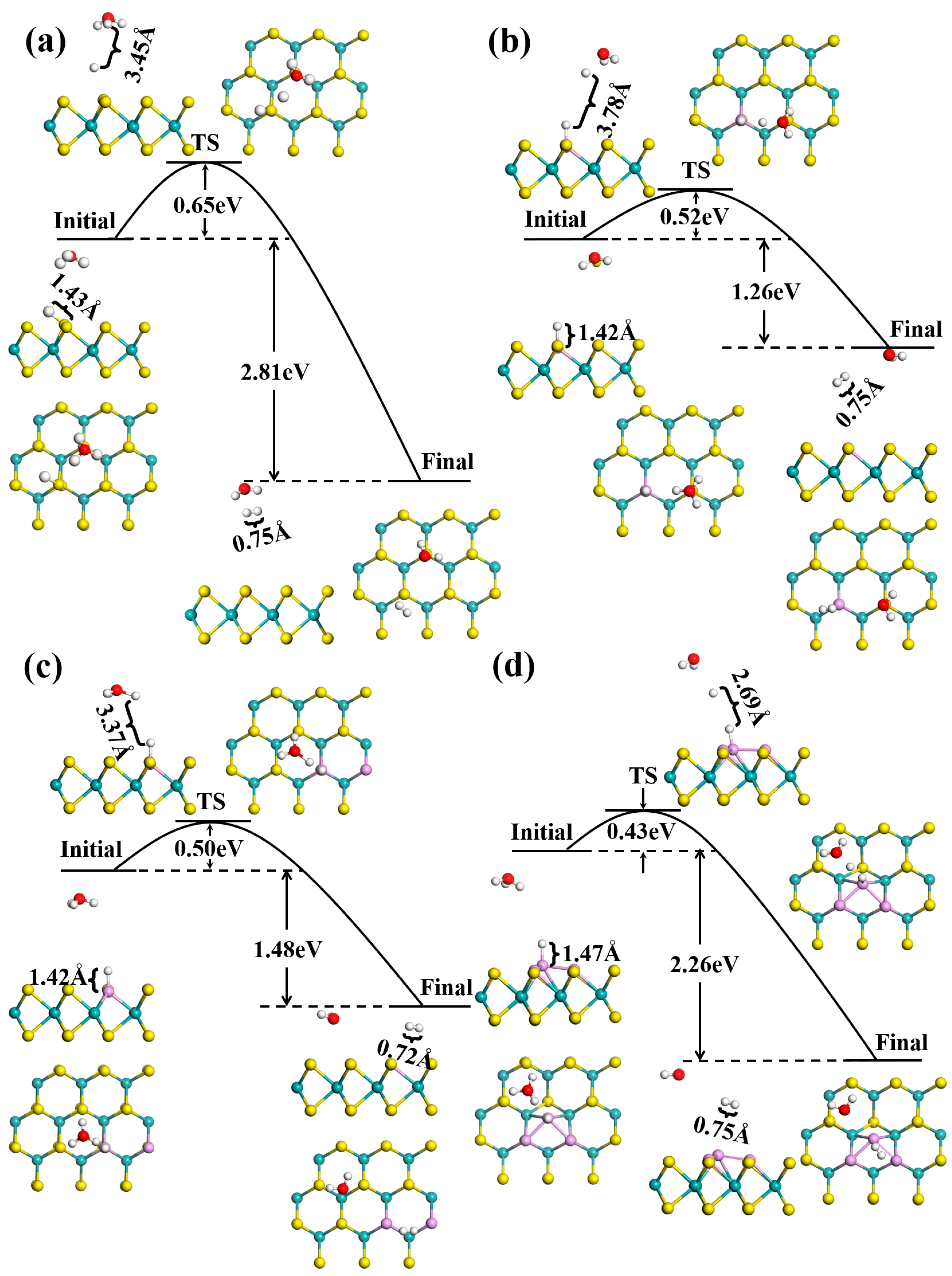
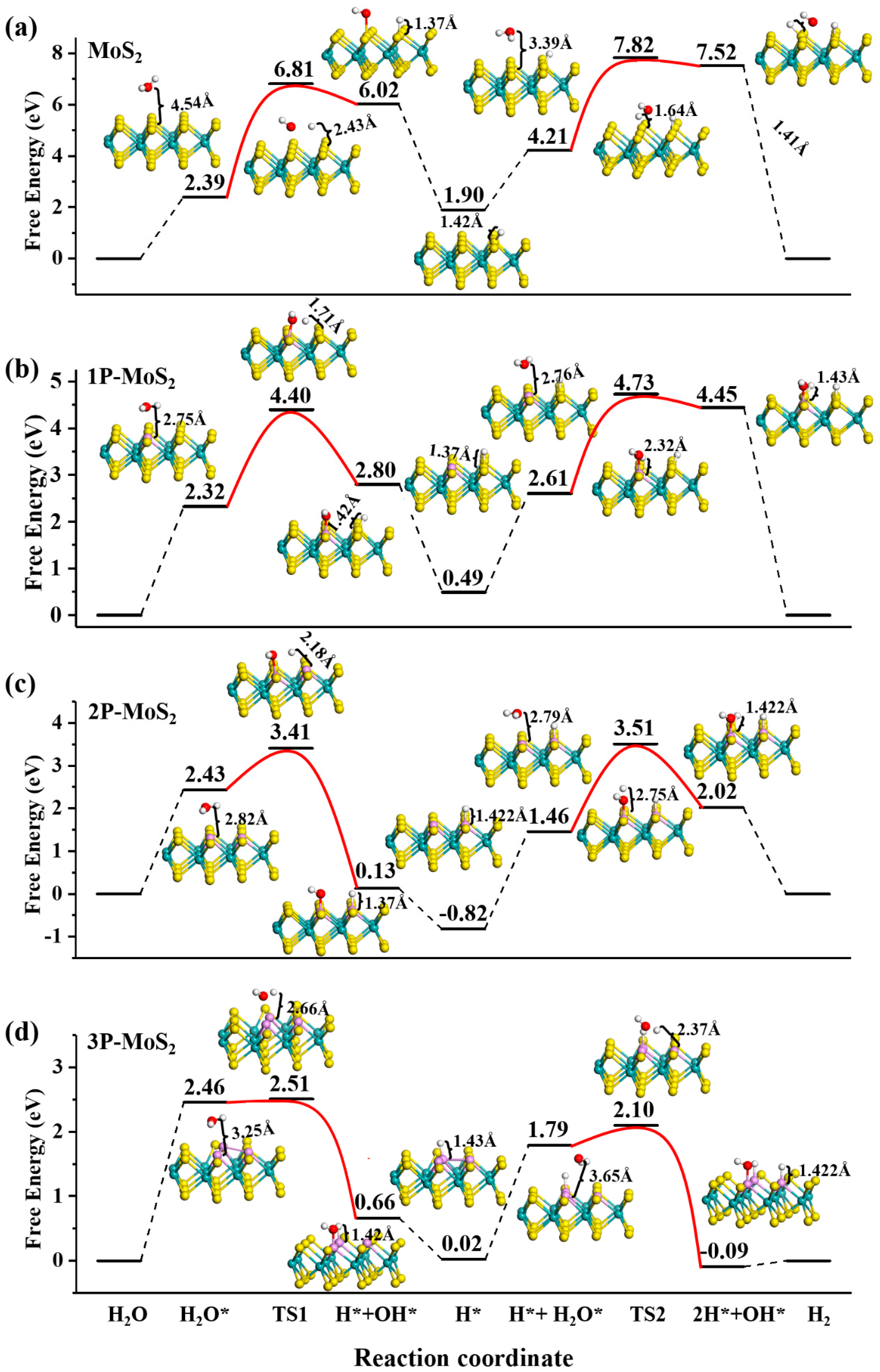
3.2. Acid HER on MoS2 Surface
3.3. Alkaline HER on MoS2 Surface
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Z.; Li, C.; Domen, K. Recent Developments in Heterogeneous Photocatalysts for Solar-Driven Overall Water Splitting. Chem. Soc. Rev. 2019, 48, 2109–2125. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hisatomi, T.; Suzuki, Y.; Pan, Z.; Seo, J.; Katayama, M.; Minegishi, T.; Nishiyama, H.; Takata, T.; Seki, K.; et al. Particulate Photocatalyst Sheets Based on Carbon Conductor Layer for Efficient Z-Scheme Pure-Water Splitting at Ambient Pressure. J. Am. Chem. Soc. 2017, 139, 1675–1683. [Google Scholar] [CrossRef]
- Hou, Y.; Abrams, B.L.; Vesborg, P.C.K.; Björketun, M.E.; Herbst, K.; Bech, L.; Setti, A.M.; Damsgaard, C.D.; Pedersen, T.; Hansen, O.; et al. Bioinspired Molecular Co-Catalysts Bonded to a Silicon Photocathode for Solar Hydrogen Evolution. Nat. Mater. 2011, 10, 434–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greeley, J.; Jaramillo, T.F.; Bonde, J.; Chorkendorff, I.; Nørskov, J.K. Computational High-Throughput Screening of Electrocatalytic Materials for Hydrogen Evolution. Nat. Mater. 2006, 5, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.R.; Liang, J.X.; Zheng, Y.R.; Xu, Y.F.; Jiang, J.; Gao, Q.; Li, J.; Yu, S.H. An Efficient Molybdenum Disulfide/Cobalt Diselenide Hybrid Catalyst for Electrochemical Hydrogen Generation. Nat. Commun. 2015, 6, 5982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greeley, J.; Stephens, I.E.L.; Bondarenko, A.S.; Johansson, T.P.; Hansen, H.A.; Jaramillo, T.F.; Rossmeisl, J.; Chorkendorff, I.; Nørskov, J.K. Alloys of Platinum and Early Transition Metals as Oxygen Reduction Electrocatalysts. Nat. Chem. 2009, 1, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Zheng, Y.; Jaroniec, M.; Qiao, S.Z. Design of Electrocatalysts for Oxygen- and Hydrogen-Involving Energy Conversion Reactions. Chem. Soc. Rev. 2015, 44, 2060–2086. [Google Scholar] [CrossRef]
- Abbas, M.A.; Bang, J.H. Rising Again: Opportunities and Challenges for Platinum-Free Electrocatalysts. Chem. Mater. 2015, 27, 7218–7235. [Google Scholar] [CrossRef]
- Wang, J.; Xu, F.; Jin, H.; Chen, Y.; Wang, Y. Non-Noble Metal-Based Carbon Composites in Hydrogen Evolution Reaction: Fundamentals to Applications. Adv. Mater. 2017, 29, 1605838. [Google Scholar] [CrossRef]
