
A First-Principle Study of Interactions between Magnesium and Metal-Atom-Doped Graphene



Abstract
:1. Introduction
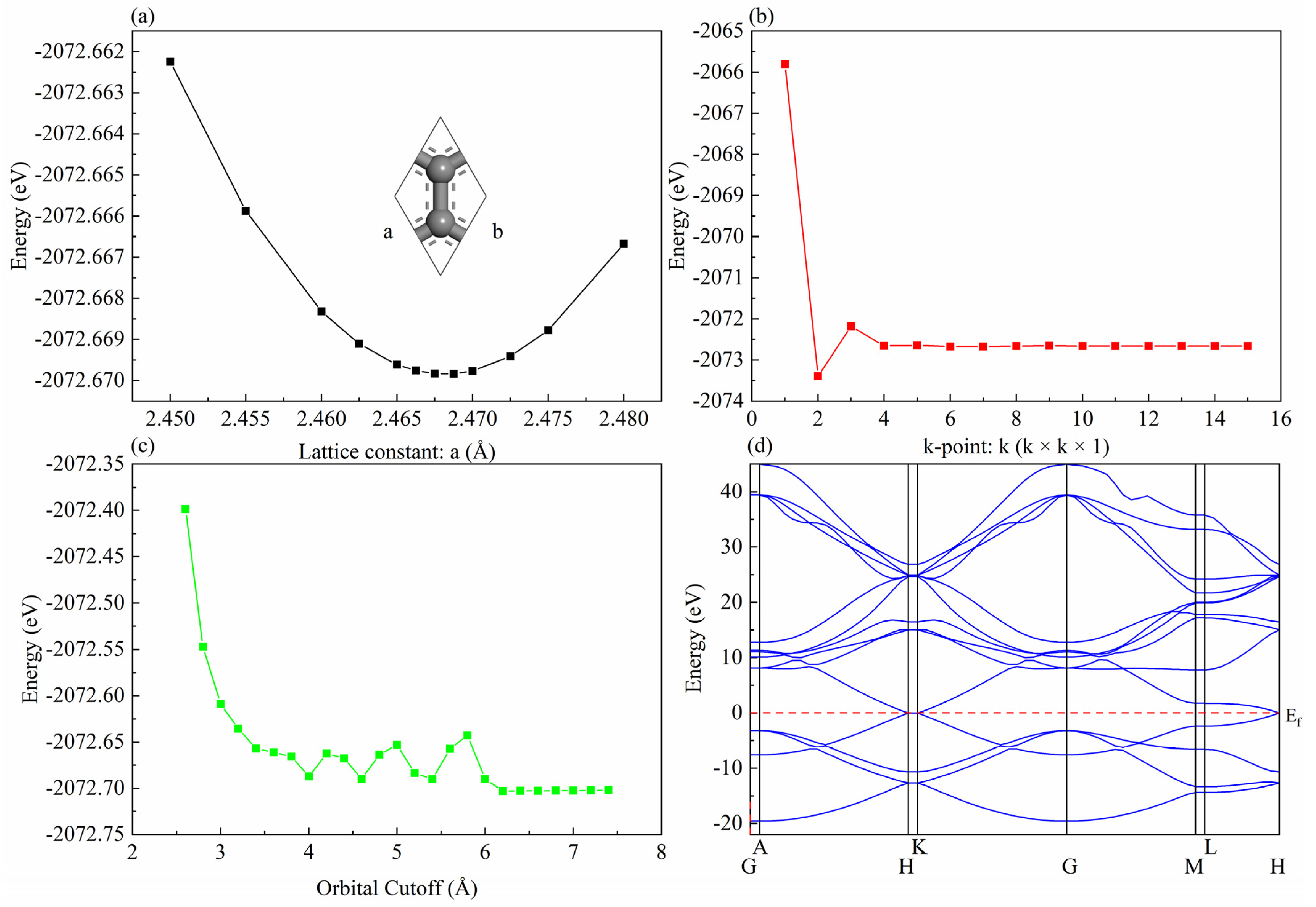
2. Computational Details
3. Results and Discussion
3.1. Interaction of Magnesium Atom with Intrinsic and Doped Graphene
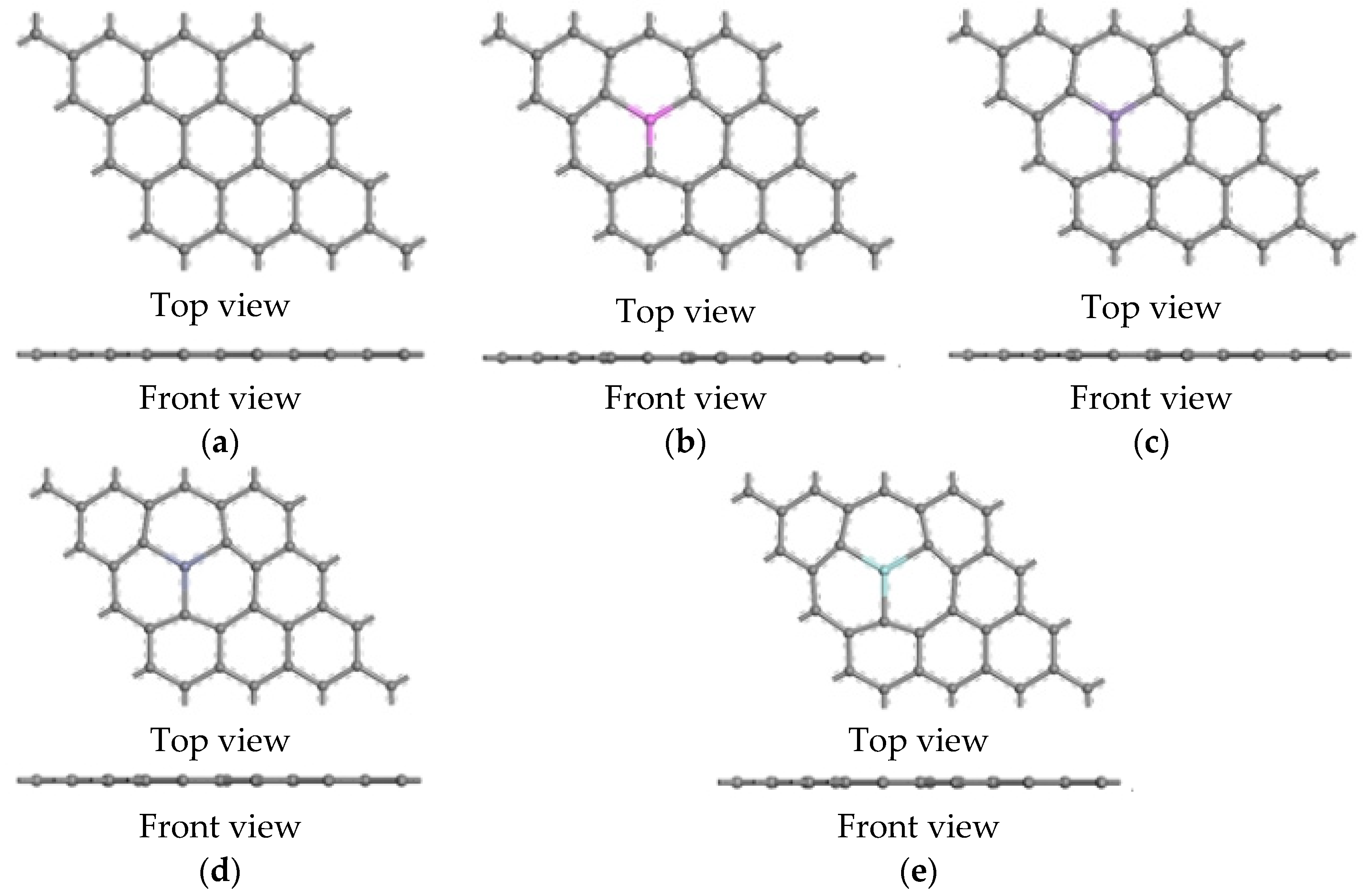
