
Light-Triggered Polymersome-Based Anticancer Therapeutics Delivery





Abstract
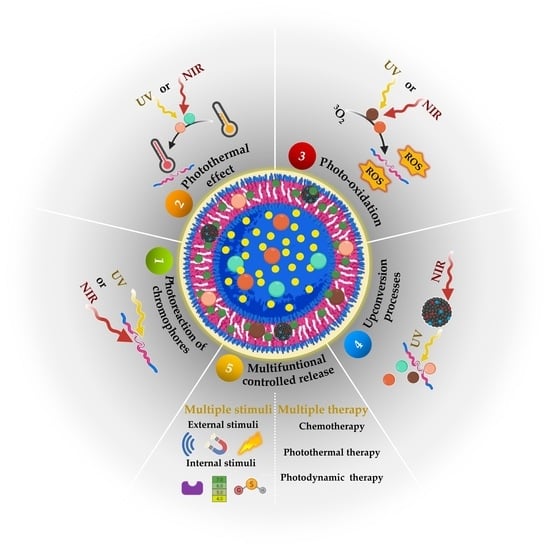
1. Introduction
2. Influence of Biological Barriers on Drug Delivery Systems
2.1. Nanocarrier Stability and Clearance inside Biological Environments
2.2. Nanocarrier Transport by Enhanced Permeability and Retention-Based Passive Tumor Targeting
2.3. Nanocarrier Uptake
2.4. Active Tumor Targeting
3. Polymersomes as a Platform for Anticancer Therapeutic Delivery
3.1. Amphiphilic Copolymer Types and Synthesis
3.1.1. Amphiphilic Block Copolymers
3.1.2. Amphiphilic Graft Copolymers
3.1.3. Amphiphilic Random Copolymers
3.1.4. Amphiphilic Alternate Copolymers
3.1.5. Amphiphilic Dendronized Copolymers
3.1.6. Amphiphilic Gradient Copolymers
3.2. Amphiphilic Copolymer Self-Assembly and Cargo Encapsulation
3.2.1. Solvent-Free Methods
3.2.2. Solvent Displacement Methods
3.3. Surface Functionalization of Polymersomes
3.3.1. Conjugation of Functional Ligands to Preformed Polymersomes
3.3.2. Self-Assembly of End-Group Functionalized Copolymers
3.3.3. Polymers with Biofunctional Hydrophilic Blocks
3.3.4. Prodrugs
3.4. Characterization of Polymersomes
4. Polymersomes for Stimuli-Responsive Anticancer Therapeutic Delivery
5. Light-Responsive Polymersomes for Anticancer Therapeutic Delivery
5.1. Controlled Release Induced by Photoreaction of Chromophores
5.2. Controlled Release Induced by the Photothermal Effect
5.3. Controlled Release Induced by Photo-Oxidation
5.4. Controlled Release Induced by the Upconversion Processes
5.5. Multifunctional Controlled Release
5.5.1. Release Controlled by Multiple Stimuli
5.5.2. Controlled Release for Multiple Therapy Polymersomes
6. Concluding Remarks and Outlooks
Author Contributions
Funding
Conflicts of Interest
References
- LoPresti, C.; Lomas, H.; Massignani, M.; Smart, T.; Battaglia, G. Polymersomes: Nature inspired nanometer sized compartments. J. Mater. Chem. 2009, 19, 3576–3590. [Google Scholar] [CrossRef]
- Sharma, A.K.; Prasher, P.; Aljabali, A.A.; Mishra, V.; Gandhi, H.; Kumar, S.; Mutalik, S.; Chellappan, D.K.; Tambuwala, M.M.; Dua, K.; et al. Emerging era of “somes”: Polymersomes as versatile drug delivery carrier for cancer diagnostics and therapy. Drug Deliv. Transl. Res. 2020, 10, 1171–1190. [Google Scholar] [CrossRef] [PubMed]
- Mattia, E.; Otto, S. Supramolecular systems chemistry. Nat. Nanotechnol. 2015, 10, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yu, G.; Huang, F. Supramolecular chemotherapy based on host-guest molecular recognition: A novel strategy in the battle against cancer with a bright future. Chem. Soc. Rev. 2017, 46, 7021–7053. [Google Scholar] [CrossRef]
- Ghosh, I.; Nau, W.M. The strategic use of supramolecular pKa shifts to enhance the bioavailability of drugs. Adv. Drug Deliv. Rev. 2012, 64, 764–783. [Google Scholar] [CrossRef]
- Cao, L.; Hettiarachchi, G.; Briken, V.; Isaacs, L. Cucurbit[7]uril containers for targeted delivery of oxaliplatin to cancer cells. Angew. Chem.-Int. Ed. 2013, 52, 12033–12037. [Google Scholar] [CrossRef]
- Gu, W.X.; Li, Q.L.; Lu, H.; Fang, L.; Chen, Q.; Yang, Y.W.; Gao, H. Construction of stable polymeric vesicles based on azobenzene and beta-cyclodextrin grafted poly(glycerol methacrylate)s for potential applications in colon-specific drug delivery. Chem. Commun. 2015, 51, 4715–4718. [Google Scholar] [CrossRef]
- Tonga, G.Y.; Jeong, Y.; Duncan, B.; Mizuhara, T.; Mout, R.; Das, R.; Kim, S.T.; Yeh, Y.C.; Yan, B.; Hou, S.; et al. Supramolecular regulation of bioorthogonal catalysis in cells using nanoparticle-embedded transition metal catalysts. Nat. Chem. 2015, 7, 597–603. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Cao, Y.; Yang, Y.; Chen, J.T.; Liu, Y. A small-sized graphene oxide supramolecular assembly for targeted delivery of camptothecin. Chem. Commun. 2014, 50, 13066–13069. [Google Scholar] [CrossRef]
- Kim, C.; Agasti, S.S.; Zhu, Z.; Isaacs, L.; Rotello, V.M. Recognition-mediated activation of therapeutic gold nanoparticles inside living cells. Nat. Chem. 2010, 2, 962–966. [Google Scholar] [CrossRef]
- Zheng, Y.R.; Suntharalingam, K.; Johnstone, T.C.; Lippard, S.J. Encapsulation of Pt(IV) prodrugs within a Pt(II) cage for drug delivery. Chem. Sci. 2015, 6, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Horcajada, P.; Chalati, T.; Serre, C.; Gillet, B.; Sebrie, C.; Baati, T.; Eubank, J.F.; Heurtaux, D.; Clayette, P.; Kreuz, C.; et al. Porous metal-organic-framework nanoscale carriers as a potential platform for drug deliveryand imaging. Nat. Mater. 2010, 9, 172–178. [Google Scholar] [CrossRef]
- Einfalt, T.; Witzigmann, D.; Edlinger, C.; Sieber, S.; Goers, R.; Najer, A.; Spulber, M.; Onaca-Fischer, O.; Huwyler, J.; Palivan, C.G. Biomimetic artificial organelles with in vitro and in vivo activity triggered by reduction in microenvironment. Nat. Commun. 2018, 9, 1127. [Google Scholar] [CrossRef] [PubMed]
