
Novel Tools to Measure Single Molecules Colocalization in Fluorescence Nanoscopy by Image Cross Correlation Spectroscopy

 ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Fluorescence Nanodiamonds (FNDs)
2.3. Immunostaining
2.4. dStorm Imaging Buffer
2.5. Confocal Imaging
2.6. dSTORM Imaging
2.7. Colocalization Analysis
3. Results
3.1. Streptavidin Conjugated Nano-Diamonds as Intracellular Reference Markers
3.2. Image Cross-Correlation Spectroscopy to Evaluate Colocalization of Molecular Species in SMLM
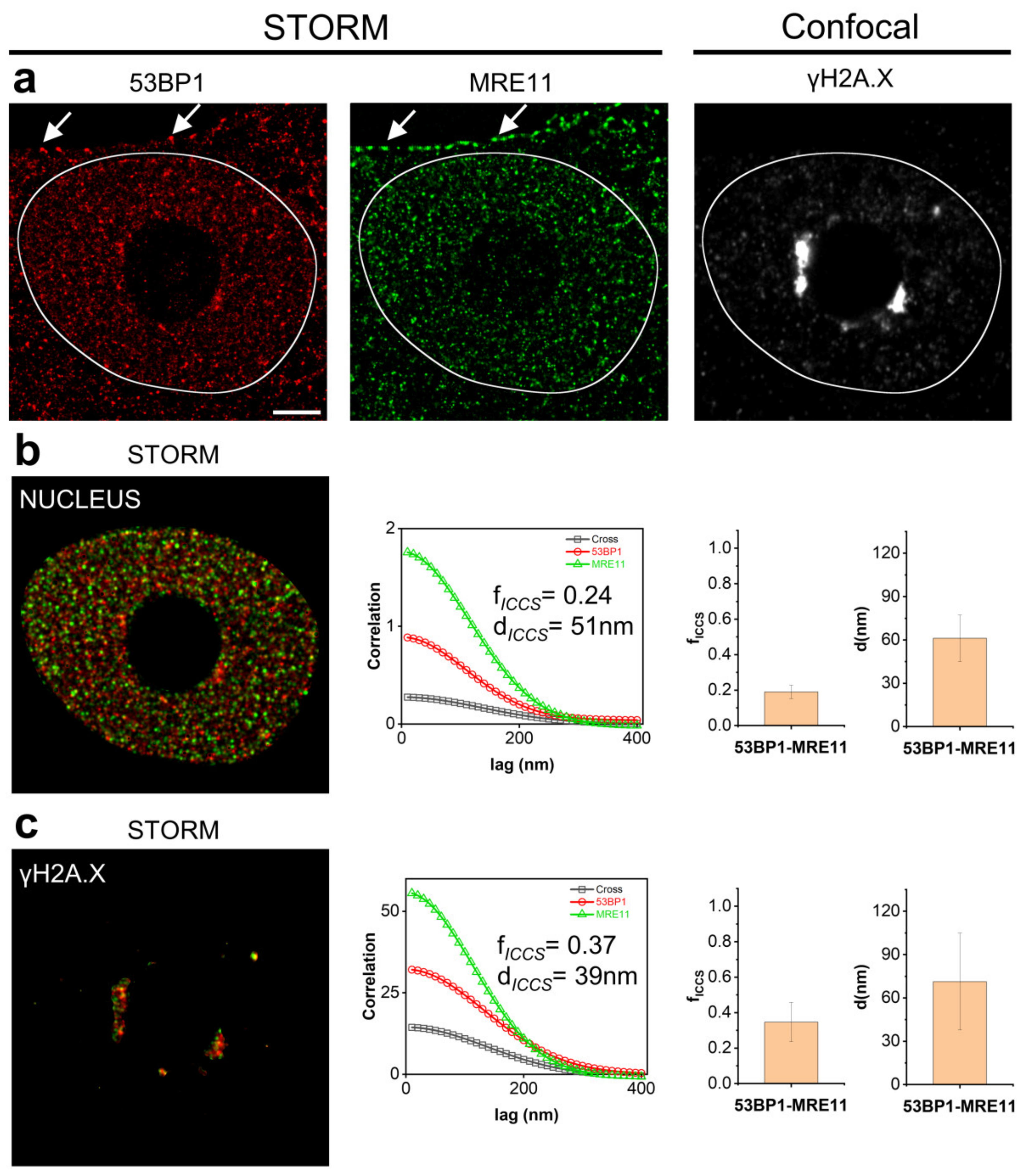
3.3. Correlative Microscopy and Image Cross-Correlation Spectroscopy as a Tool to Measure Compartmentalization of Biomolecular Interactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zinchuk, V.; Grossenbacher-Zinchuk, O. Quantitative Colocalization Analysis of Fluorescence Microscopy Images. Curr. Protoc. Cell Biol. 2014, 62, 4.19.1–4.19.14. [Google Scholar] [CrossRef] [PubMed]
- Veeraraghavan, R.; Gourdie, R.G. Stochastic optical reconstruction microscopy-based relative localization analysis (STORM-RLA) for quantitative nanoscale assessment of spatial protein organization. Mol. Biol. Cell 2016, 27, 3583–3590. [Google Scholar] [CrossRef] [PubMed]
- Pageon, S.V.; Nicovich, P.R.; Mollazade, M.; Tabarin, T.; Gaus, K. Clus-DoC: A combined cluster detection and colocalization analysis for single-molecule localization microscopy data. Mol. Biol. Cell 2016, 27, 3627–3636. [Google Scholar] [CrossRef] [PubMed]
- Piston, D.W.; Kremers, G.-J. Fluorescent protein FRET: The good, the bad and the ugly. Trends Biochem. Sci. 2007, 32, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Pelicci, S.; Diaspro, A.; Lanzanò, L. Chromatin nanoscale compaction in live cells visualized by acceptor-to-donor ratio corrected Förster resonance energy transfer between DNA dyes. J. Biophotonics 2019, 12, e201900164. [Google Scholar] [CrossRef] [Green Version]
- Lou, J.; Scipioni, L.; Wright, B.K.; Bartolec, T.K.; Zhang, J.; Masamsetti, V.P.; Gaus, K.; Gratton, E.; Cesare, A.J.; Hinde, E. Phasor histone FLIM-FRET microscopy quantifies spatiotemporal rearrangement of chromatin architecture during the DNA damage response. Proc. Natl. Acad. Sci. USA 2019, 116, 7323. [Google Scholar] [CrossRef] [Green Version]
- Furia, L.; Pelicci, P.G.; Faretta, M. A computational platform for robotized fluorescence microscopy (II): DNA damage, replication, checkpoint activation, and cell cycle progression by high-content high-resolution multiparameter image-cytometry. Cytom. Part A 2013, 83A, 344–355. [Google Scholar] [CrossRef]
- Söderberg, O.; Leuchowius, K.-J.; Gullberg, M.; Jarvius, M.; Weibrecht, I.; Larsson, L.-G.; Landegren, U. Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 2008, 45, 227–232. [Google Scholar] [CrossRef]