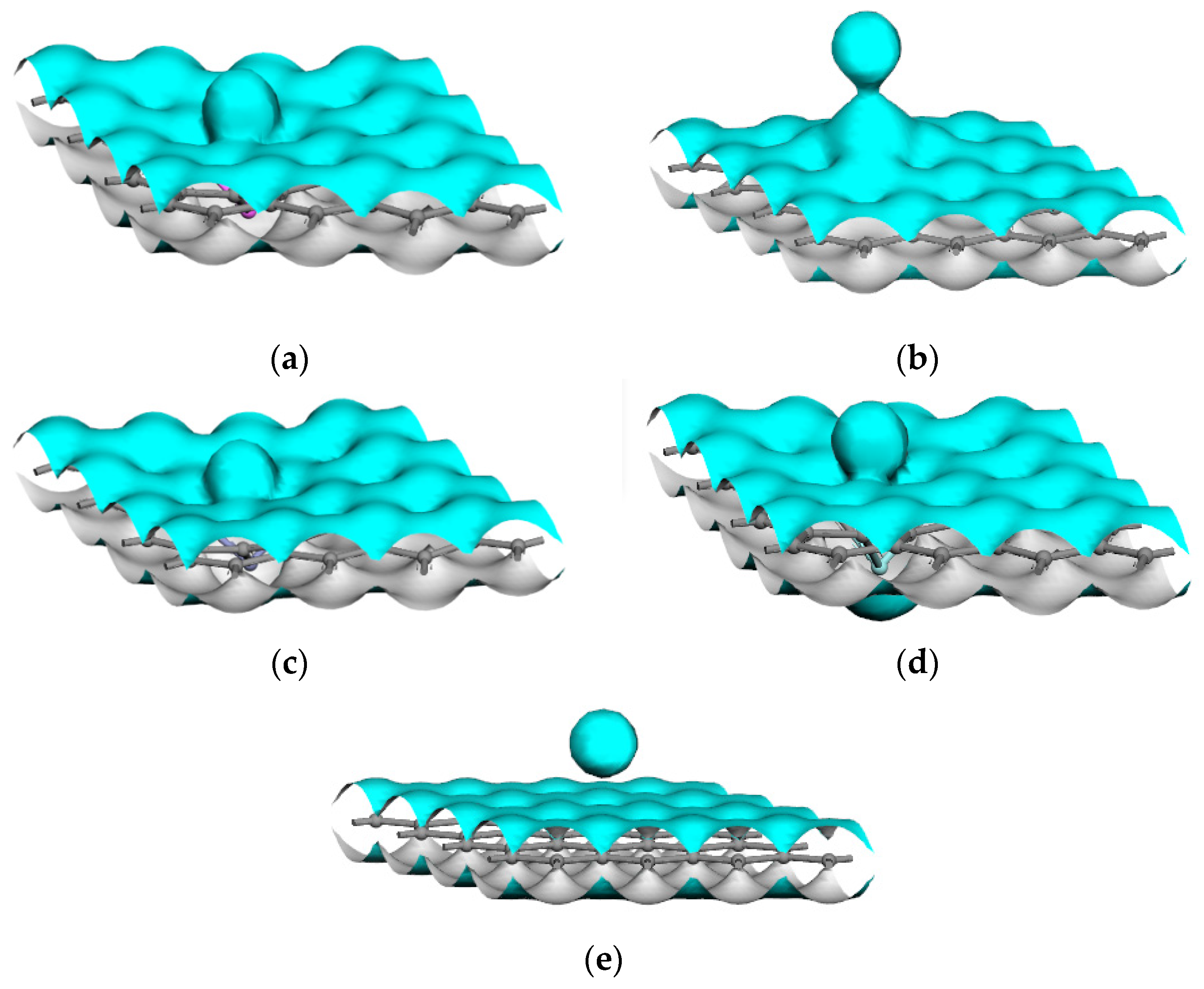
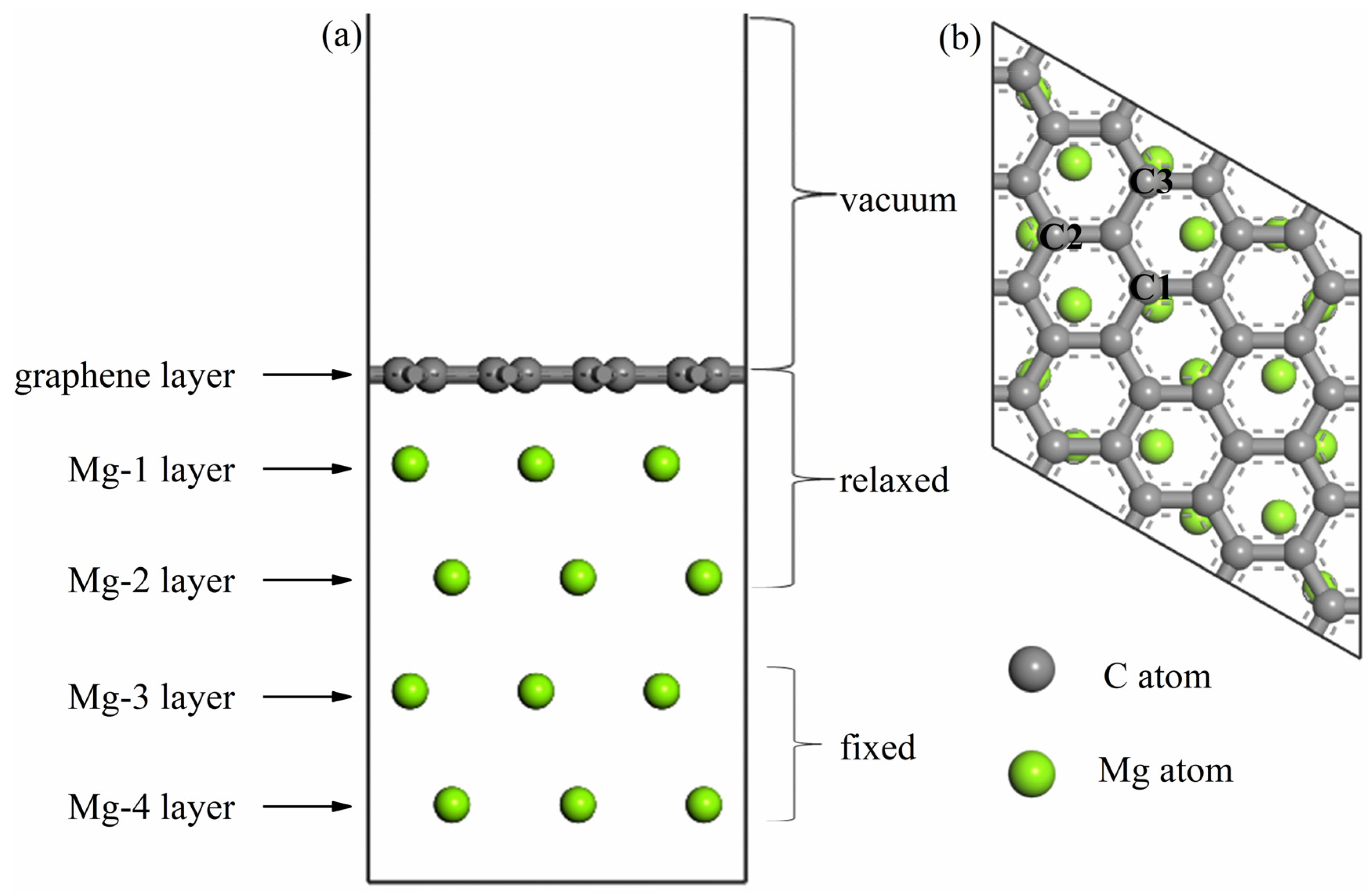
3.1.1. The Adsorbed Structural Properties
3.1.2. Interaction Energy of Magnesium with Doped and Intrinsic Graphene
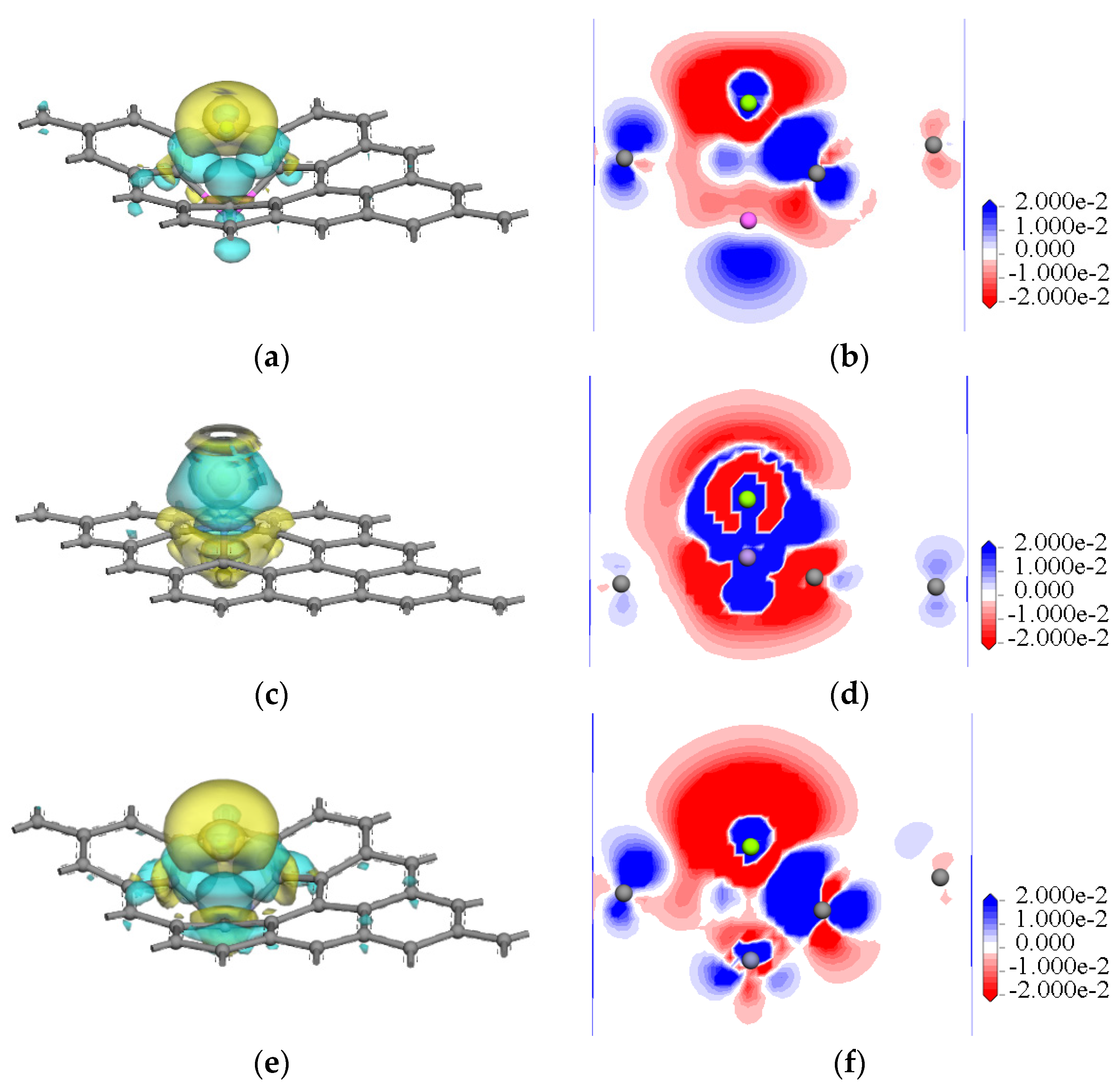
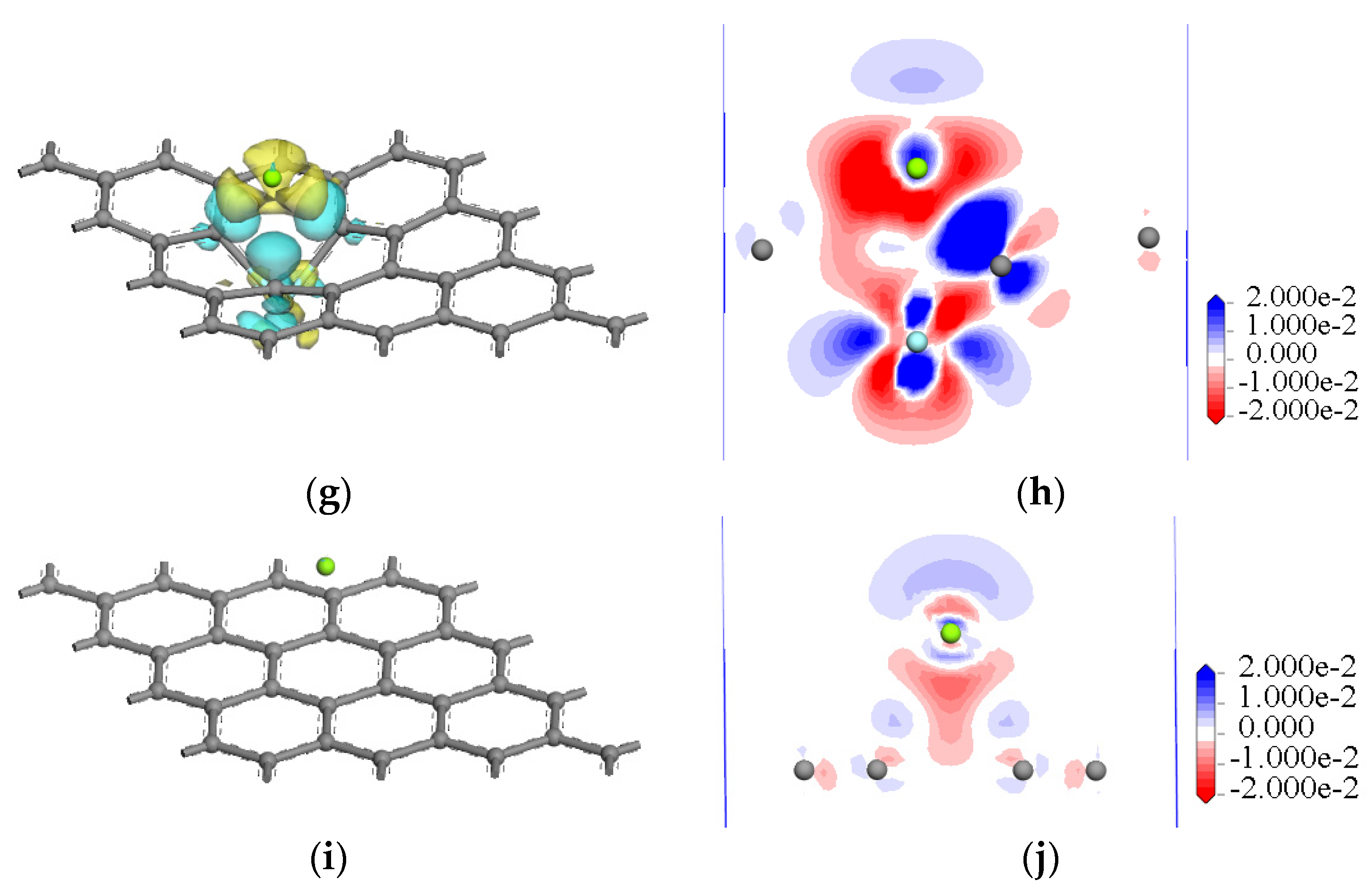
3.1.3. Electronic Properties of Magnesium with Doped and Intrinsic Graphene
3.2. Interaction of Mg(001) Surface with Intrinsic and Doped Graphene
3.2.1. Interface Structures
3.2.2. Interface Adhesion Work of Mg(001) with Doped and Intrinsic Graphene
3.2.3. Electronic Structure of Mg(001) with Doped and Intrinsic Graphene
4. Conclusions
- For the interactions of a magnesium atom with Al-, Mn-, Zn-, and Zr-doped and intrinsic graphene, the bond lengths of the doped metallic atoms with the surrounding C atoms were stretched or compressed, which greatly changed the local structure of graphene. The interaction energy of Zn-doped graphene (−3.833 eV) was higher than that of Mn-doped graphene (−2.590 eV). The result indicated that the Zn-doped graphene had the largest capability to capture the Mg atom, whereas the Mn-doped graphene displayed the lowest capability to capture the Mg atom. Additionally, the electron transfers between the doped graphene and the Mg atom were examined. The results indicated that the interaction between Zn-doped graphene and magnesium atoms was strongest.