- Mason, A.F.; Thordarson, P. Polymersomes as protocellular constructs. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 3817–3825. [Google Scholar] [CrossRef]
- Rideau, E.; Dimova, R.; Schwille, P.; Wurm, F.R.; Landfester, K. Liposomes and polymersomes: A comparative review towards cell mimicking. Chem. Soc. Rev. 2018, 47, 8572–8610. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, H.; Thevenot, J.; Lecommandoux, S. Smart polymersomes for therapy and diagnosis: Fast progress toward multifunctional biomimetic nanomedicines. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2012, 4, 525–546. [Google Scholar] [CrossRef] [PubMed]
- Leong, J.; Teo, J.Y.; Aakalu, V.K.; Yang, Y.Y.; Kong, H. Engineering Polymersomes for Diagnostics and Therapy. Adv. Healthc. Mater. 2018, 7, 1701276. [Google Scholar] [CrossRef]
- Martins, C.; Chauhan, V.M.; Araújo, M.; Abouselo, A.; Barrias, C.C.; Aylott, J.W.; Sarmento, B. Advanced polymeric nanotechnology to augment therapeutic delivery and disease diagnosis. Nanomedicine 2020, 15, 2287–2309. [Google Scholar] [CrossRef]
- Mohammadi, M.; Ramezani, M.; Abnous, K.; Alibolandi, M. Biocompatible polymersomes-based cancer theranostics: Towards multifunctional nanomedicine. Int. J. Pharm. 2017, 519, 287–303. [Google Scholar] [CrossRef]
- Bixner, O.; Gal, N.; Zaba, C.; Scheberl, A.; Reimhult, E. Fluorescent magnetopolymersomes: A theranostic platform to track intracellular delivery. Materials 2017, 10, 1303. [Google Scholar] [CrossRef]
- Nair, P.; Christian, D.; Discher, D.E. Polymersomes. In The Giant Vesicle Book; CRC Press: Boca Raton, FL, USA, 2020; pp. 537–550. [Google Scholar]
- World Health Organization. Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 14 January 2022).
- Stone, J.B.; DeAngelis, L.M. Cancer-treatment-induced neurotoxicity-focus on newer treatments. Nat. Rev. Clin. Oncol. 2016, 13, 92–105. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.; Mellotte, G.; Ryan, B.; O’Connor, A. Gastrointestinal side effects of cancer treatments. Ther. Adv. Chronic Dis. 2020, 11, 204062232097035. [Google Scholar] [CrossRef] [PubMed]
- AlQahtani, A.D.; O’Connor, D.; Domling, A.; Goda, S.K. Strategies for the production of long-acting therapeutics and efficient drug delivery for cancer treatment. Biomed. Pharmacother. 2019, 113, 108750. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rao, L.; Yu, G.; Cook, T.R.; Chen, X.; Huang, F. Supramolecular cancer nanotheranostics. Chem. Soc. Rev. 2021, 50, 2839–2891. [Google Scholar] [CrossRef]
- Ghorbanizamani, F.; Moulahoum, H.; Zihnioglu, F.; Timur, S. Nanohybrid carriers: The yin-yang equilibrium between natural and synthetic in biomedicine. Biomater. Sci. 2020, 8, 3237–3247. [Google Scholar] [CrossRef]
- Araste, F.; Aliabadi, A.; Abnous, K.; Taghdisi, S.M.; Ramezani, M.; Alibolandi, M. Self-assembled polymeric vesicles: Focus on polymersomes in cancer treatment. J. Control. Release 2021, 330, 502–528. [Google Scholar] [CrossRef]
- Aubert, S.; Bezagu, M.; Spivey, A.C.; Arseniyadis, S. Spatial and temporal control of chemical processes. Nat. Rev. Chem. 2019, 3, 706–722. [Google Scholar] [CrossRef]
- Benoit, D.S.; Overby, C.T.; Sims, K.R., Jr; Ackun-farmmer, M.A. Drug Delivery Systems. In Biomaterials Science: An Introduction to Materials in Medicine; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1237–1266. [Google Scholar]
- Liao, Z.; Wong, S.W.; Yeo, H.L.; Zhao, Y. Smart nanocarriers for cancer treatment: Clinical impact and safety. NanoImpact 2020, 20, 100253. [Google Scholar] [CrossRef]
- Sun, T.; Zhang, Y.S.; Pang, B.; Hyun, D.C.; Yang, M.; Xia, Y. Engineered nanoparticles for drug delivery in cancer therapy. Angew. Chem.-Int. Ed. 2014, 53, 12320–12364. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Xu, H.; Gao, M.; Tang, X.; Zhang, W.; Luo, D.; Chen, M. Micro/Nano Technology for Next-Generation Diagnostics. Small Methods 2020, 4, 1900506. [Google Scholar] [CrossRef]
- Din, F.U.; Aman, W.; Ullah, I.; Qureshi, O.S.; Mustapha, O.; Shafique, S.; Zeb, A. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int. J. Nanomed. 2017, 12, 7291–7309. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, D.; Kiselev, M.A.; Caccamo, M.T. Smart Nanoparticles for Drug Delivery Application: Development of Versatile Nanocarrier Platforms in Biotechnology and Nanomedicine. J. Nanomater. 2019, 2019, 3702518. [Google Scholar] [CrossRef]
- Bamrungsap, S.; Zhao, Z.; Chen, T.; Wang, L.; Li, C.; Fu, T.; Tan, W. Nanotechnology in therapeutics: A focus on nanoparticles as a drug delivery system. Carbohydr. Polym. 2016, 1, 71–88. [Google Scholar] [CrossRef]
- Shah, A.; Aftab, S.; Nisar, J.; Ashiq, M.N.; Iftikhar, F.J. Nanocarriers for targeted drug delivery. J. Drug Deliv. Sci. Technol. 2021, 62, 102426. [Google Scholar] [CrossRef]