- Forster, T. Experimental and theoretical investigation of intermolecular transfer of electron activation energy. Z. Naturforsch. 1949, 4a, 321–327. [Google Scholar]
- Weibrecht, I.; Leuchowius, K.-J.; Clausson, C.-M.; Conze, T.; Jarvius, M.; Howell, W.M.; Kamali-Moghaddam, M.; Söderberg, O. Proximity ligation assays: A recent addition to the proteomics toolbox. Expert Rev. Proteom. 2010, 7, 401–409. [Google Scholar] [CrossRef]
- Leuchowius, K.-J.; Weibrecht, I.; Söderberg, O. In Situ Proximity Ligation Assay for Microscopy and Flow Cytometry. Curr. Protoc. Cytom. 2011, 56, 9.36.1–9.36.15. [Google Scholar] [CrossRef]
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780–782. [Google Scholar] [CrossRef]
- Gustafsson, M.G.L. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-schwartz, J.; Hess, H.F. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science 2006, 313, 1642–1646. [Google Scholar] [CrossRef] [Green Version]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793. [Google Scholar] [CrossRef] [Green Version]
- Van de Linde, S.; Löschberger, A.; Klein, T.; Heidbreder, M.; Wolter, S.; Heilemann, M.; Sauer, M. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat. Protoc. 2011, 6, 991. [Google Scholar] [CrossRef]
- Sauer, M.; Heilemann, M. Single-Molecule Localization Microscopy in Eukaryotes. Chem. Rev. 2017, 117, 7478–7509. [Google Scholar] [CrossRef]
- Lelek, M.; Gyparaki, M.T.; Beliu, G.; Schueder, F.; Griffié, J.; Manley, S.; Jungmann, R.; Sauer, M.; Lakadamyali, M.; Zimmer, C. Single-molecule localization microscopy. Nat. Rev. Methods Prim. 2021, 1, 39. [Google Scholar] [CrossRef]
- Deschout, H.; Zanacchi, F.C.; Mlodzianoski, M.; Diaspro, A.; Bewersdorf, J.; Hess, S.T.; Braeckmans, K. Precisely and accurately localizing single emitters in fluorescence microscopy. Nat. Methods 2014, 11, 253–266. [Google Scholar] [CrossRef]
- Pertsinidis, A.; Zhang, Y.; Chu, S. Subnanometre single-molecule localization, registration and distance measurements. Nature 2010, 466, 647–651. [Google Scholar] [CrossRef]
- Bates, M.; Jones, S.A.; Zhuang, X. Stochastic optical reconstruction microscopy (STORM): A method for superresolution fluorescence imaging. Cold Spring Harb. Protoc. 2013, 8, 498–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGorty, R.; Kamiyama, D.; Huang, B. Active microscope stabilization in three dimensions using image correlation. Opt. Nanoscopy 2013, 2, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Baday, M.; Tjioe, M.; Simonson, P.D.; Zhang, R.; Cai, E.; Selvin, P.R. Using fixed fiduciary markers for stage drift correction. Opt. Express 2012, 20, 12177–12183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balinovic, A.; Albrecht, D.; Endesfelder, U. Spectrally red-shifted fluorescent fiducial markers for optimal drift correction in localization microscopy. J. Phys. D Appl. Phys. 2019, 52, 204002. [Google Scholar] [CrossRef]