- Li, S.; Sirisomboonchai, S.; An, X.; Ma, X.; Li, P.; Ling, L.; Hao, X.; Abudula, A.; Guan, G. Engineering Interfacial Structures to Accelerate Hydrogen Evolution Efficiency of MoS2 over a Wide PH Range. Nanoscale 2020, 12, 6810–6820. [Google Scholar] [CrossRef]
- Men, Y.; Li, P.; Yang, F.; Cheng, G.; Chen, S.; Luo, W. Nitrogen-Doped CoP as Robust Electrocatalyst for High-Efficiency PH-Universal Hydrogen Evolution Reaction. Appl. Catal. B Environ. 2019, 253, 21–27. [Google Scholar] [CrossRef]
- Tan, C.A.; Topper, S.; del Gaudio, D.; Nelakuditi, V.; Shchelochkov, O.; Nowaczyk, M.J.M.; Zeesman, S.; Brady, L.; Russell, L.; Meeks, N.; et al. Characterization of Patients Referred for Non-Specific Intellectual Disability Testing: The Importance of Autosomal Genes for Diagnosis. Clin. Genet. 2016, 89, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Wang, W.; Ge, R.; Hao, S.; Qu, F.; Du, G.; Asiri, A.M.; Wei, Q.; Chen, L.; Sun, X. An Amorphous FeMoS4 Nanorod Array toward Efficient Hydrogen Evolution Electrocatalysis under Neutral Conditions. Chem. Commun. 2017, 53, 9000–9003. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, F.; Zhang, S.; Liang, Y.; Wang, R. Engineering MoS2 Basal Planes for Hydrogen Evolution via Synergistic Ruthenium Doping and Nanocarbon Hybridization. Adv. Sci. 2019, 6, 2198–3844. [Google Scholar] [CrossRef] [Green Version]
- Chhowalla, M.; Shin, H.S.; Eda, G.; Li, L.J.; Loh, K.P.; Zhang, H. The Chemistry of Two-Dimensional Layered Transition Metal Dichalcogenide Nanosheets. Nat. Chem. 2013, 5, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhou, Y.; Yang, D.R.; Xu, W.X.; Wang, C.; Wang, F.B.; Xu, J.J.; Xia, X.H.; Chen, H.Y. Energy Level Engineering of MoS2 by Transition-Metal Doping for Accelerating Hydrogen Evolution Reaction. J. Am. Chem. Soc. 2017, 139, 15479–15485. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Han, J.; Zhang, Y.; Zhang, X.; Xu, P.; Yuan, Q.; Samad, L.; Wang, X.; Wang, Y.; Zhang, Z.; et al. Contributions of Phase, Sulfur Vacancies, and Edges to the Hydrogen Evolution Reaction Catalytic Activity of Porous Molybdenum Disulfide Nanosheets. J. Am. Chem. Soc. 2016, 138, 7965–7972. [Google Scholar] [CrossRef]
- Huang, X.; Zeng, Z.; Zhang, H. Metal Dichalcogenide Nanosheets: Preparation, Properties and Applications. Chem. Soc. Rev. 2013, 42, 1934–1946. [Google Scholar] [CrossRef]