- Hassan, S.; Prakash, G.; Bal Ozturk, A.; Saghazadeh, S.; Farhan Sohail, M.; Seo, J.; Remzi Dokmeci, M.; Zhang, Y.S.; Khademhosseini, A. Evolution and clinical translation of drug delivery nanomaterials. Nano Today 2017, 15, 91–106. [Google Scholar] [CrossRef]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef]
- Chamundeeswari, M.; Jeslin, J.; Verma, M.L. Nanocarriers for drug delivery applications. Environ. Chem. Lett. 2019, 17, 849–865. [Google Scholar] [CrossRef]
- Hossen, S.; Hossain, M.K.; Basher, M.K.; Mia, M.N.H.; Rahman, M.T.; Uddin, M.J. Smart nanocarrier-based drug delivery systems for cancer therapy and toxicity studies: A review. J. Adv. Res. 2019, 15, 1–18. [Google Scholar] [CrossRef]
- Huda, S.; Alam, M.A.; Sharma, P.K. Smart nanocarriers-based drug delivery for cancer therapy: An innovative and developing strategy. J. Drug Deliv. Sci. Technol. 2020, 60, 102018. [Google Scholar] [CrossRef]
- Shi, Z.; Zhou, Y.; Fan, T.; Lin, Y.; Zhang, H.; Mei, L. Inorganic nano-carriers based smart drug delivery systems for tumor therapy. Smart Mater. Med. 2020, 1, 32–47. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Mena-Giraldo, P.; Pérez-Buitrago, S.; Londoño-Berrío, M.; Ortiz-Trujillo, I.C.; Hoyos-Palacio, L.M.; Orozco, J. Photosensitive nanocarriers for specific delivery of cargo into cells. Sci. Rep. 2020, 10, 2110. [Google Scholar] [CrossRef]
- Mejía, S.P.; Sánchez, A.; Vásquez, V.; Orozco, J. Functional Nanocarriers for Delivering Itraconazole Against Fungal Intracellular Infections. Front. Pharmacol. 2021, 12, 685391. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, A.; Mejía, S.P.; Orozco, J. Recent advances in polymeric nanoparticle-encapsulated drugs against intracellular infections. Molecules 2020, 25, 3760. [Google Scholar] [CrossRef]
- Sun, Y.; Zheng, L.; Yang, Y.; Qian, X.; Fu, T.; Li, X.; Yang, Z.; Yan, H.; Cui, C.; Tan, W. Metal–Organic Framework Nanocarriers for Drug Delivery in Biomedical Applications. Nano-Micro Lett. 2020, 12, 103. [Google Scholar] [CrossRef]
- He, S.; Wu, L.; Li, X.; Sun, H.; Xiong, T.; Liu, J.; Huang, C.; Xu, H.; Sun, H.; Chen, W.; et al. Metal-organic frameworks for advanced drug delivery. Acta Pharm. Sin. B 2021, 11, 2362–2395. [Google Scholar] [CrossRef] [PubMed]
- van der Meel, R.; Sulheim, E.; Shi, Y.; Kiessling, F.; Mulder, W.J.M.; Lammers, T. Smart cancer nanomedicine. Nat. Nanotechnol. 2019, 14, 1007–1017. [Google Scholar] [CrossRef]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef]
- Kalyane, D.; Raval, N.; Maheshwari, R.; Tambe, V.; Kalia, K.; Tekade, R.K. Employment of enhanced permeability and retention effect (EPR): Nanoparticle-based precision tools for targeting of therapeutic and diagnostic agent in cancer. Mater. Sci. Eng. C 2019, 98, 1252–1276. [Google Scholar] [CrossRef]
- Greish, K. Enhanced permeability and retention effect for selective targeting of anticancer nanomedicine: Are we there yet? Drug Discov. Today Technol. 2012, 9, e161–e166. [Google Scholar] [CrossRef] [PubMed]
- Donahue, N.D.; Acar, H.; Wilhelm, S. Concepts of nanoparticle cellular uptake, intracellular trafficking, and kinetics in nanomedicine. Adv. Drug Deliv. Rev. 2019, 143, 68–96. [Google Scholar] [CrossRef] [PubMed]
- Piktel, E.; Niemirowicz, K.; Watek, M.; Wollny, T.; Deptuła, P.; Bucki, R. Recent insights in nanotechnology-based drugs and formulations designed for effective anti-cancer therapy. J. Nanobiotechnol. 2016, 14, 39. [Google Scholar] [CrossRef]
- Fernández, M.; Orozco, J. Advances in functionalized photosensitive polymeric nanocarriers. Polymers 2021, 13, 2464. [Google Scholar] [CrossRef] [PubMed]
- Colorado, D.; Fernandez, M.; Orozco, J.; Lopera, Y.; Muñoz, D.L.; Acín, S.; Balcazar, N. Metabolic Activity of Anthocyanin Extracts Loaded into Non-ionic Niosomes in Diet-Induced Obese Mice. Pharm. Res. 2020, 37, 152. [Google Scholar] [CrossRef]
- Dan, N. Vesicle-based Drug carriers: Liposomes, polymersomes, and niosomes. In Design and Development of New Nanocarriers; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1–55. [Google Scholar]
- Trucillo, P. Drug carriers: Classification, administration, release profiles, and industrial approach. Processes 2021, 9, 470. [Google Scholar] [CrossRef]
- Bleul, R.; Thiermann, R.; Maskos, M. Techniques to Control Polymersome Size. Macromolecules 2015, 48, 7396–7409. [Google Scholar] [CrossRef]
- Aibani, N.; Khan, T.N.; Callan, B. Liposome mimicking polymersomes; A comparative study of the merits of polymersomes in terms of formulation and stability. Int. J. Pharm. X 2020, 2, 100040. [Google Scholar] [CrossRef]
- Alexandridis, P. Amphiphilic copolymers and their applications. Curr. Opin. Colloid Interface Sci. 1996, 1, 490–501. [Google Scholar] [CrossRef]
- Bates, F.S.; Hillmyer, M.A.; Lodge, T.P.; Bates, C.M.; Delaney, K.T.; Fredrickson, G.H. Multiblock polymers: Panacea or Dandora’s box? Science 2012, 336, 434–440. [Google Scholar] [CrossRef]
- Sadeghi, G.M.M.; Sayaf, M. Nanostructure Formation in Block Copolymers. Nanostruct. Polym. Blends 2014, 195–271. [Google Scholar] [CrossRef]
