
Size-Dependent Thermal Stability and Optical Properties of Ultra-Small Nanodiamonds Synthesized under High Pressure

 , ,
, ,  and
and 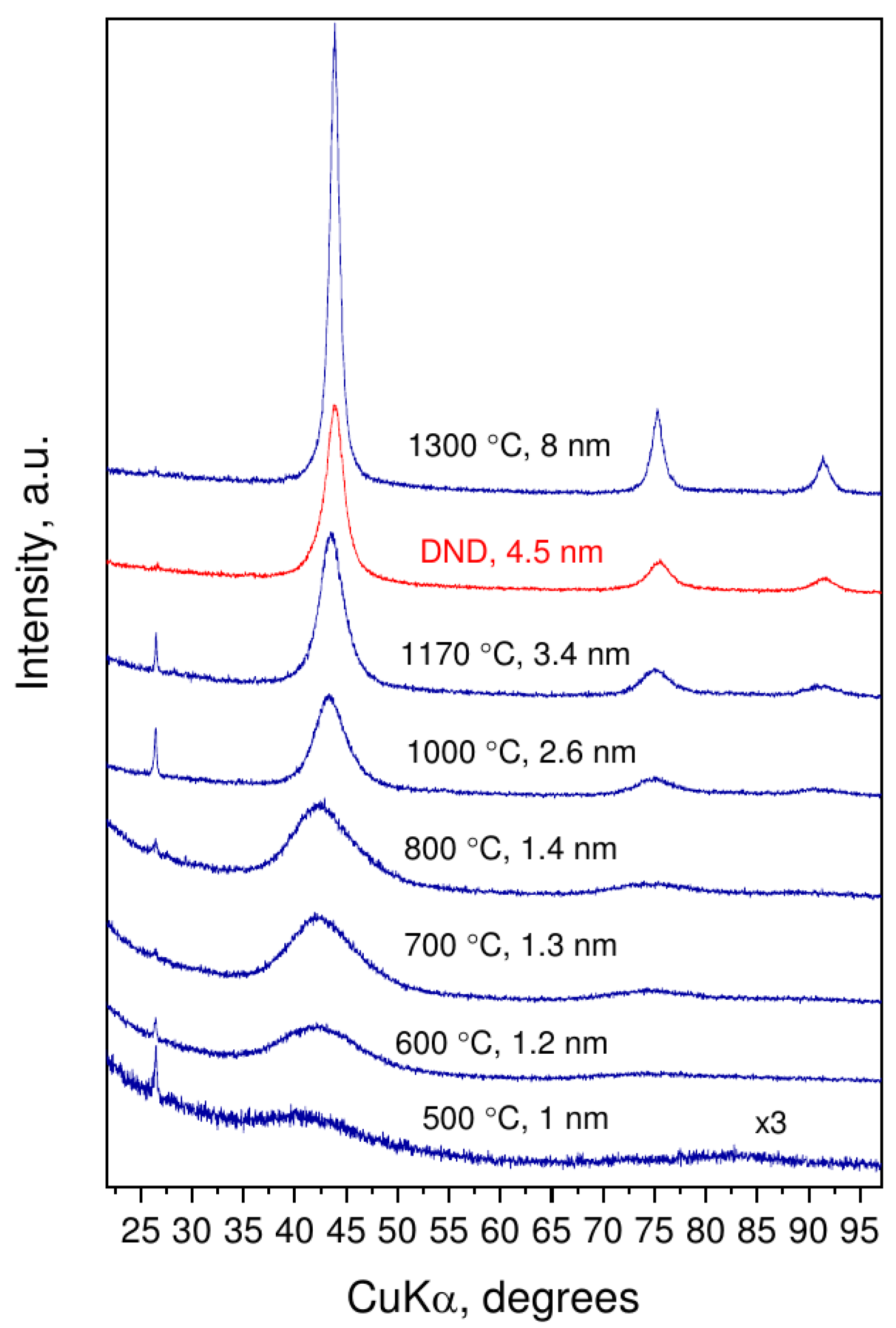{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
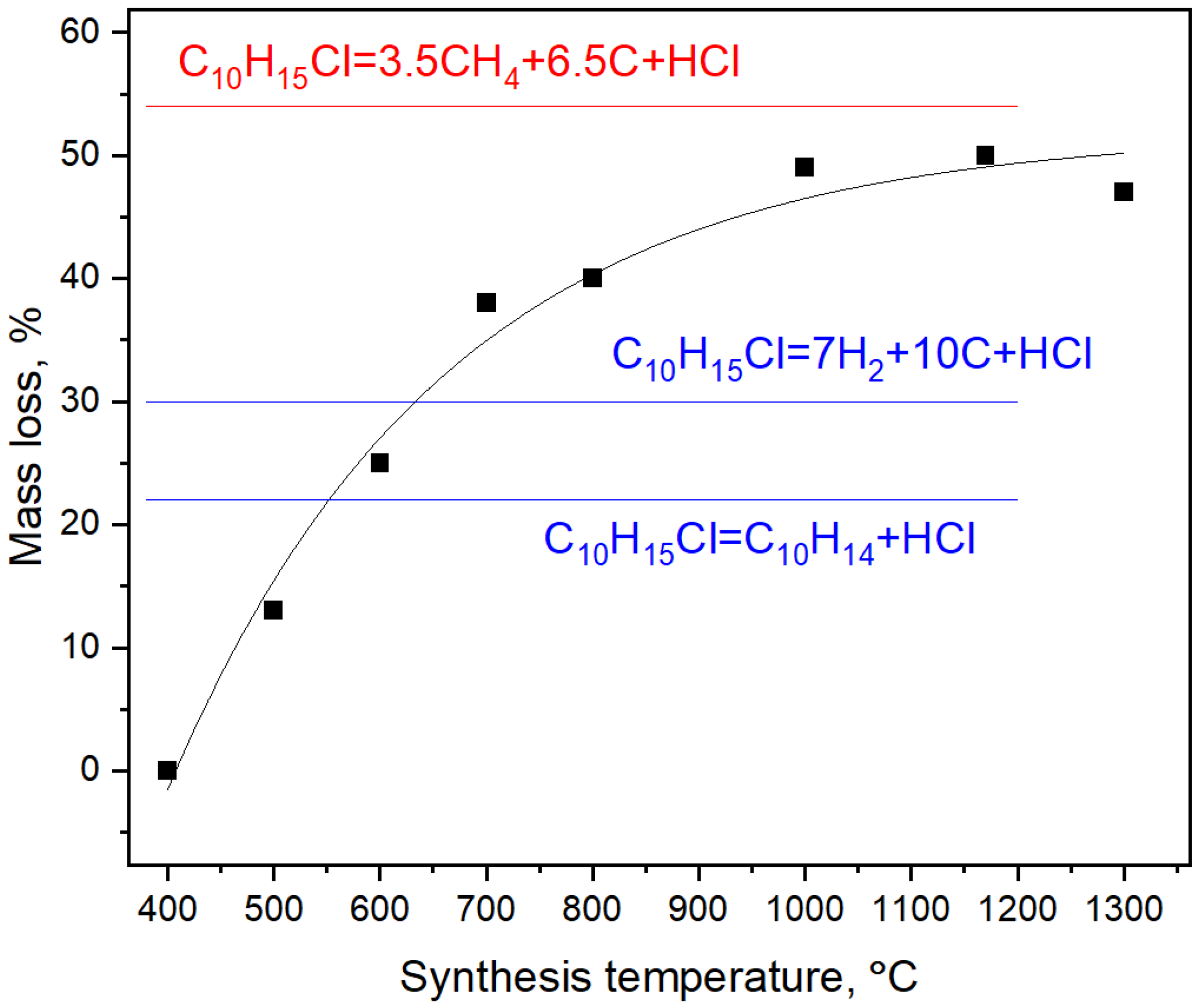
3.1. Synthesis
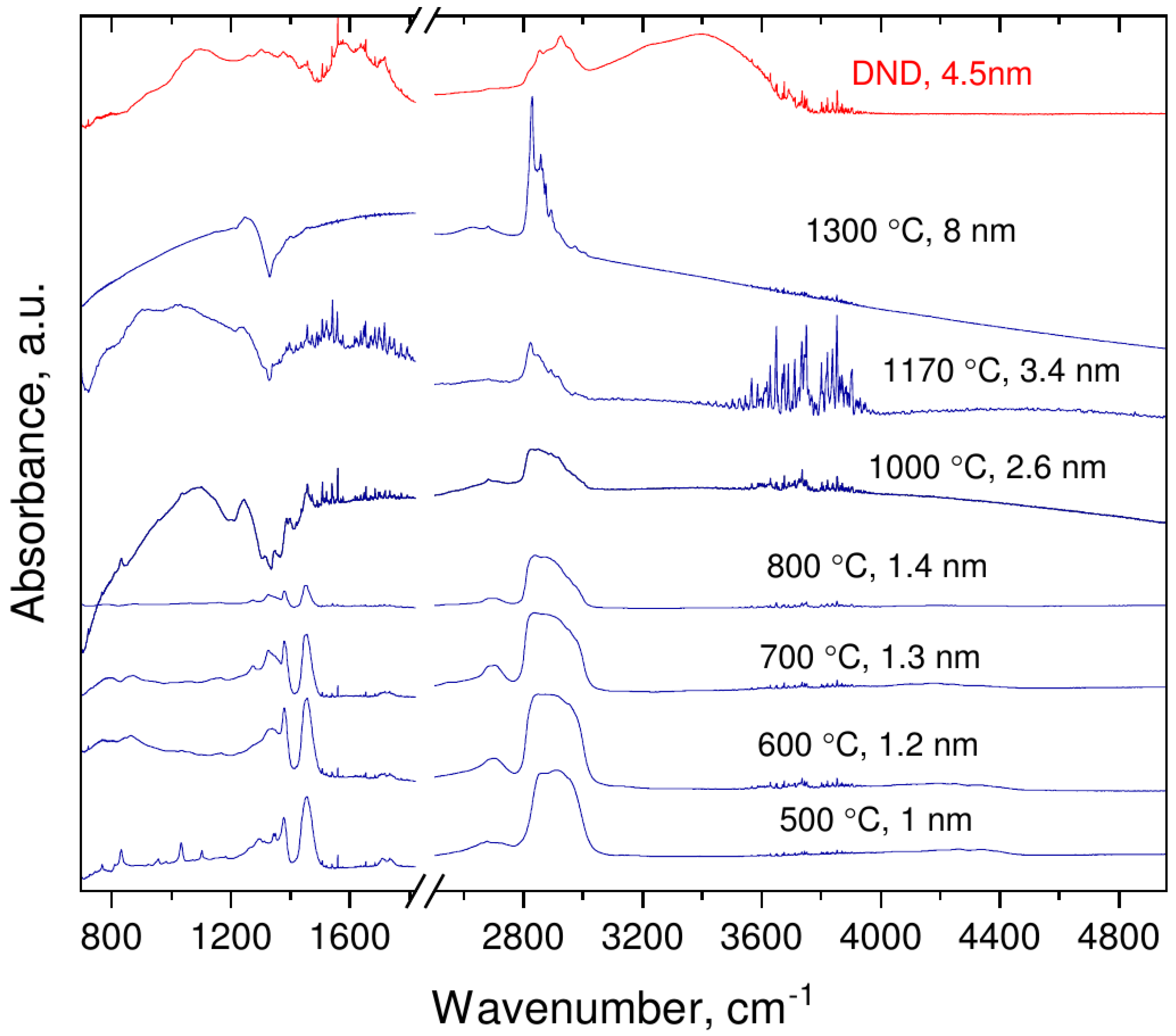
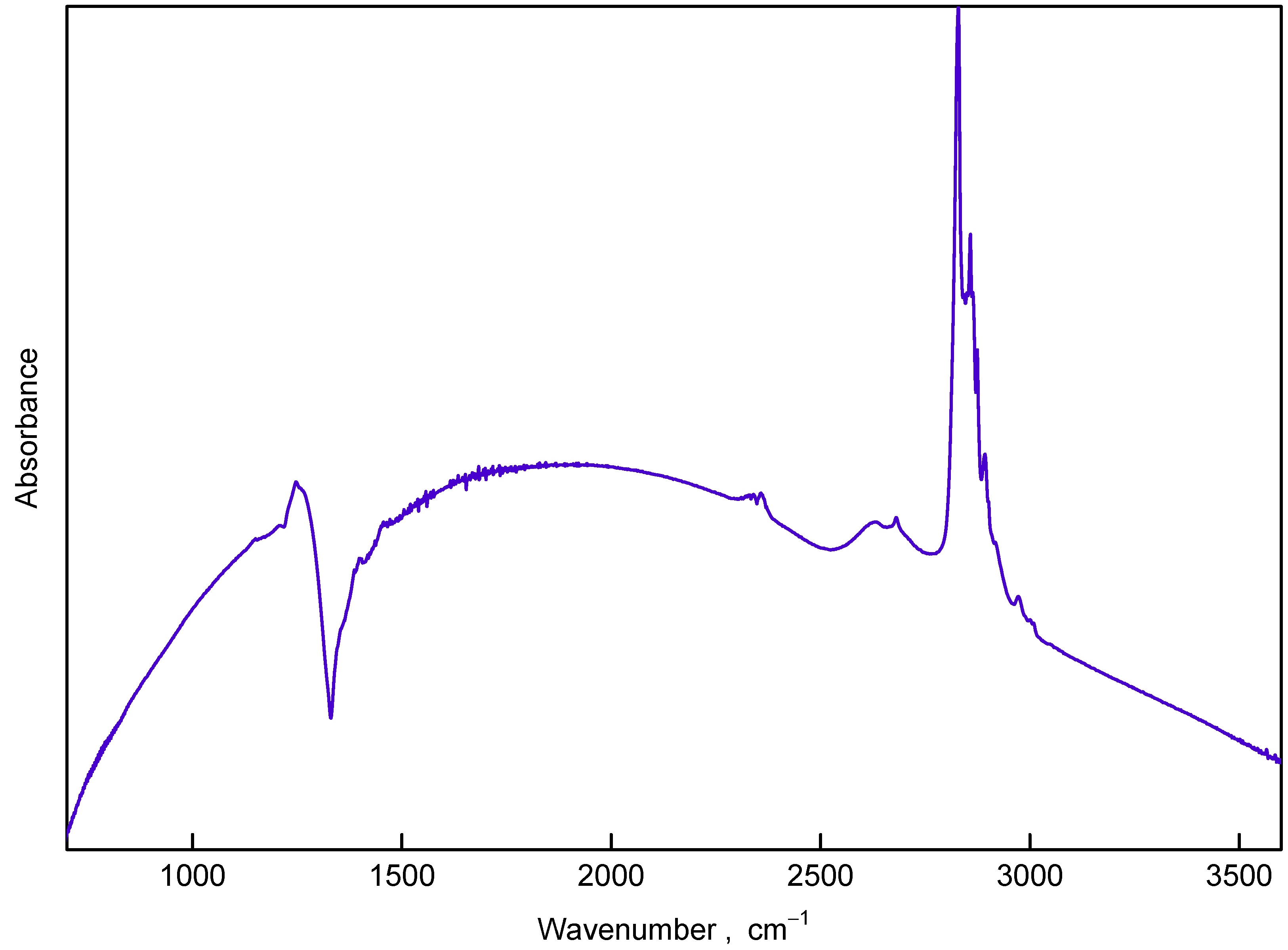
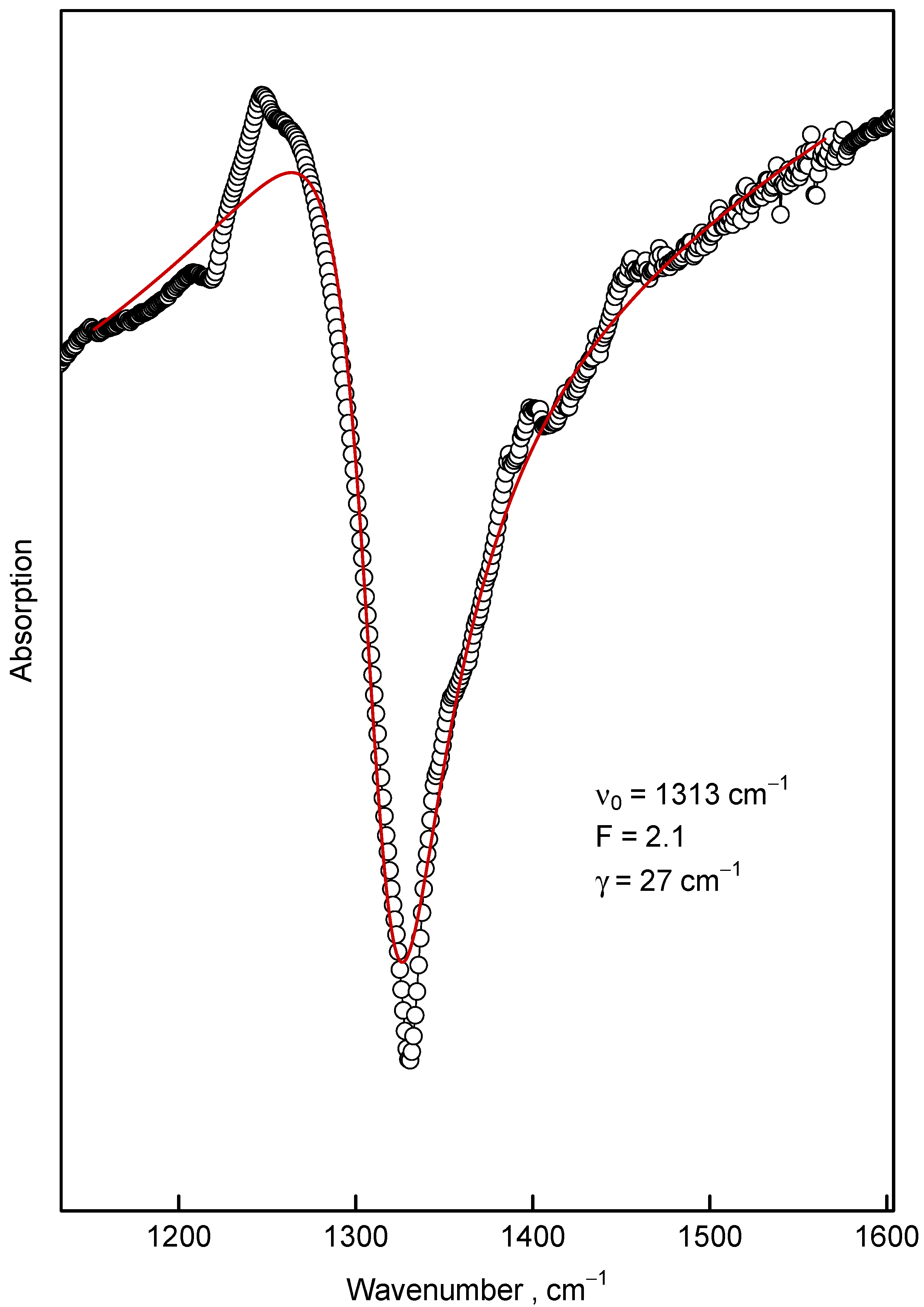
3.2. Infrared Spectroscopy
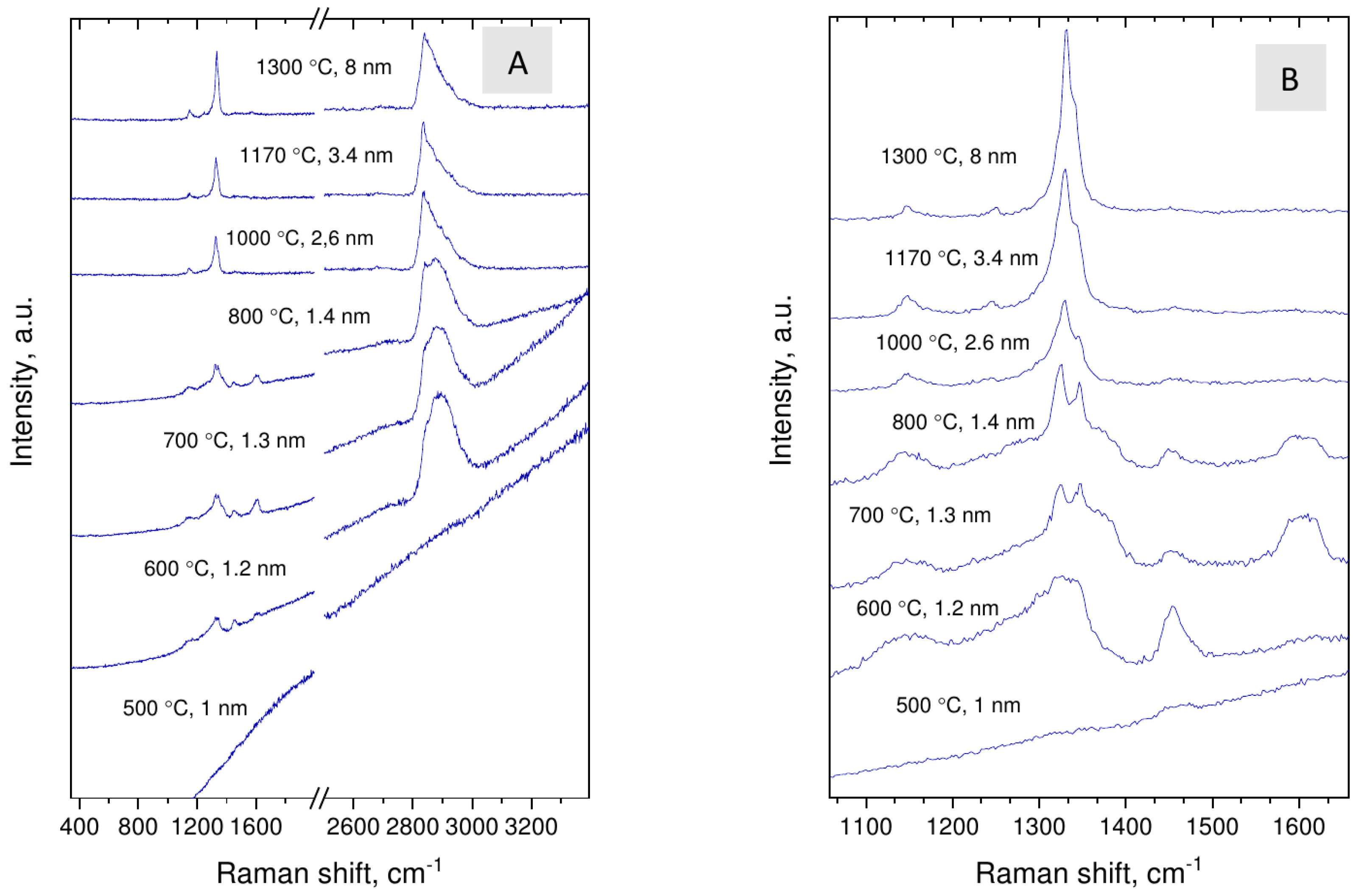
3.3. Raman Spectroscopy
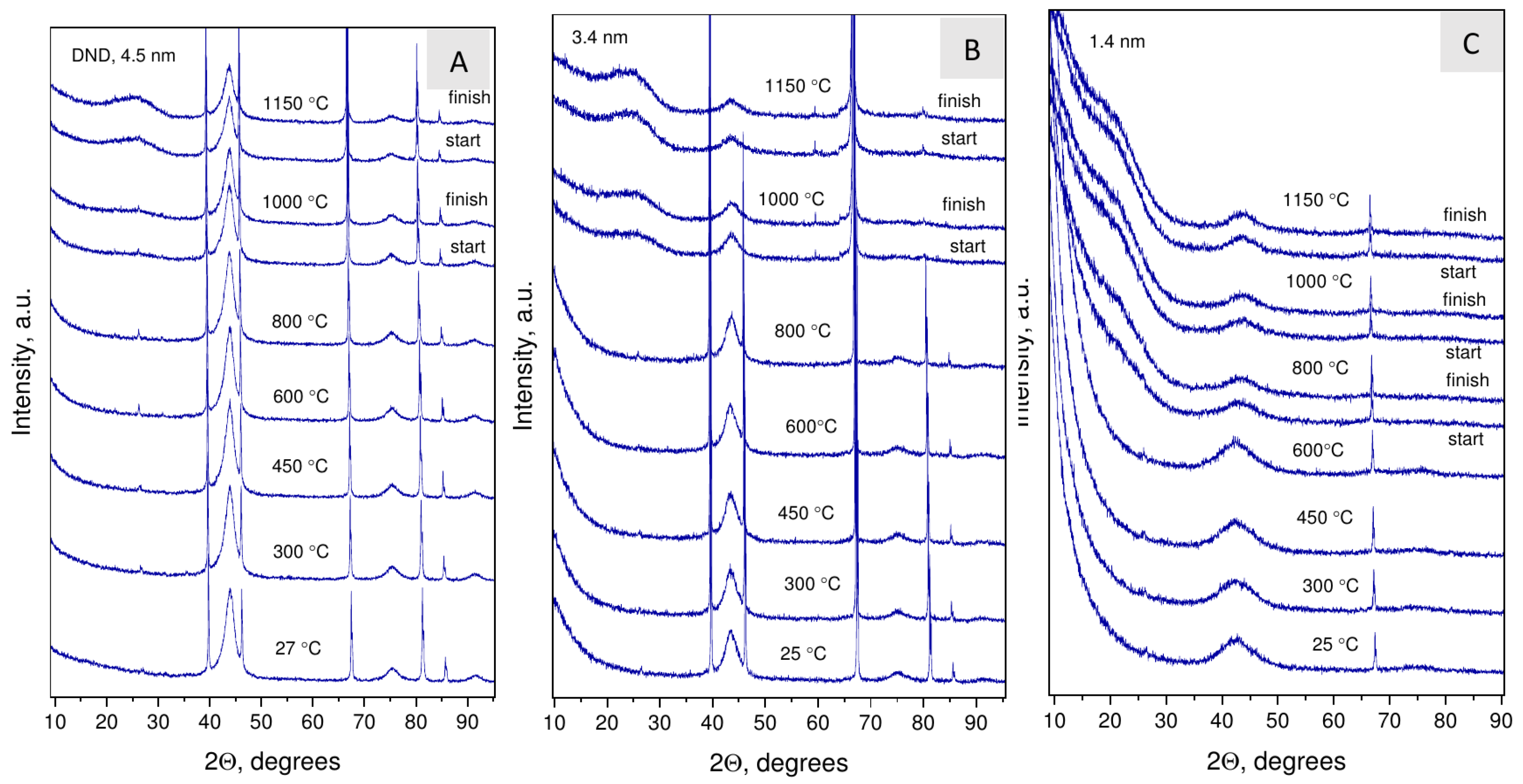
3.4. Thermal Behavior of Nanodiamond Samples: Graphitization
4. Discussion
4.1. Synthesis Mechanism
4.2. Fano Effect in IR and Raman Spectra
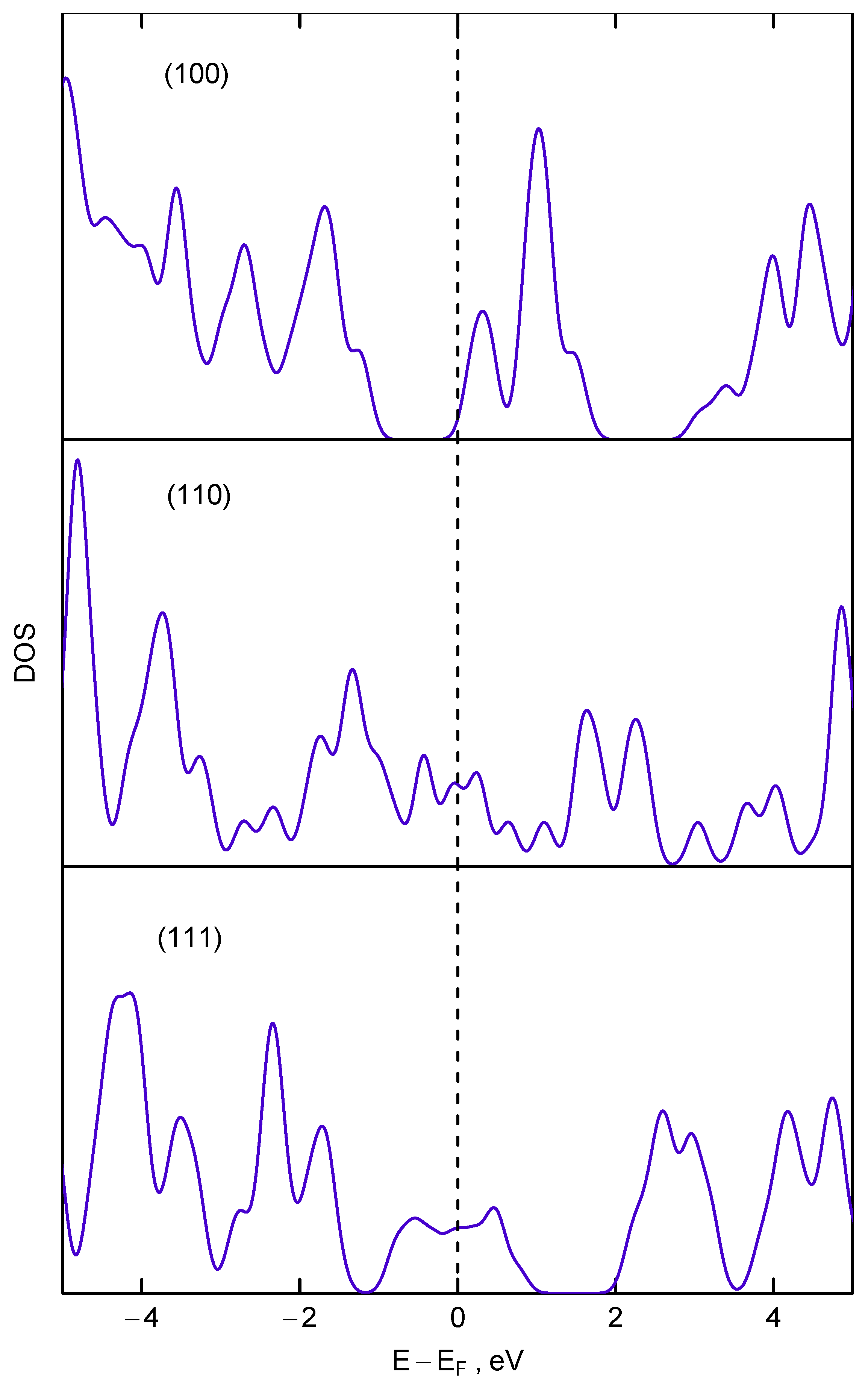
4.3. Electrical Conductivity Measurements
4.4. Thermal Annealing
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stehlik, S.; Varga, M.; Ledinsky, M.; Jirasek, V.; Artemenko, A.; Kozak, H.; Ondic, L.; Skakalova, V.; Argentero, G.; Pennycook, T.; et al. Size and Purity Control of HPHT Nanodiamonds down to 1 nm. J. Phys. Chem. C 2015, 119, 27708–27720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stehlik, S.; Varga, M.; Stenclova, P.; Ondic, L.; Ledinsky, M.; Pangrac, J.; Vanek, O.; Lipov, J.; Kromka, A.; Rezek, B. Ultrathin Nanocrystalline Diamond Films with Silicon Vacancy Color Centers via Seeding by 2 nm Detonation Nanodiamonds. ACS Appl. Mater. Interfaces 2017, 9, 38842–38853. [Google Scholar] [CrossRef] [PubMed]
- Shenderova, O.A.; Shames, A.I.; Nunn, N.A.; Torelli, M.D.; Vlasov, I.; Zaitsev, A. Review Article: Synthesis, properties, and applications of fluorescent diamond particles. J. Vac. Sci. Technol. B 2019, 37, 030802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekimov, E.A.; Kondrin, M.V. Vacancy-impurity centers in diamond: Perspectives of synthesis and applications. Phys. Uspekhi 2017, 60, 539–558. [Google Scholar] [CrossRef]