- For the interactions of a Mg(001) surface with Al-, Mn-, Zn-, and Zr-doped and intrinsic graphene (intrinsic and doped graphene/Mg interface), the doped atoms are far away from or near the Mg atoms because of the influence of several Mg atoms on the Mg(001) surface, and the constraint of the doped atom–C chemical bond results in a great change in the local structure around the doped atoms, resulting in the wrinkles of the graphene layer, which also increases the specific surface area of the graphene layer. The results indicated that the doped atoms could improve the interface adhesion work and thus make the interface more stable. There was no chemical bond at the intrinsic graphene/Mg interface due to the maximum interface spacing (3.8801 Å). Mg–C chemical bonds were formed at the Al-, Zn-, and Zr-doped interface, and a Mg–Mn chemical bond was formed at the Mn-doped interface.
- Compared with the interactions of a magnesium atom with intrinsic and doped graphene, the movement directions of doped atoms are consistent in the interface model. For example, an Al atom, Zn atom, and Zr atom are far away from Mg atoms due to the gain of charge, and Mn is close to Mg atoms due to the loss of charge.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Abergel, D.S.L.; Apalkov, V.; Berashevich, J.; Ziegler, K.; Chakraborty, T. Properties of graphene: A theoretical perspective. Adv. Phys. 2010, 59, 261–482. [Google Scholar] [CrossRef] [Green Version]
- Rani, P.; Jindal, V.K. Designing band gap of graphene by B and N dopant atoms. RSC Adv. 2013, 3, 802–812. [Google Scholar] [CrossRef] [Green Version]
- Rani, P.; Dubey, G.S.; Jindal, V.K. DFT study of optical properties of pure and doped graphene. Physica E 2014, 62, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Rani, P.; Bhandari, R.; Jindal, V.K. Band gap modulation of graphene with increasing concentration of Li/B doping. Adv. Sci. Lett. 2015, 21, 2826–2829. [Google Scholar] [CrossRef]
- Mann, S.; Rani, P.; Kumar, R.; Dubey, G.S.; Jindal, V.K. Thermodynamic properties of pure and doped (B, N) graphene. RSC Adv. 2016, 6, 12158–12168. [Google Scholar] [CrossRef] [Green Version]
- Caragiu, M.; Finberg, S. Alkali metal adsorption on graphite: A review. J. Phys. Condens. Matter 2005, 17, R995–R1024. [Google Scholar] [CrossRef]
- Song, C.L.; Sun, B.; Wang, Y.L.; Jiang, Y.P.; Wang, L.; He, K.; Chen, X.; Zhang, P.; Ma, X.C.; Xue, Q.K. Charge-transfer-induced cesium superlattices on graphene. Phys. Rev. Lett. 2012, 108, 489–492. [Google Scholar] [CrossRef] [Green Version]
- Renard, J.; Lundeberg, M.B.; Folk, J.A.; Pennec, Y. Real-time imaging of K atoms on graphite: Interactions and diffusion. Phys. Rev. Lett. 2011, 106, 256–264. [Google Scholar] [CrossRef] [Green Version]
- Olsson, E.; Chai, G.; Dove, M.; Cai, Q. Adsorption and migration of alkali metals (Li, Na, and K) on pristine and defective graphene surfaces. Nanoscale 2019, 11, 5274–5284. [Google Scholar] [CrossRef] [Green Version]
- Wehling, T.O.; Balatsky, A.V.; Katsnelson, M.I.; Lichtenstein, A.I.; Rosch, A. The orbitally controlled kondo effect of Co adatoms on graphene. Phys. Rev. B 2010, 81, 115427. [Google Scholar] [CrossRef] [Green Version]
- Porter, C.D.; Stroud, D. The clustering and magnetic anisotropy of Fe adatoms on graphene. Phys. Rev. B 2012, 85, 913–919. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Gari, R.R.S.; Li, Z.; Craig, M.M.; Hou, S. Graphene-supported platinum and platinum-ruthenium nanoparticles with high electro-catalytic activity for methanol and ethanol oxidation. Carbon 2010, 48, 781–787. [Google Scholar] [CrossRef]
- Hu, L.; Hu, X.; Wu, X.; Du, C.; Dai, Y.; Deng, J. The density functional calculation of the transition metal adatom adsorption on graphene. J. Phys. Condens. Matter 2010, 405, 3337–3341. [Google Scholar] [CrossRef]