- Storey, R.F. CHAPTER 2. Fundamental Aspects of Living Polymerization. In Fundamentals of Controlled/Living Radical Polymerization; Royal Society of Chemistry: Hattiesburg, MS, USA, 2013; pp. 60–77. [Google Scholar]
- Biais, P.; Colombani, O.; Bouteiller, L.; Stoffelbach, F.; Rieger, J. Unravelling the formation of BAB block copolymer assemblies during PISA in water. Polym. Chem. 2020, 11, 4568–4578. [Google Scholar] [CrossRef]
- Zheng, W.; Wang, Z.-G. Morphology of ABC Triblock Copolymers. Macromolecules 2002, 28, 7215–7223. [Google Scholar] [CrossRef]
- Lefley, J.; Waldron, C.; Becer, C.R. Macromolecular design and preparation of polymersomes. Polym. Chem. 2020, 11, 7124–7136. [Google Scholar] [CrossRef]
- Feng, H.; Lu, X.; Wang, W.; Kang, N.-G.; Mays, J. Block Copolymers: Synthesis, Self-Assembly, and Applications. Polymers 2017, 9, 494. [Google Scholar] [CrossRef] [PubMed]
- Deb, P.K.; Kokaz, S.F.; Abed, S.N.; Paradkar, A.; Tekade, R.K. Chapter 6—Pharmaceutical and biomedical applications of polymers. In Basic Fundamentals of Drug Delivery; Academic Press: Gujarat, India, 2018; pp. 203–267. ISBN 9780128179093. [Google Scholar]
- Nuyken, O.; Pask, S.D. Ring-opening polymerization-An introductory review. Polymers 2013, 5, 361–403. [Google Scholar] [CrossRef]
- Semsarilar, M.; Perrier, S. “Green” reversible addition-fragmentation chain-transfer (RAFT) polymerization. Nat. Chem. 2010, 2, 811–820. [Google Scholar] [CrossRef]
- Layek, R.K.; Nandi, A.K. A review on synthesis and properties of polymer functionalized graphene. Polymer 2013, 54, 5087–5103. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Xia, J. Atom transfer radical polymerization. Chem. Rev. 2001, 101, 2921–2990. [Google Scholar] [CrossRef]
- Zhang, L.; Eisenberg, A. Multiple morphologies of “crew-cut” aggregates of polystyrene-b-poly(acrylic acid) block copolymers. Science 1995, 268, 1728–1731. [Google Scholar] [CrossRef]
- Discher, B.M.; Won, Y.Y.; Ege, D.S.; Lee, J.C.-M.; Bates, F.S.; Discher, D.E.; Hammer, D.A. Polymersomes: Tough vesicles made from diblock copolymers. Science 1999, 284, 1143–1146. [Google Scholar] [CrossRef] [PubMed]
- Rösler, A.; Vandermeulen, G.W.M.; Klok, H.A. Advanced drug delivery devices via self-assembly of amphiphilic block copolymers. Adv. Drug Deliv. Rev. 2012, 64, 270–279. [Google Scholar] [CrossRef]
- Hou, W.; Liu, R.; Bi, S.; He, Q.; Wang, H.; Gu, J. Photo-Responsive Polymersomes as Drug Delivery System for Potential Medical Applications. Molecules 2020, 25, 5147. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Xia, Y.; Shao, J.; Guo, B.; Dong, Y.; Pijpers, I.A.B.; Zhong, Z.; Meng, F.; Abdelmohsen, L.K.E.A.; Williams, D.S.; et al. Biodegradable Polymersomes with Structure Inherent Fluorescence and Targeting Capacity for Enhanced Photo-Dynamic Therapy. Angew. Chem. Int. Ed. 2021, 60, 17629–17637. [Google Scholar] [CrossRef]
- Uchida, S. Graft Copolymer Synthesis. In Encyclopedia of Polymeric Nanomaterials; Springer: Berlin/Heidelberg, Germany, 2013; pp. 1–4. [Google Scholar]
- Maity, N.; Dawn, A. Conducting polymer grafting: Recent and key developments. Polymers 2020, 12, 709. [Google Scholar] [CrossRef]
- Babinot, J.; Guigner, J.M.; Renard, E.; Langlois, V. Poly(3-hydroxyalkanoate)-derived amphiphilic graft copolymers for the design of polymersomes. Chem. Commun. 2012, 48, 5364–5366. [Google Scholar] [CrossRef]
- Zheng, C.; Qiu, L.; Zhu, K. Novel polymersomes based on amphiphilic graft polyphosphazenes and their encapsulation of water-soluble anti-cancer drug. Polymer 2009, 50, 1173–1177. [Google Scholar] [CrossRef]
- Naolou, T.; Meister, A.; Schöps, R.; Pietzsch, M.; Kressler, J. Synthesis and characterization of graft copolymers able to form polymersomes and worm-like aggregates. Soft Matter 2013, 9, 10364–10372. [Google Scholar] [CrossRef]
- Homyak, C.; Anson, F.; Thayumanavan, S. Supramolecular Polymers in Nanomedicine. In Comprehensive Supramolecular Chemistry II; Elsevier: Cambridge, MA, USA, 2017; Volume 5, pp. 227–254. ISBN 9780128031988. [Google Scholar]
- Rudin, A.; Choi, P. Introductory Concepts and Definitions. In The Elements of Polymer Science & Engineering; Academic Press: Cambridge, MA, USA, 2013; pp. 1–62. ISBN 9780123821782. [Google Scholar]
- Li, L.; Raghupathi, K.; Song, C.; Prasad, P.; Thayumanavan, S. Self-assembly of Random Copolymers. Chem. Commun. 2014, 50, 13417–13432. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, M. Self-assembly and morphology control of new l-glutamic acid-based amphiphilic random copolymers: Giant vesicles, vesicles, spheres, and honeycomb Film. Langmuir 2011, 27, 12844–12850. [Google Scholar] [CrossRef]
- Li, N.; Li, Y.; Wang, X. Photoresponsive submicron-sized hollow spheres obtained from amphiphilic azobenzene-containing random copolymer. Polymer 2012, 53, 3975–3985. [Google Scholar] [CrossRef]