- Stehlik, S.; Mermoux, M.; Schummer, B.; Vanek, O.; Kolarova, K.; Stenclova, P.; Vlk, A.; Ledinsky, M.; Pfeifer, R.; Romanyuk, O.; et al. Size Effects on Surface Chemistry and Raman Spectra of Sub-5 nm Oxidized High-Pressure High-Temperature and Detonation Nanodiamonds. J. Phys. Chem. C 2021, 125, 5647–5669. [Google Scholar] [CrossRef]
- Kuznetsov, V.L.; Butenko, Y.V. 13—Diamond Phase Transitions at Nanoscale. In Ultrananocrystalline Diamond; Shenderova, O.A., Gruen, D.M., Eds.; William Andrew Publishing: Norwich, NY, USA, 2006; pp. 405–475. [Google Scholar] [CrossRef]
- Zhao, D.; Zhao, M.; Jiang, Q. Size and temperature dependence of nanodiamond–nanographite transition related with surface stress. Diam. Relat. Mater. 2002, 11, 234–236. [Google Scholar] [CrossRef]
- Sun, Y.; Kvashnin, A.G.; Sorokin, P.B.; Yakobson, B.I.; Billups, W.E. Radiation-Induced Nucleation of Diamond from Amorphous Carbon: Effect of Hydrogen. J. Phys. Chem. Lett. 2014, 5, 1924–1928. [Google Scholar] [CrossRef]
- Barnard, A.S.; Russo, S.P.; Snook, I.K. Size dependent phase stability of carbon nanoparticles: Nanodiamond versus fullerenes. J. Chem. Phys. 2003, 118, 5094–5097. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Chen, J.; Yang, G.; Xu, N. Thermodynamic Stability and Ultrasmall-Size Effect of Nanodiamonds. Angew. Chem. Int. Ed. 2005, 44, 7414–7418. [Google Scholar] [CrossRef]
- Butenko, Y.V.; Kuznetsov, V.L.; Chuvilin, A.L.; Kolomiichuk, V.N.; Stankus, S.V.; Khairulin, R.A.; Segall, B. Kinetics of the graphitization of dispersed diamonds at “low” temperatures. J. Appl. Phys. 2000, 88, 4380–4388. [Google Scholar] [CrossRef]
- Xu, N.; Chen, J.; Deng, S. Effect of heat treatment on the properties of nano-diamond under oxygen and argon ambient. Diam. Relat. Mater. 2002, 11, 249–256. [Google Scholar] [CrossRef]
- Qiao, Z.; Li, J.; Zhao, N.; Shi, C.; Nash, P. Graphitization and microstructure transformation of nanodiamond to onion-like carbon. Scr. Mater. 2006, 54, 225–229. [Google Scholar] [CrossRef]