- Schnitzbauer, J.; Strauss, M.T.; Schlichthaerle, T.; Schueder, F.; Jungmann, R. Super-resolution microscopy with DNA-PAINT. Nat. Protoc. 2017, 12, 1198–1228. [Google Scholar] [CrossRef] [PubMed]
- Colomb, W.; Czerski, J.; Sau, J.D.; Sarkar, S.K. Estimation of microscope drift using fluorescent nanodiamonds as fiducial markers. J. Microsc. 2017, 266, 298–306. [Google Scholar] [CrossRef]
- Bumb, A.; Sarkar, S.K.; Billington, N.; Brechbiel, M.W.; Neuman, K.C. Silica encapsulation of fluorescent nanodiamonds for colloidal stability and facile surface functionalization. J. Am. Chem. Soc. 2013, 135, 7815–7818. [Google Scholar] [CrossRef] [Green Version]
- Jelezko, F.; Wrachtrup, J. Single defect centres in diamond: A review. Phys. Status Solidi 2006, 203, 3207–3225. [Google Scholar] [CrossRef]
- Yi, J.; Manna, A.; Barr, V.A.; Hong, J.; Neuman, K.C.; Samelson, L.E. MadSTORM: A superresolution technique for large-scale multiplexing at single-molecule accuracy. Mol. Biol. Cell 2016, 27, 3591–3600. [Google Scholar] [CrossRef] [Green Version]
- Lagache, T.; Sauvonnet, N.; Danglot, L.; Olivo-Marin, J.C. Statistical analysis of molecule colocalization in bioimaging. Cytom. Part A 2015, 87, 568–579. [Google Scholar] [CrossRef]
- Levet, F.; Julien, G.; Galland, R.; Butler, C.; Beghin, A.; Chazeau, A.; Hoess, P.; Ries, J.; Giannone, G.; Sibarita, J.-B. A tessellation-based colocalization analysis approach for single-molecule localization microscopy. Nat. Commun. 2019, 10, 2379. [Google Scholar] [CrossRef] [Green Version]
- Andronov, L.; Orlov, I.; Lutz, Y.; Vonesch, J.-L.; Klaholz, B.P. ClusterViSu, a method for clustering of protein complexes by Voronoi tessellation in super-resolution microscopy. Sci. Rep. 2016, 6, 24084. [Google Scholar] [CrossRef] [Green Version]
- Owen, D.M.; Rentero, C.; Rossy, J.; Magenau, A.; Williamson, D.; Rodriguez, M.; Gaus, K. PALM imaging and cluster analysis of protein heterogeneity at the cell surface. J. Biophotonics 2010, 3, 446–454. [Google Scholar] [CrossRef]
- Bolte, S.; Cordelières, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef]
- Wiseman, P.W. Image Correlation Spectroscopy: Principles and Applications. Cold Spring Harb. Protoc. 2015, 336–349. [Google Scholar] [CrossRef]
- Di Bona, M.; Mancini, M.; Mazza, D.; Vicidominii, G.; Diaspro, A.; Lanzanò, L. Measuring mobility in chromatin by intensity sorted FCS. Biophys. J. 2019, 116, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Digman, M.A.; Gratton, E. Lessons in Fluctuation Correlation Spectroscopy. Annu. Rev. Phys. Chem. 2011, 62, 645–668. [Google Scholar] [CrossRef] [Green Version]
- Foo, Y.H.; Naredi-Rainer, N.; Lamb, D.C.; Ahmed, S.; Wohland, T. Factors affecting the quantification of biomolecular interactions by fluorescence cross-correlation spectroscopy. Biophys. J. 2012, 102, 1174–1183. [Google Scholar] [CrossRef] [Green Version]