- Lee, H.J.; Yang, S.R.; An, E.J.; Kim, J.D. Biodegradable polymersomes from poly(2-hydroxyethyl aspartamide) grafted with lactic acid oligomers in aqueous solution. Macromolecules 2006, 39, 4938–4940. [Google Scholar] [CrossRef]
- Lai, M.H.; Jeong, J.H.; Devolder, R.J.; Brockman, C.; Schroeder, C.; Kong, H. Ellipsoidal polyaspartamide polymersomes with enhanced cell-targeting ability. Adv. Funct. Mater. 2012, 22, 3239–3246. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Cha, C.; Kaczmarowski, A.; Haan, J.; Oh, S.; Kong, H. Polyaspartamide vesicle induced by metallic nanoparticles. Soft Matter 2012, 8, 2237–2242. [Google Scholar] [CrossRef][Green Version]
- Lai, M.H.; Lee, S.; Smith, C.E.; Kim, K.; Kong, H. Tailoring polymersome bilayer permeability improves enhanced permeability and retention effect for bioimaging. ACS Appl. Mater. Interfaces 2014, 6, 10821–10829. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Yu, Y.; Wang, C.; Yang, S. Consecutive morphological transitions in nanoaggregates assembled from amphiphilic random copolymer via water-driven micellization and light-triggered dissociation. Macromolecules 2008, 41, 3585–3588. [Google Scholar] [CrossRef]
- Dan, K.; Bose, N.; Ghosh, S. Vesicular assembly and thermo-responsive vesicle-to-micelle transition from an amphiphilic random copolymer. Chem. Commun. 2011, 47, 12491–12493. [Google Scholar] [CrossRef]
- Deshpande, N.U.; Jayakannan, M. Biotin-Tagged Polysaccharide Vesicular Nanocarriers for Receptor-Mediated Anticancer Drug Delivery in Cancer Cells. Biomacromolecules 2018, 19, 3572–3585. [Google Scholar] [CrossRef]
- Huang, J.; Turner, S.R. Recent advances in alternating copolymers: The synthesis, modification, and applications of precision polymers. Polymer 2017, 116, 572–586. [Google Scholar] [CrossRef]
- Painter, P.C.; Coleman, M.M. Fundamentals of Polymer Science; Taylor & Francis: Boca Raton, FL, USA, 2019; ISBN 9780203755211. [Google Scholar]
- Goswami, K.G.; Mete, S.; Chaudhury, S.S.; Sar, P.; Ksendzov, E.; Das Mukhopadhyay, C.; Kostjuk, S.V.; De, P. Self-Assembly of Amphiphilic Copolymers with Sequence-Controlled Alternating Hydrophilic-Hydrophobic Pendant Side Chains. ACS Appl. Polym. Mater. 2020, 2, 2035–2045. [Google Scholar] [CrossRef]
- Xu, Q.; Li, S.; Yu, C.; Zhou, Y. Self-assembly of Amphiphilic Alternating Copolymers. Chem.–A Eur. J. 2019, 25, 4255–4264. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Abezgauz, L.; Danino, D.; Ho, C.C.; Co, C.C. Alternating polymer vesicles. Soft Matter 2008, 4, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Ray, D.; Aswal, V.K.; Ghosh, S. Multi-Stimuli-Responsive Directional Assembly of an Amphiphilic Donor–Acceptor Alternating Supramolecular Copolymer. Chem.–A Eur. J. 2018, 24, 16379–16387. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, C.; Li, S.; Rasheed, T.; Huang, P.; Huang, T.; Zhang, Y.; Huang, W.; Zhou, Y. Self-assembly and functionalization of alternating copolymer vesicles. Polym. Chem. 2017, 8, 4688–4695. [Google Scholar] [CrossRef]
- Lv, Y.; Wang, L.; Liu, F.; Feng, W.; Wei, J.; Lin, S. Self-assembly of amphiphilic alternating copolymers with stimuli-responsive rigid pendant groups. Polym. Chem. 2020, 11, 4798–4806. [Google Scholar] [CrossRef]
- Chen, Y.; Xiong, X. Tailoring dendronized polymers. Chem. Commun. 2010, 46, 5049–5060. [Google Scholar] [CrossRef]
- Van Hest, J.C.M.; Delnoye, D.A.P.; Baars, M.W.P.L.; Van Genderen, M.H.P.; Meijer, E.W. Polystyrene-dendrimer amphiphilic block copolymers with a generation-dependent aggregation. Science 1995, 268, 1592–1595. [Google Scholar] [CrossRef]
- Del Barrios, J.; Oriol, L.; Sánchez, C.; Luis Serrano, J.; Di Cicco, A.; Keller, P.; Li, M.H. Self-assembly of linear-dendritic diblock copolymers: From nanofibers to polymersomes. J. Am. Chem. Soc. 2010, 132, 3762–3769. [Google Scholar] [CrossRef]
- Lin, Y.L.; Chang, H.Y.; Sheng, Y.J.; Tsao, H.K. Photoresponsive polymersomes formed by amphiphilic linear-dendritic block copolymers: Generation-dependent aggregation behavior. Macromolecules 2012, 45, 7143–7156. [Google Scholar] [CrossRef]
- Medina-Torres, L.; Núñez-Ramírez, D.M.; Calderas, F.; Bernad-Bernad, M.J.; Gracia-Mora, J.; Rodríguez-Ramírez, J.; González-Laredo, R.F.; Gallegos-Infante, J.A.; Manero, O. Curcumin encapsulation by spray drying using Aloe vera mucilage as encapsulating agent. J. Food Process Eng. 2019, 42, e12972. [Google Scholar] [CrossRef]
- Abad, M.; Martínez-Bueno, A.; Mendoza, G.; Arruebo, M.; Oriol, L.; Sebastián, V.; Piñol, M. Supramolecular functionalizable linear–dendritic block copolymers for the preparation of nanocarriers by microfluidics. Polymers 2021, 13, 684. [Google Scholar] [CrossRef] [PubMed]
- Kryszewski, M. Gradient polymers and copolymers. Polym. Adv. Technol. 1998, 9, 244–259. [Google Scholar] [CrossRef]
- Beginn, U. Gradient copolymers. Colloid Polym. Sci. 2008, 286, 1465–1474. [Google Scholar] [CrossRef]