- Kuznetsov, V.; Butenko, Y. Nanodiamond Graphitization and Properties of Onion-Like Carbon. In Synthesis, Properties and Applications of Ultrananocrystalline Diamond; Gruen, D.M., Shenderova, O.A., Vul’, A.Y., Eds.; Springer: Dordrecht, The Netherlands, 2005; pp. 199–216. [Google Scholar]
- Tomita, S.; Sakurai, T.; Ohta, H.; Fujii, M.; Hayashi, S. Structure and electronic properties of carbon onions. J. Chem. Phys. 2001, 114, 7477–7482. [Google Scholar] [CrossRef]
- Kuznetsov, V.L.; Zilberberg, I.L.; Butenko, Y.V.; Chuvilin, A.L.; Segall, B. Theoretical study of the formation of closed curved graphite-like structures during annealing of diamond surface. J. Appl. Phys. 1999, 86, 863–870. [Google Scholar] [CrossRef]
- Davydov, V.A.; Rakhmanina, A.V.; Lyapin, S.G.; Ilichev, I.D.; Boldyrev, K.N.; Shiryaev, A.A.; Agafonov, V.N. Production of nano- and microdiamonds with Si-V and N-V luminescent centers at high pressures in systems based on mixtures of hydrocarbon and fluorocarbon compounds. JETP Lett. 2014, 99, 585–589. [Google Scholar] [CrossRef]
- Ekimov, E.; Lyapin, S.; Grigoriev, Y.; Zibrov, I.; Kondrina, K. Size-controllable synthesis of ultrasmall diamonds from halogenated adamantanes at high static pressure. Carbon 2019, 150, 436–438. [Google Scholar] [CrossRef]
- Ekimov, E.; Kondrin, M.; Lyapin, S.; Grigoriev, Y.; Razgulov, A.; Krivobok, V.; Gierlotka, S.; Stelmakh, S. High-pressure synthesis and optical properties of nanodiamonds obtained from halogenated adamantanes. Diam. Relat. Mater. 2020, 103, 107718. [Google Scholar] [CrossRef]
- Ekimov, E.; Kondrina, K.; Zibrov, I.; Lyapin, S.; Lovygin, M.; Kazanskiy, P. Iodine-mediated high-pressure high-temperature carbonization of hydrocarbons and synthesis of nanodiamonds. Mater. Res. Bull. 2021, 137, 111189. [Google Scholar] [CrossRef]
- Kondrin, M.V.; Zibrov, I.P.; Lyapin, S.G.; Grigoriev, Y.V.; Khmelnitskiy, R.A.; Ekimov, E.A. A New Pressure-induced Mechanism of Polyvinyl Chloride Pyrolysis with Formation of Nanodiamonds. ChemNanoMat 2021, 7, 17–26. [Google Scholar] [CrossRef]