- Mamiyev, Z.; Balayeva, N.O. Metal Sulfide Photocatalysts for Hydrogen Generation: A Review of Recent Advances. Catalysts 2022, 12, 1316. [Google Scholar] [CrossRef]
- Tsai, C.; Abild-Pedersen, F.; Nørskov, J.K. Tuning the MoS2 Edge-Site Activity for Hydrogen Evolution via Support Interactions. Nano Lett. 2014, 14, 1381–1387. [Google Scholar] [CrossRef]
- Hinnemann, B.; Moses, P.G.; Bonde, J.; Jørgensen, K.P.; Nielsen, J.H.; Horch, S.; Chorkendorff, I.; Nørskov, J.K. Biomimetic Hydrogen Evolution: MoS2 Nanoparticles as Catalyst for Hydrogen Evolution. J. Am. Chem. Soc. 2005, 127, 5308–5309. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Jiao, Y.; Jaroniec, M.; Qiao, S.Z. Advancing the Electrochemistry of the Hydrogen- Evolution Reaction through Combining Experiment. Angew. Chem.-Int. Ed. 2015, 54, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.E. Faraday Discussions: Preface. Faraday Discuss. 2008, 140, 9–10. [Google Scholar] [CrossRef]
- Pan, H.; Zhang, Y.W. Tuning the electronic and magnetic properties of MoS2 nanoribbons by strain engineering. J. Phys. Chem. C 2012, 116, 11752–11757. [Google Scholar] [CrossRef]
- Pan, J.; Wang, R.; Zhou, X.; Zhong, J.; Xu, X.; Hu, J. Transition-Metal Doping Induces the Transition of Electronic and Magnetic Properties in Armchair MoS2 Nanoribbons. Phys. Chem. Chem. Phys. 2017, 19, 24594–24604. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Tsai, C.; Koh, A.L.; Cai, L.; Contryman, A.W.; Fragapane, A.H.; Zhao, J.; Han, H.S.; Manoharan, H.C.; Abild-Pedersen, F.; et al. Erratum: Activating and Optimizing MoS2 Basal Planes for Hydrogen Evolution through the Formation of Strained Sulphur Vacancies. Nat. Mater. 2016, 15, 364. [Google Scholar] [CrossRef] [Green Version]
- Zang, Y.; Niu, S.; Wu, Y.; Zheng, X.; Cai, J.; Ye, J.; Xie, Y.; Liu, Y.; Zhou, J.; Zhu, J.; et al. Tuning Orbital Orientation Endows Molybdenum Disulfide with Exceptional Alkaline Hydrogen Evolution Capability. Nat. Commun. 2019, 10, 1217. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Zhang, M.; Yang, H.; Li, G.; Xing, S.; Li, M.; Xu, Y.; Zhang, Q.; Hu, S.; Liao, H.; et al. Creating Fluorine-Doped MoS2 Edge Electrodes with Enhanced Hydrogen Evolution Activity. Small Methods 2021, 5, 2100612. [Google Scholar] [CrossRef]
- Xiao, W.; Liu, P.; Zhang, J.; Song, W.; Feng, Y.P.; Gao, D.; Ding, J. Dual-Functional N Dopants in Edges and Basal Plane of MoS2 Nanosheets Toward Efficient and Durable Hydrogen Evolution. Adv. Energy Mater. 2017, 7, 1602086. [Google Scholar] [CrossRef]