- Oneto, M.; Scipioni, L.; Sarmento, M.J.; Cainero, I.; Pelicci, S.; Furia, L.; Pelicci, P.G.; Dellino, G.I.; Bianchini, P.; Faretta, M.; et al. Nanoscale Distribution of Nuclear Sites by Super-Resolved Image Cross-Correlation Spectroscopy. Biophys. J. 2019, 117, 2054–2065. [Google Scholar] [CrossRef] [Green Version]
- Cainero, I.; Cerutti, E.; Faretta, M.; Dellino, G.I.; Pelicci, P.G.; Diaspro, A.; Lanzanò, L. Measuring nanoscale distances by structured illumination microscopy and image cross-correlation spectroscopy (Sim-iccs). Sensors 2021, 21, 2010. [Google Scholar] [CrossRef]
- Hess, S.T.; Girirajan, T.P.K.; Mason, M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar] [CrossRef] [Green Version]
- Egner, A.; Geisler, C.; Von Middendorff, C.; Bock, H.; Wenzel, D.; Medda, R.; Andresen, M.; Stiel, A.C.; Jakobs, S.; Eggeling, C.; et al. Fluorescence nanoscopy in whole cells by asynchronous localization of photoswitching emitters. Biophys. J. 2007, 93, 3285–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, P.J.; Evans, F.C. Distance to Nearest Neighbor as a Measure of Spatial Relationships in Populations. Ecology 1954, 35, 445–453. [Google Scholar] [CrossRef]
- Ester, M.; Kriegel, H.P.; Sander, J.; Xiaowei, X. A Density-Based Algorithm for Discovering Clusters in Large Spatial Databases with Noise; AAAI Press: Menlo Park, CA, USA, 1996. [Google Scholar]
- Ripley, B.D. The second-order analysis of stationary point processes. J. Appl. Probab. 1976, 13, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Scipioni, L.; Gratton, E.; Diaspro, A.; Lanzanò, L. Phasor Analysis of Local ICS Detects Heterogeneity in Size and Number of Intracellular Vesicles. Biophys. J. 2016, 111, 619–629. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pelicci, S.; Furia, L.; Scanarini, M.; Pelicci, P.G.; Lanzanò, L.; Faretta, M. Novel Tools to Measure Single Molecules Colocalization in Fluorescence Nanoscopy by Image Cross Correlation Spectroscopy. Nanomaterials 2022, 12, 686. https://doi.org/10.3390/nano12040686
Pelicci S, Furia L, Scanarini M, Pelicci PG, Lanzanò L, Faretta M. Novel Tools to Measure Single Molecules Colocalization in Fluorescence Nanoscopy by Image Cross Correlation Spectroscopy. Nanomaterials. 2022; 12(4):686. https://doi.org/10.3390/nano12040686
Chicago/Turabian StylePelicci, Simone, Laura Furia, Mirco Scanarini, Pier Giuseppe Pelicci, Luca Lanzanò, and Mario Faretta. 2022. "Novel Tools to Measure Single Molecules Colocalization in Fluorescence Nanoscopy by Image Cross Correlation Spectroscopy" Nanomaterials 12, no. 4: 686. https://doi.org/10.3390/nano12040686
APA StylePelicci, S., Furia, L., Scanarini, M., Pelicci, P. G., Lanzanò, L., & Faretta, M. (2022). Novel Tools to Measure Single Molecules Colocalization in Fluorescence Nanoscopy by Image Cross Correlation Spectroscopy. Nanomaterials, 12(4), 686. https://doi.org/10.3390/nano12040686