- Liu, X.; Wang, C.Z.; Yao, Y.X.; Lu, W.C.; Hupalo, M.; Tringides, M.C.; Ho, K.M. The bonding and charge transfer by metal adatom adsorption on graphene. Phys. Rev. B 2011, 83, 1971–1980. [Google Scholar] [CrossRef]
- Liu, X.; Wang, C.Z.; Hupalo, M.; Lin, H.Q.; Ho, K.M.; Tringides, M.C. Metals on graphene: Interactions, growth morphology, and thermal stability. Crystals 2013, 3, 79–111. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Peng, Y. A study of the structural, electronic, and magnetic properties of Sn atoms adsorbed on defective graphene using first-principle calculations. Appl. Surf. Sci. 2014, 307, 158–164. [Google Scholar] [CrossRef]
- Zhou, Y.G.; Zu, X.T.; Gao, F.; Lv, H.F.; Xiao, H.Y. The adsorption-induced magnetic properties and metallic behavior of graphene. Appl. Phys. Lett. 2009, 95, 123119. [Google Scholar] [CrossRef]
- Li, W.; Zhao, M.; Xia, Y.; Zhang, R.; Mu, Y. Covalent-adsorption induced magnetism in graphene. J. Mater. Chem. 2009, 19, 9274–9282. [Google Scholar] [CrossRef]
- Chen, L.; Ouyang, Y.; Pan, H.; Sun, Y.; Li, D. Adsorption-induced magnetism properties in graphene. J. Magn. Magn. Mater. 2011, 323, 547–551. [Google Scholar] [CrossRef]
- Dai, X.; Li, Y.; Xie, M.; Hu, G.; Zhao, J.; Zhao, B. The structural stability, electronic, and magnetic properties of Ge adsorption on defected graphene: A first-principles study. Physica E 2011, 43, 1461–1464. [Google Scholar] [CrossRef]
- Ning, Z.; Chen, Z.; Du, X.; Ran, R.; Dong, W.; Chen, C. The structural stability, electronic, and magnetic properties of Cu adsorption on defected graphene: A first-principles study. J. Supercond. Nov. Magn. 2014, 27, 115–120. [Google Scholar] [CrossRef]
- Abazari, S.; Shamsipur, A.; Bakhsheshi-Rad, H.R.; Ismail, A.F.; Sharif, S.; Razzaghi, M.; Ramakrishna, S.; Berto, F. Carbon nanotubes (CNTs)-reinforced magnesium-based matrix composites: A comprehensive review. Materials 2020, 13, 442. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Iyama, T.; Tachikawa, H. A density functional theory study of the interactions of magnesium ions with graphene surfaces. Jpn. J. Appl. Phys. 2014, 53, 110–121. [Google Scholar] [CrossRef]
- Kato, K.; Iyama, T.; Tachikawa, H. A density functional theory study of the interactions of magnesium ions with graphene chips. Jpn. J. Appl. Phys. 2011, 50, 01BJ01. [Google Scholar] [CrossRef]
- Tachikawa, H.; Iyama, T.; Kawabata, H. A MD simulation of the interactions of magnesium with graphene. Thin Solid Films 2009, 518, 877–879. [Google Scholar] [CrossRef]
- Delley, B. Hardness conserving semi-local pseudopotentials. Phys. Rev. B 2002, 66, 155125. [Google Scholar] [CrossRef]
- Grimme, S. A semi-empirical GA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Ding, Y.; Wen, C.; Hodgson, P.; Li, Y. Effects of alloying elements on the corrosion behavior and biocompatibility of biodegradable magnesium alloys: A review. J. Mater. Chem. B 2014, 2, 1912–1933. [Google Scholar] [CrossRef]
- Zhang, H.P.; Luo, X.G.; Lin, X.Y.; Lu, X.; Leng, Y. The density functional theory calculations of hydrogen adsorption on Ti-, Zn-, Zr-, Al-, and N-doped and intrinsic graphene sheets. Int. J. Hydrogen Energy 2013, 38, 14269–14275. [Google Scholar] [CrossRef]
- Xiang, S.L.; Gupta, M.; Wang, X.J.; Wang, L.D.; Hu, X.S.; Wu, K. Enhanced overall strength and ductility of magnesium matrix composites by low content of graphene nanoplatelets. Compos. Part A Appl. Sci. Manuf. 2017, 100, 183–193. [Google Scholar] [CrossRef]
- Li, X.; Hui, Q.; Shao, D.; Chen, J.; Wang, P.; Jia, Z.; Li, C.; Chen, Z.; Cheng, N. First-principles study on the stability and electronic structure of Mg/ZrB2 interfaces. Sci. China Mater. 2016, 59, 28–37. [Google Scholar] [CrossRef] [Green Version]