- Xie, J.; Zhang, J.; Li, S.; Grote, F.; Zhang, X.; Zhang, H.; Wang, R.; Lei, Y.; Pan, B.; Xie, Y. Controllable Disorder Engineering in Oxygen-Incorporated MoS2 Ultrathin Nanosheets for Efficient Hydrogen Evolution. J. Am. Chem. Soc. 2013, 135, 17881–17888. [Google Scholar] [CrossRef]
- Zhao, B.; Shang, C.; Qi, N.; Chen, Z.Y.; Chen, Z.Q. Stability of Defects in Monolayer MoS2 and Their Interaction with O2 Molecule: A First-Principles Study. Appl. Surf. Sci. 2017, 412, 385–393. [Google Scholar] [CrossRef]
- Xie, J.; Zhang, H.; Li, S.; Wang, R.; Sun, X.; Zhou, M.; Zhou, J.; Wen, X.; Lou, D.; Xie, Y. Defect-Rich MoS2 Ultrathin Nanosheets with Additional Active Edge Sites for Enhanced Electrocatalytic Hydrogen Evolution. Adv. Mater. 2013, 25, 5807–5813. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, J.A.; Klysubun, W.; Wang, G.G.; Sattayaporn, S.; Li, F.; Cai, Y.W.; Zhang, F.; Yu, J.; Yang, Y. Interfacial Electronic Structure Engineering on Molybdenum Sulfide for Robust Dual-PH Hydrogen Evolution. Nat. Commun. 2021, 12, 5260. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Zhu, Y.; Gu, X.; Dai, S.; Mao, Q.; Bao, L.; Li, W.; Liu, S.; Bao, J.; Mu, S. Awakening the Oxygen Evolution Activity of MoS2 by Oxophilic-Metal Induced Surface Reorganization Engineering. J. Energy Chem. 2021, 62, 546–551. [Google Scholar] [CrossRef]
- Liang, K.; Pakhira, S.; Yang, Z.; Nijamudheen, A.; Ju, L.; Wang, M.; Aguirre-Velez, C.I.; Sterbinsky, G.E.; Du, Y.; Feng, Z.; et al. S-Doped MoP Nanoporous Layer Toward High-Efficiency Hydrogen Evolution in PH-Universal Electrolyte. ACS Catal. 2019, 9, 651–659. [Google Scholar] [CrossRef]
- Liu, P.; Zhu, J.; Zhang, J.; Xi, P.; Tao, K.; Gao, D.; Xue, D. P Dopants Triggered New Basal Plane Active Sites and Enlarged Interlayer Spacing in MoS2 Nanosheets toward Electrocatalytic Hydrogen Evolution. ACS Energy Lett. 2017, 2, 745–752. [Google Scholar] [CrossRef]
- Bian, L.; Gao, W.; Sun, J.; Han, M.; Li, F.; Gao, Z.; Shu, L.; Han, N.; Yang, Z.X.; Song, A.; et al. Phosphorus-Doped MoS2 Nanosheets Supported on Carbon Cloths as Efficient Hydrogen-Generation Electrocatalysts. ChemCatChem 2018, 10, 1571–1577. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Xu, H.; Cao, D.; Cheng, D. Interface Construction of P-Substituted MoS2 as Efficient and Robust Electrocatalyst for Alkaline Hydrogen Evolution Reaction. Nano Energy 2020, 78, 105253. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B-Condens. Matter Mater. Phys. 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B-Condens. Matter Mater. Phys. 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