- Kudryavtsev, O.S.; Bagramov, R.H.; Satanin, A.M.; Shiryaev, A.A.; Lebedev, O.I.; Romshin, A.M.; Pasternak, D.G.; Nikolaev, A.V.; Filonenko, V.P.; Vlasov, I.I. Fano-type effect in hydrogen-terminated pure nanodiamond. arXiv 2021, arXiv:physics.optics/2106.03230. [Google Scholar]
- Kondrina, K.; Kudryavtsev, O.; Vlasov, I.; Khmelnitskiy, R.; Ekimov, E. High-pressure synthesis of microdiamonds from polyethylene terephthalate. Diam. Relat. Mater. 2018, 83, 190–195. [Google Scholar] [CrossRef]
- Mermoux, M.; Crisci, A.; Petit, T.; Girard, H.A.; Arnault, J.C. Surface Modifications of Detonation Nanodiamonds Probed by Multiwavelength Raman Spectroscopy. J. Phys. Chem. C 2014, 118, 23415–23425. [Google Scholar] [CrossRef]
- Shenderova, O.; Panich, A.M.; Moseenkov, S.; Hens, S.C.; Kuznetsov, V.; Vieth, H.M. Hydroxylated Detonation Nanodiamond: FTIR, XPS, and NMR Studies. J. Phys. Chem. C 2011, 115, 19005–19011. [Google Scholar] [CrossRef]
- Stehlik, S.; Glatzel, T.; Pichot, V.; Pawlak, R.; Meyer, E.; Spitzer, D.; Rezek, B. Water interaction with hydrogenated and oxidized detonation nanodiamonds—Microscopic and spectroscopic analyses. Diam. Relat. Mater. 2016, 63, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Stehlik, S.; Henych, J.; Stenclova, P.; Kral, R.; Zemenova, P.; Pangrac, J.; Vanek, O.; Kromka, A.; Rezek, B. Size and nitrogen inhomogeneity in detonation and laser synthesized primary nanodiamond particles revealed via salt-assisted deaggregation. Carbon 2021, 171, 230–239. [Google Scholar] [CrossRef]
- Petit, T.; Puskar, L.; Dolenko, T.; Choudhury, S.; Ritter, E.; Burikov, S.; Laptinskiy, K.; Brzustowski, Q.; Schade, U.; Yuzawa, H.; et al. Unusual Water Hydrogen Bond Network around Hydrogenated Nanodiamonds. J. Phys. Chem. C 2017, 121, 5185–5194. [Google Scholar] [CrossRef] [Green Version]