- Zaremski, M.Y.; Kalugin, D.I.; Golubev, V.B. Gradient copolymers: Synthesis, structure, and properties. Polym. Sci.-Ser. A 2009, 51, 103–122. [Google Scholar] [CrossRef]
- Alam, M.M.; Jack, K.S.; Hill, D.J.T.; Whittaker, A.K.; Peng, H. Gradient copolymers–Preparation, properties and practice. Eur. Polym. J. 2019, 116, 394–414. [Google Scholar] [CrossRef]
- Lynn, D.M.; Mohr, B.; Grubbs, R.H. Living Ring-Opening Metathesis Polymerization in Water. J. Am. Chem. Soc. 1998, 120, 1627–1628. [Google Scholar] [CrossRef]
- Buchmeiser, M.R. Current and Forthcoming Applications of ROMP-Derived Polymers: Functional Surfaces and Supports. In Polymer Science: A Comprehensive Reference, 10 Volume Set; Elsevier B.V.: Amsterdam, The Netherlands, 2012; Volume 4, pp. 695–717. ISBN 9780080878621. [Google Scholar]
- Kowsari, E. Polymer Synthesis; Nova Science Pub Inc.: New York, NY, USA, 2011; ISBN 9781613246726. [Google Scholar]
- Lamontagne, H.R.; Lessard, B.H. Nitroxide-Mediated Polymerization: A Versatile Tool for the Engineering of Next Generation Materials. ACS Appl. Polym. Mater. 2020, 2, 5327–5344. [Google Scholar] [CrossRef]
- Nicolas, J.; Guillaneuf, Y.; Lefay, C.; Bertin, D.; Gigmes, D.; Charleux, B. Nitroxide-mediated polymerization. Prog. Polym. Sci. 2013, 38, 63–235. [Google Scholar] [CrossRef]
- Milonaki, Y.; Kaditi, E.; Pispas, S.; Demetzos, C. Amphiphilic gradient copolymers of 2-methyl- and 2-phenyl-2-oxazoline: Self-organization in aqueous media and drug encapsulation. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 1226–1237. [Google Scholar] [CrossRef]
- Zhu, C.; Yao, R.; Chen, Y.; Feng, M.; Ma, S.; Zhang, C. Self-assembly of fluorinated gradient copolymer in three-dimensional co-flow focusing microfluidic. J. Colloid Interface Sci. 2018, 526, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Kravchenko, V.S.; Abetz, V.; Potemkin, I.I. Self-assembly of gradient copolymers in a selective solvent. New structures and comparison with diblock and statistical copolymers. Polymer 2021, 235, 124288. [Google Scholar] [CrossRef]
- Jia, F.; Li, Y.; Lu, J.; Deng, X.; Wu, Y. Amphiphilic Block Copolymers-Guided Strategies for Assembling Nanoparticles: From Basic Construction Methods to Bioactive Agent Delivery Applications. ACS Appl. Bio Mater. 2020, 3, 6546–6555. [Google Scholar] [CrossRef] [PubMed]
- Kunzler, C.; Handschuh-Wang, S.; Roesener, M.; Schönherr, H. Giant Biodegradable Poly(ethylene glycol)-block-Poly(ε-caprolactone) Polymersomes by Electroformation. Macromol. Biosci. 2020, 20, 2000014. [Google Scholar] [CrossRef]
- Pan, X.Q.; Gong, Y.C.; Li, Z.L.; Li, Y.P.; Xiong, X.Y. Folate-conjugated pluronic/polylactic acid polymersomes for oral delivery of paclitaxel. Int. J. Biol. Macromol. 2019, 139, 377–386. [Google Scholar] [CrossRef]
- Ghorbanizamani, F.; Moulahoum, H.; Sanli, S.; Bayir, E.; Zihnioglu, F.; Timur, S. pH-bioresponsive poly(ε-caprolactone)-based polymersome for effective drug delivery in cancer and protein glycoxidation prevention. Arch. Biochem. Biophys. 2020, 695, 108643. [Google Scholar] [CrossRef]
- Lebleu, C.; Rodrigues, L.; Guigner, J.M.; Brûlet, A.; Garanger, E.; Lecommandoux, S. Self-Assembly of PEG-b-PTMC Copolymers: Micelles and Polymersomes Size Control. Langmuir 2019, 35, 13364–13374. [Google Scholar] [CrossRef]
- Liang, H.; Zhou, Q.; Long, Y.; Wei, W.; Feng, S.; Liang, G.; Zhu, F. Synthesis and self-assembly of a novel amphiphilic diblock copolymer consisting of isotactic polystyrene and 1,4-trans-polybutadiene-graft-poly(ethylene oxide). RSC Adv. 2018, 8, 12752–12759. [Google Scholar] [CrossRef]
- Rijpkema, S.J.; Langens, S.G.H.A.; van der Kolk, M.R.; Gavriel, K.; Toebes, B.J.; Wilson, D.A. Modular Approach to the Functionalization of Polymersomes. Biomacromolecules 2020, 21, 1853–1864. [Google Scholar] [CrossRef]
- Polymeropoulos, G.; Zapsas, G.; Ntetsikas, K.; Bilalis, P.; Gnanou, Y.; Hadjichristidis, N. 50th Anniversary Perspective: Polymers with Complex Architectures. Macromolecules 2017, 50, 1253–1290. [Google Scholar] [CrossRef]
- Discher, D.E.; Ortiz, V.; Srinivas, G.; Klein, M.L.; Kim, Y.; Christian, D.; Cai, S.; Photos, P.; Ahmed, F. Emerging applications of polymersomes in delivery: From molecular dynamics to shrinkage of tumors. Prog. Polym. Sci. 2007, 32, 838–857. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, K.; Khatua, P.; Shinoda, W.; Loverde, S.M. Domain Formation in Charged Polymer Vesicles. Macromolecules 2021, 54, 9258–9267. [Google Scholar] [CrossRef]
- Chakraborty, K.; Shinoda, W.; Loverde, S.M. Molecular simulation of the shape deformation of a polymersome. Soft Matter 2020, 16, 3234–3244. [Google Scholar] [CrossRef] [PubMed]
- Ryzhkov, A.; Raikher, Y. Size-Dependent Properties of Magnetosensitive Polymersomes: Computer Modelling. Sensors 2019, 19, 5266. [Google Scholar] [CrossRef]