- Ye, R.; Del Angel-Vicente, P.; Liu, Y.; Arellano-Jimenez, M.J.; Peng, Z.; Wang, T.; Li, Y.; Yakobson, B.I.; Wei, S.H.; Yacaman, M.J.; et al. High-Performance Hydrogen Evolution from MoS2(1−x)Px Solid Solution. Adv. Mater. 2016, 28, 1427–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, K.F.; Lee, C.; Hone, J.; Shan, J.; Heinz, T.F. Atomically Thin MoS2: A New Direct-Gap Semiconductor. Phys. Rev. Lett. 2010, 105, 2–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böker, T.; Severin, R.; Müller, A.; Janowitz, C.; Manzke, R.; Voß, D.; Krüger, P.; Mazur, A.; Pollmann, J. Band Structure of (Formula Presented) (Formula Presented) and (Formula Presented) Angle-Resolved Photoelectron Spectroscopy and Ab Initio Calculations. Phys. Rev. B-Condens. Matter Mater. Phys. 2001, 64, 235305. [Google Scholar] [CrossRef] [Green Version]
- Cui, Z.; Sa, R.; Du, W.; Xiao, C.; Li, Q.; Ma, Z. Theoretical Screening of Group IIIA-VIIA Elements Doping to Promote Hydrogen Evolution of MoS2 Basal Plane. Appl. Surf. Sci. 2021, 542, 148535. [Google Scholar] [CrossRef]
- Tang, Q.; Jiang, D.E. Mechanism of Hydrogen Evolution Reaction on 1T-MoS2 from First Principles. ACS Catal. 2016, 6, 4953–4961. [Google Scholar] [CrossRef]
- Lai, J.; Huang, B.; Chao, Y.; Chen, X.; Guo, S. Strongly Coupled Nickel–Cobalt Nitrides/Carbon Hybrid Nanocages with Pt-Like Activity for Hydrogen Evolution Catalysis. Adv. Mater. 2019, 31, 1805541. [Google Scholar] [CrossRef]
- Lv, F.; Feng, J.; Wang, K.; Dou, Z.; Zhang, W.; Zhou, J.; Yang, C.; Luo, M.; Yang, Y.; Li, Y.; et al. Iridium-Tungsten Alloy Nanodendrites as PH-Universal Water-Splitting Electrocatalysts. ACS Cent. Sci. 2018, 4, 1244–1252. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Wang, R.; Xu, X.; Hu, J.; Ma, L. Transition Metal Doping Activated Basal-Plane Catalytic Activity of Two-Dimensional 1T’-ReS2 for Hydrogen Evolution Reaction: A First-Principles Calculation Study. Nanoscale 2019, 11, 10402–10409. [Google Scholar] [CrossRef]
- Pi, C.; Huang, C.; Yang, Y.; Song, H.; Zhang, X.; Zheng, Y.; Gao, B.; Fu, J.; Chu, P.K.; Huo, K. In Situ Formation of N-Doped Carbon-Coated Porous MoP Nanowires: A Highly Efficient Electrocatalyst for Hydrogen Evolution Reaction in a Wide PH Range. Appl. Catal. B Environ. 2020, 263, 118358. [Google Scholar] [CrossRef]
- Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. Crystal Orbital Hamilton Population (COHP) Analysis as Projected from Plane-Wave Basis Sets. J. Phys. Chem. A 2011, 115, 5461–5466. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, K.; Li, Y.; Yuan, S.; Huang, G.; Li, X.; Li, N. Activation Engineering on Metallic 1T-MoS2 by Constructing In-Plane Heterostructure for Efficient Hydrogen Generation. Appl. Catal. B Environ. 2022, 300, 120696. [Google Scholar] [CrossRef]
- Liang, X.; Wu, C.M.L. Metal-Free Two-Dimensional Phosphorus Carbide as an Efficient Electrocatalyst for Hydrogen Evolution Reaction Comparable to Platinum. Nano Energy 2020, 71, 104603. [Google Scholar] [CrossRef]
- Dronskowski, R.; Festkbrperforschung, M.; Blochl, P.E. Crystal Orbital Hamilton Populations (COHP). Energy-Resolved Visualization of Chemical Bonding in Solids Based on Density-Functional Calculations. J. Phys. Chem. 1993, 97, 8617–8624. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | The First Step | The Second Step | The Third Step | The Fourth Step | The Fifth Step | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2O* | ΔG | H* | OH* | ΔE | H* | ΔG | H* | H2O* | ΔG | H1* | H2* | HO* | ΔE | |
| pure | S | 2.39 | S | S | 4.42 | S | 1.90 | S | S | 2.31 | S | S | S | 3.61 |
| 1P | P | 2.32 | S | P | 2.08 | S | 0.49 | S | P | 2.12 | S | S | P | 2.12 |
| 2P | P | 2.43 | P | P | 0.98 | P | –0.82 | P | P | 2.28 | P | S | P | 2.05 |
| 3P | P | 2.46 | P | P | 0.13 | P | 0.02 | P | P | 1.77 | P | P | P | 0.31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, F.; Zhang, M.; Xu, X.; Pan, J.; Zhu, L.; Hu, J. Orbital Modulation with P Doping Improves Acid and Alkaline Hydrogen Evolution Reaction of MoS2. Nanomaterials 2022, 12, 4273. https://doi.org/10.3390/nano12234273
Dong F, Zhang M, Xu X, Pan J, Zhu L, Hu J. Orbital Modulation with P Doping Improves Acid and Alkaline Hydrogen Evolution Reaction of MoS2. Nanomaterials. 2022; 12(23):4273. https://doi.org/10.3390/nano12234273
Chicago/Turabian StyleDong, Fuyu, Minghao Zhang, Xiaoyong Xu, Jing Pan, Liyan Zhu, and Jingguo Hu. 2022. "Orbital Modulation with P Doping Improves Acid and Alkaline Hydrogen Evolution Reaction of MoS2" Nanomaterials 12, no. 23: 4273. https://doi.org/10.3390/nano12234273