- Beltán, M.; Marcilla, A. Fourier Transform Infrared Spectroscopy Applied to the Study of PVC Decomposition. Eur. Polym. J. 1997, 33, 1135–1142. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Raman spectroscopy of amorphous, nanostructured, diamond-like carbon, and nanodiamond. Philos. Trans. R. Soc. Lond. Ser. A Math. Phys. Eng. Sci. 2004, 362, 2477–2512. [Google Scholar] [CrossRef]
- Chang, H.C.; Lin, J.C.; Wu, J.Y.; Chen, K.H. Infrared spectroscopy and vibrational relaxation of CHx and CDx stretches on synthetic diamond nanocrystal surfaces. J. Phys. Chem. 1995, 99, 11081–11088. [Google Scholar] [CrossRef]
- Cheng, C.L.; Chen, C.F.; Shaio, W.C.; Tsai, D.S.; Chen, K.H. The CH stretching features on diamonds of different origins. Diam. Relat. Mater. 2005, 14, 1455–1462. [Google Scholar] [CrossRef]
- Sheppard, N. Some Characteristic Frequencies in the Raman Spectra of Saturated Aliphatic Hydrocarbons. J. Chem. Phys. 1948, 16, 690–697. [Google Scholar] [CrossRef]
- Harada, I.; Miura, T.; Takeuchi, H. Origin of the doublet at 1360 and 1340 cm-1 in the Raman spectra of tryptophan and related compounds. Spectrochim. Acta Part A Mol. Spectrosc. 1986, 42, 307–312. [Google Scholar] [CrossRef]
- Barnard, A.S.; Russo, S.P.; Snook, I.K. Coexistence of bucky diamond with nanodiamond and fullerene carbon phases. Phys. Rev. B 2003, 68, 073406. [Google Scholar] [CrossRef]
- Chang, S.L.Y.; Dwyer, C.; Osawa, E.; Barnard, A.S. Size dependent surface reconstruction in detonation nanodiamonds. Nanoscale Horiz. 2018, 3, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Ohfuji, H.; Irifune, T. A Novel Technique for the Synthesis of Nanodiamond Powder. J. Nanomater. 2013, 2013, 201845. [Google Scholar] [CrossRef]
- Korepanov, V.I.; Hamaguchi, H.; Osawa, E.; Ermolenkov, V.; Lednev, I.K.; Etzold, B.J.; Levinson, O.; Zousman, B.; Epperla, C.P.; Chang, H.C. Carbon structure in nanodiamonds elucidated from Raman spectroscopy. Carbon 2017, 121, 322–329. [Google Scholar] [CrossRef]
- Kondrin, M.V.; Brazhkin, V.V. Diamond monohydride: The most stable three-dimensional hydrocarbon. Phys. Chem. Chem. Phys. 2015, 17, 17739–17744. [Google Scholar] [CrossRef] [Green Version]