- Ortiz, V.; Nielsen, S.O.; Discher, D.E.; Klein, M.L.; Lipowsky, R.; Shillcock, J. Dissipative particle dynamics simulations of polymersomes. J. Phys. Chem. B 2005, 109, 17708–17714. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, S.; Yu, C.; Zhou, Y. Computational design of Janus polymersomes with controllable fission from double emulsions. Phys. Chem. Chem. Phys. 2020, 22, 24934–24942. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, Y.; Liu, H.; Yu, C.; Zhou, Y.; Yan, D. Asymmetric Polymersomes from an Oil-in-Oil Emulsion: A Computer Simulation Study. Langmuir 2017, 33, 10084–10093. [Google Scholar] [CrossRef]
- Sorrell, I.; Shipley, R.J.; Hearnden, V.; Colley, H.E.; Thornhill, M.H.; Murdoch, C.; Webb, S.D. Combined mathematical modelling and experimentation to predict polymersome uptake by oral cancer cells. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 339–348. [Google Scholar] [CrossRef]
- Fraaije, J.G.E.M.; Van Sluis, C.A.; Kros, A.; Zvelindovsky, A.V.; Sevink, G.J.A. Design of chimaeric polymersomes. Faraday Discuss. 2005, 128, 355–361. [Google Scholar] [CrossRef]
- Ryzhkov, A.; Raikher, Y. Coarse-Grained Molecular Dynamics Modelling of a Magnetic Polymersome. Nanomaterials 2018, 8, 763. [Google Scholar] [CrossRef]
- Blanazs, A.; Armes, S.P.; Ryan, A.J. Self-Assembled Block Copolymer Aggregates: From Micelles to Vesicles and their Biological Applications. Macromol. Rapid Commun. 2009, 30, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Mosley, G.L.; Yamanishi, C.D.; Kamei, D.T. Mathematical modeling of vesicle drug delivery systems 1: Vesicle formation and stability along with drug loading and release. J. Lab. Autom. 2013, 18, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Rangelov, S.; Pispas, S. Polymer and Polymer-Hybrid Nanoparticles: From Synthesis to Biomedical Applications; CRC Press: Boca Raton, FL, USA, 2013; ISBN 9781439869093. [Google Scholar]
- Chang, A.B.; Bates, F.S. The ABCs of Block Polymers. Macromolecules 2020, 53, 2765–2768. [Google Scholar] [CrossRef]
- Atanase, L.I.; Riess, G. Self-assembly of block and graft copolymers in organic solvents: An overview of recent advances. Polymers 2018, 10, 62. [Google Scholar] [CrossRef]
- Lv, F.; An, Z.; Wu, P. Scalable preparation of alternating block copolymer particles with inverse bicontinuous mesophases. Nat. Commun. 2019, 10, 1397. [Google Scholar] [CrossRef]
- Chandrasiri, I.; Abebe, D.G.; Loku Yaddehige, M.; Williams, J.S.D.; Zia, M.F.; Dorris, A.; Barker, A.; Simms, B.L.; Parker, A.; Vinjamuri, B.P.; et al. Self-Assembling PCL-PAMAM Linear Dendritic Block Copolymers (LDBCs) for Bioimaging and Phototherapeutic Applications. ACS Appl. Bio Mater. 2020, 3, 5664–5677. [Google Scholar] [CrossRef]
- Ahmed, M. Nanomaterial synthesis. In Polymer Science and Nanotechnology; Elsevier: Amsterdam, The Netherlands, 2020; pp. 361–399. [Google Scholar]
- Lee, J.S.; Feijen, J. Polymersomes for drug delivery: Design, formation and characterization. J. Control. Release 2012, 161, 473–483. [Google Scholar] [CrossRef]
- Chidanguro, T.; Ghimire, E.; Liu, C.H.; Simon, Y.C. Polymersomes: Breaking the Glass Ceiling? Small 2018, 14, 1802734. [Google Scholar] [CrossRef]
- Martínez-Muñoz, O.I.; Ospina-Giraldo, L.F.; Mora-Huertas, C.E. Nanoprecipitation: Applications for Entrapping Active Molecules of Interest in Pharmaceutics. In Nano- and Microencapsulation-Techniques and Applications; IntechOpen: London, UK, 2021; ISBN 978-1-83968-349-7. [Google Scholar]
- Massignani, M.; Lomas, H.; Battaglia, G. Polymersomes: A Synthetic Biological Approach to Encapsulation and Delivery. In Modern Techniques for Nano-and Microreactors/-Reactions; Springer: Berlin/Heidelberg, Germany, 2010; pp. 115–154. [Google Scholar]
- Messager, L.; Gaitzsch, J.; Chierico, L.; Battaglia, G. Novel aspects of encapsulation and delivery using polymersomes. Curr. Opin. Pharmacol. 2014, 18, 104–111. [Google Scholar] [CrossRef]
- Ahmed, F.; Pakunlu, R.L.; Brannan, A.; Bates, F.; Minko, T.; Discher, D.E. Biodegradable polymersomes loaded with both paclitaxel and doxorubicin permeate and shrink tumors, inducing apoptosis in proportion to accumulated drug. J. Control. Release 2006, 116, 150–158. [Google Scholar] [CrossRef]
- Colley, H.E.; Hearnden, V.; Avila-Olias, M.; Cecchin, D.; Canton, I.; Madsen, J.; Macneil, S.; Warren, N.; Hu, K.; McKeating, J.A.; et al. Polymersome-mediated delivery of combination anticancer therapy to head and neck cancer cells: 2D and 3D in vitro evaluation. Mol. Pharm. 2014, 11, 1176–1188. [Google Scholar] [CrossRef] [PubMed]
- Fetsch, C.; Gaitzsch, J.; Messager, L.; Battaglia, G.; Luxenhofer, R. Self-assembly of amphiphilic block copolypeptoids-Micelles, worms and polymersomes. Sci. Rep. 2016, 6, 33491. [Google Scholar] [CrossRef] [PubMed]
- Matoori, S.; Leroux, J.C. Twenty-five years of polymersomes: Lost in translation? Mater. Horiz. 2020, 7, 1297–1309. [Google Scholar] [CrossRef]
- dos Santos, E.C.; Angelini, A.; Hürlimann, D.; Meier, W.; Palivan, C.G. Giant Polymer Compartments for Confined Reactions. Chemistry 2020, 2, 470–489. [Google Scholar] [CrossRef]