- Han, F.; Yuan, M.; Wei, Z.; Yao, Y.; Yao, L.; Xin, L.; Shen, X. First-principles study of the Ti/Al3Ti interfacial properties. Appl. Surf. Sci. 2021, 544, 148960. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Graphene | Intrinsic | Al-Doped | Mn-Doped | Zn-Doped | Zr-Doped |
|---|---|---|---|---|---|
| Energy | −33,162.54 | −38,713.16 | −35,433.05 | −39,039.06 | −34,056.45 |
| Deviation | - | 16.74% | 6.85% | 17.72% | 2.70% |
| Bond Length | B1 | B2 | B3 | |||
|---|---|---|---|---|---|---|
| Before Interaction | After Interaction | Before Interaction | After Interaction | Before Interaction | After Interaction | |
| Intrinsic | 1.425 | 1.423 | 1.425 | 1.427 | 1.425 | 1.427 |
| Al-doped | 1.707 | 1.920 | 1.707 | 1.920 | 1.707 | 1.920 |
| Mn-doped | 1.671 | 1.790 | 1.671 | 1.790 | 1.671 | 1.790 |
| Zn-doped | 1.722 | 1.974 | 1.722 | 1.974 | 1.722 | 1.974 |
| Zr-doped | 1.845 | 2.088 | 1.845 | 2.088 | 1.845 | 2.088 |
| Bond Length | Doped Atom-Mg | C1-Mg | C2-Mg | C3-Mg |
|---|---|---|---|---|
| Intrinsic | 3.032 | - | 3.029 | 3.031 |
| Al-doped | 2.716 | 2.268 | 2.268 | 2.268 |
| Mn-doped | 2.788 | 4.028 | 4.028 | 4.028 |
| Zn-doped | 2.564 | 2.161 | 2.161 | 2.161 |
| Zr-doped | 3.204 | 2.376 | 2.376 | 2.376 |
| Graphene | Intrinsic | Al-Doped | Mn-Doped | Zn-Doped | Zr-Doped |
|---|---|---|---|---|---|
| Interaction energy | −1.900 | −3.715 | −2.590 | −3.833 | −2.858 |
| Graphene System | Mg | Doped Atom | C1 | C2 | C3 | |
|---|---|---|---|---|---|---|
| MAC of intrinsic graphene | Before interaction | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| After interaction | 0.125 | −0.032 | 0.014 | −0.030 | −0.031 | |
| MAC of Al-doped graphene | Before interaction | 0.000 | 0.574 | −0.340 | −0.328 | −0.362 |
| After interaction | 0.619 | 0.575 | −0.495 | −0.496 | −0.486 | |
| MAC of Mn-doped graphene | Before interaction | 0.000 | −0.279 | −0.037 | −0.029 | −0.059 |
| After interaction | 0.232 | −0.434 | −0.016 | −0.012 | −0.025 | |
| MAC of Zn-doped graphene | Before interaction | 0.000 | −0.238 | −0.115 | −0.071 | 0.059 |
| After interaction | 0.806 | 0.200 | −0.386 | −0.388 | −0.371 | |
| MAC of Zr-doped graphene | Before interaction | 0.000 | 0.758 | −0.377 | −0.381 | −0.355 |
| After interaction | 0.479 | 0.575 | −0.486 | −0.484 | −0.471 | |
| Interface System | Intrinsic | Al-Doped | Mn-Doped | Zn-Doped | Zr-Doped |
|---|---|---|---|---|---|
| Wad (J/m2) | 0.0631 | 0.1163 | 0.7537 | 0.2823 | 1.2801 |
| d0 (Å) | 3.8801 | 2.8604 | 3.2576 | 2.9036 | 3.1092 |
| Interface System | Mg-1 Layer | Mg-2 Layer | Doped Atom | C1 | C2 | C3 |
|---|---|---|---|---|---|---|
| Intrinsic | 0.052–0.053 | −0.026 | −0.007 | −0.007 | −0.007 | −0.007 |
| Al-doped | 0.074–0.244 | −0.015–0.012 | 0.607 | −0.282 | −0.282 | −0.662 |
| Mn-doped | 0.070–0.120 | −0.021–−0.016 | −0.696 | −0.064 | −0.071 | −0.065 |
| Zn-doped | 0.055–0.226 | −0.019–0.005 | 0.145 | 0.145 | −0.483 | −0.493 |
| Zr-doped | 0.063–0.231 | −0.017–0.008 | 0.622 | 0.622 | −0.226 | −0.481 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Pei, X.; Zhang, H.; Yuan, M. A First-Principle Study of Interactions between Magnesium and Metal-Atom-Doped Graphene. Nanomaterials 2022, 12, 834. https://doi.org/10.3390/nano12050834
Li Y, Pei X, Zhang H, Yuan M. A First-Principle Study of Interactions between Magnesium and Metal-Atom-Doped Graphene. Nanomaterials. 2022; 12(5):834. https://doi.org/10.3390/nano12050834
Chicago/Turabian StyleLi, Yaoming, Xin Pei, Huang Zhang, and Meini Yuan. 2022. "A First-Principle Study of Interactions between Magnesium and Metal-Atom-Doped Graphene" Nanomaterials 12, no. 5: 834. https://doi.org/10.3390/nano12050834
APA StyleLi, Y., Pei, X., Zhang, H., & Yuan, M. (2022). A First-Principle Study of Interactions between Magnesium and Metal-Atom-Doped Graphene. Nanomaterials, 12(5), 834. https://doi.org/10.3390/nano12050834