- Kondrin, M.V.; Brazhkin, V.V. Is graphane the most stable carbon monohydride? Nanosyst. Phys. Chem. Math. 2016, 7, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Kondrin, M.V.; Lebed, Y.B.; Brazhkin, V.V. Structure and topology of three-dimensional hydrocarbon polymers. Acta Crystallogr. Sect. B 2016, 72, 634–641. [Google Scholar] [CrossRef]
- Park, S.; Abate, I.I.; Liu, J.; Wang, C.; Dahl, J.E.P.; Carlson, R.M.K.; Yang, L.; Prakapenka, V.B.; Greenberg, E.; Devereaux, T.P.; et al. Facile diamond synthesis from lower diamondoids. Sci. Adv. 2020, 6, eaay9405. [Google Scholar] [CrossRef] [Green Version]
- Lapointe, F.; Gaufrès, E.; Tremblay, I.; Tang, N.Y.W.; Martel, R.; Desjardins, P. Fano Resonances in the Midinfrared Spectra of Single-Walled Carbon Nanotubes. Phys. Rev. Lett. 2012, 109, 097402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fano, U. Effects of Configuration Interaction on Intensities and Phase Shifts. Phys. Rev. 1961, 124, 1866–1878. [Google Scholar] [CrossRef]
- Luk’yanchuk, B.; Zheludev, N.I.; Maier, S.A.; Halas, N.J.; Nordlander, P.; Giessen, H.; Chong, C.T. The Fano resonance in plasmonic nanostructures and metamaterials. Nat. Mater. 2010, 9, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Gallinet, B.; Martin, O.J.F. Ab initio theory of Fano resonances in plasmonic nanostructures and metamaterials. Phys. Rev. B 2011, 83, 235427. [Google Scholar] [CrossRef] [Green Version]
- Joe, Y.S.; Satanin, A.M.; Kim, C.S. Classical analogy of Fano resonances. Physica Scripta 2006, 74, 259–266. [Google Scholar] [CrossRef]
- Limonov, M.F.; Rybin, M.V.; Poddubny, A.N.; Kivshar, Y.S. Fano resonances in photonics. Nat. Photonics 2017, 11, 543–554. [Google Scholar] [CrossRef]
- Lax, M.; Burstein, E. Infrared Lattice Absorption in Ionic and Homopolar Crystals. Phys. Rev. 1955, 97, 39–52. [Google Scholar] [CrossRef]
- Birman, J.L. Theory of Crystal Space Groups and Infra-Red and Raman Lattice Processes of Insulating Crystals. In Theory of Crystal Space Groups and Lattice Dynamics: Infra-Red and Raman Optical Processes of Insulating Crystals; Springer: Berlin/Heidelberg, Germany, 1974; pp. 1–521. [Google Scholar] [CrossRef]
- Gu, P.; Cai, X.; Wu, G.; Xue, C.; Chen, J.; Zhang, Z.; Yan, Z.; Liu, F.; Tang, C.; Du, W.; et al. Ultranarrow and Tunable Fano Resonance in Ag Nanoshells and a Simple Ag Nanomatryushka. Nanomaterials 2021, 11, 2039. [Google Scholar] [CrossRef]
- Maier, F.; Riedel, M.; Mantel, B.; Ristein, J.; Ley, L. Origin of Surface Conductivity in Diamond. Phys. Rev. Lett. 2000, 85, 3472–3475. [Google Scholar] [CrossRef]
- Chakrapani, V.; Angus, J.C.; Anderson, A.B.; Wolter, S.D.; Stoner, B.R.; Sumanasekera, G.U. Charge Transfer Equilibria Between Diamond and an Aqueous Oxygen Electrochemical Redox Couple. Science 2007, 318, 1424–1430. [Google Scholar] [CrossRef] [Green Version]
- Tribelsky, M.I.; Miroshnichenko, A.E. Resonant scattering of electromagnetic waves by small metal particles. Physics-Uspekhi in press. 2022. [Google Scholar] [CrossRef]
- Ekimov, E.; Lebed, Y.B.; Kondrin, M. Influence of surface reconstruction on elastic properties of nanosized diamond films and nanodiamonds. Carbon 2021, 171, 634–638. [Google Scholar] [CrossRef]
- De La Pierre, M.; Bruno, M.; Manfredotti, C.; Nestola, F.; Prencipe, M.; Manfredotti, C. The (100), (111) and (110) surfaces of diamond: An ab initio B3LYP study. Mol. Phys. 2014, 112, 1030–1039. [Google Scholar] [CrossRef]
- Barnard, A.; Russo, S.; Snook, I. Structural relaxation and relative stability of nanodiamond morphologies. Diam. Relat. Mater. 2003, 12, 1867–1872. [Google Scholar] [CrossRef]
- López-Ríos, T.; Sandré, E.; Leclercq, S.; Sauvain, E. Polyacetylene in Diamond Films Evidenced by Surface Enhanced Raman Scattering. Phys. Rev. Lett. 1996, 76, 4935–4938. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.C.; Robertson, J. Origin of the 1150 cm-1 Raman mode in nanocrystalline diamond. Phys. Rev. B 2001, 63, 121405. [Google Scholar] [CrossRef]
- Ito, T.; Shirakawa, H.; Ikeda, S. Thermal cis/trans isomerization and decomposition of polyacetylene. J. Polym. Sci. Polym. Chem. Ed. 1975, 13, 1943–1950. [Google Scholar] [CrossRef]
- Davydov, V.; Rakhmanina, A.; Agafonov, V.; Narymbetov, B.; Boudou, J.P.; Szwarc, H. Conversion of polycyclic aromatic hydrocarbons to graphite and diamond at high pressures. Carbon 2004, 42, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Kondrin, M.V.; Nikolaev, N.A.; Boldyrev, K.N.; Shulga, Y.M.; Zibrov, I.P.; Brazhkin, V.V. Bulk graphanes synthesized from benzene and pyridine. CrystEngComm 2017, 19, 958–966. [Google Scholar] [CrossRef] [Green Version]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ekimov, E.; Shiryaev, A.A.; Grigoriev, Y.; Averin, A.; Shagieva, E.; Stehlik, S.; Kondrin, M. Size-Dependent Thermal Stability and Optical Properties of Ultra-Small Nanodiamonds Synthesized under High Pressure. Nanomaterials 2022, 12, 351. https://doi.org/10.3390/nano12030351
Ekimov E, Shiryaev AA, Grigoriev Y, Averin A, Shagieva E, Stehlik S, Kondrin M. Size-Dependent Thermal Stability and Optical Properties of Ultra-Small Nanodiamonds Synthesized under High Pressure. Nanomaterials. 2022; 12(3):351. https://doi.org/10.3390/nano12030351
Chicago/Turabian StyleEkimov, Evgeny, Andrey A. Shiryaev, Yuriy Grigoriev, Alexey Averin, Ekaterina Shagieva, Stepan Stehlik, and Mikhail Kondrin. 2022. "Size-Dependent Thermal Stability and Optical Properties of Ultra-Small Nanodiamonds Synthesized under High Pressure" Nanomaterials 12, no. 3: 351. https://doi.org/10.3390/nano12030351
APA StyleEkimov, E., Shiryaev, A. A., Grigoriev, Y., Averin, A., Shagieva, E., Stehlik, S., & Kondrin, M. (2022). Size-Dependent Thermal Stability and Optical Properties of Ultra-Small Nanodiamonds Synthesized under High Pressure. Nanomaterials, 12(3), 351. https://doi.org/10.3390/nano12030351