- Wang, L.; Chierico, L.; Little, D.; Patikarnmonthon, N.; Yang, Z.; Azzouz, M.; Madsen, J.; Armes, S.P.; Battaglia, G. Encapsulation of Biomacromolecules within Polymersomes by Electroporation. Angew. Chem. Int. Ed. 2012, 51, 11122–11125. [Google Scholar] [CrossRef] [PubMed]
- Brož, P.; Benito, S.M.; Saw, C.L.; Burger, P.; Heider, H.; Pfisterer, M.; Marsch, S.; Meier, W.; Hunziker, P. Cell targeting by a generic receptor-targeted polymer nanocontainer platform. J. Control. Release 2005, 102, 475–488. [Google Scholar] [CrossRef]
- Chen, S.; Zhao, X.; Chen, J.; Chen, J.; Kuznetsova, L.; Wong, S.S.; Ojima, I. Mechanism-Based Tumor-Targeting Drug Delivery System. Validation of Efficient Vitamin Receptor-Mediated Endocytosis and Drug Release. Bioconjug. Chem. 2010, 21, 979. [Google Scholar] [CrossRef]
- Pawar, P.V.; Gohil, S.V.; Jain, J.P.; Kumar, N. Functionalized polymersomes for biomedical applications. Polym. Chem. 2013, 4, 3160–3176. [Google Scholar] [CrossRef]
- Petersen, M.A.; Yin, L.; Kokkoli, E.; Hillmyer, M.A. Synthesis and characterization of reactive PEO–PMCL polymersomes. Polym. Chem. 2010, 1, 1281–1290. [Google Scholar] [CrossRef]
- Varlas, S.; Foster, J.C.; Georgiou, P.G.; Keogh, R.; Husband, J.T.; Williams, D.S.; O’Reilly, R.K. Tuning the membrane permeability of polymersome nanoreactors developed by aqueous emulsion polymerization-induced self-assembly. Nanoscale 2019, 11, 12643–12654. [Google Scholar] [CrossRef]
- Domes, S.; Filiz, V.; Nitsche, J.; Frömsdorf, A.; Förster, S. Covalent Attachment of Polymersomes to Surfaces. Langmuir 2010, 26, 6927–6931. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Sasaki, Y.; Akiyoshi, K. Biotransporting Self-Assembled Nanofactories Using Polymer Vesicles with Molecular Permeability for Enzyme Prodrug Cancer Therapy. Adv. Mater. 2017, 29, 1702406. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Gu, X.; Sun, Y.; Meng, F.; Storm, G.; Zhong, Z. Transferrin-binding peptide functionalized polymersomes mediate targeted doxorubicin delivery to colorectal cancer in vivo. J. Control. Release 2020, 319, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Egli, S.; Schlaad, H.; Bruns, N.; Meier, W. Functionalization of Block Copolymer Vesicle Surfaces. Polymers 2011, 3, 252–280. [Google Scholar] [CrossRef]
- Kubilis, A.; Abdulkarim, A.; Eissa, A.M.; Cameron, N.R. Giant Polymersome Protocells Dock with Virus Particle Mimics via Multivalent Glycan-Lectin Interactions. Sci. Rep. 2016, 6, 32414. [Google Scholar] [CrossRef]
- Huang, J.; Bonduelle, C.; Thévenot, J.; Lecommandoux, S.; Heise, A. Biologically Active Polymersomes from Amphiphilic Glycopeptides. J. Am. Chem. Soc. 2011, 134, 119–122. [Google Scholar] [CrossRef]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Järvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef]
- Nicolas, J. Drug-Initiated Synthesis of Polymer Prodrugs: Combining Simplicity and Efficacy in Drug Delivery. Chem. Mater. 2016, 28, 1591–1606. [Google Scholar] [CrossRef]
- Li, J.; Li, Y.; Wang, Y.; Ke, W.; Chen, W.; Wang, W.; Ge, Z. Polymer Prodrug-Based Nanoreactors Activated by Tumor Acidity for Orchestrated Oxidation/Chemotherapy. Nano Lett. 2017, 17, 6983–6990. [Google Scholar] [CrossRef]
- Chen, H.; Zeng, X.; Tham, H.P.; Phua, S.Z.F.; Cheng, W.; Zeng, W.; Shi, H.; Mei, L.; Zhao, Y. NIR-Light-Activated Combination Therapy with a Precise Ratio of Photosensitizer and Prodrug Using a Host–Guest Strategy. Angew. Chem. Int. Ed. 2019, 58, 7641–7646. [Google Scholar] [CrossRef]
- Stetefeld, J.; McKenna, S.A.; Patel, T.R. Dynamic light scattering: A practical guide and applications in biomedical sciences. Biophys. Rev. 2016, 8, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Raval, N.; Maheshwari, R.; Kalyane, D.; Youngren-Ortiz, S.R.; Chougule, M.B.; Tekade, R.K. Importance of physicochemical characterization of nanoparticles in pharmaceutical product development. In Basic Fundamentals of Drug Delivery; Elsevier Inc.: Amsterdam, The Netherlands, 2018; pp. 369–400. ISBN 9780128179093. [Google Scholar]
- Bhattacharjee, S. DLS and zeta potential—What they are and what they are not? J. Control. Release 2016, 235, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Wright, M. Nanoparticle tracking analysis for the multiparameter characterization and counting of nanoparticle suspensions. Methods Mol. Biol. 2012, 906, 511–524. [Google Scholar] [CrossRef]
- Gardiner, C.; Ferreira, Y.J.; Dragovic, R.A.; Redman, C.W.G.; Sargent, I.L. Extracellular vesicle sizing and enumeration by nanoparticle tracking analysis. J. Extracell. Vesicles 2013, 2, 19671. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Cao, S.; Williams, D.S.; Abdelmohsen, L.K.E.A.; van Hest, J.C.M. Photoactivated Polymersome Nanomotors: Traversing Biological Barriers. Angew. Chem.-Int. Ed. 2020, 59, 16918–16925. [Google Scholar] [CrossRef]
- Yamamoto, S.; Yamada, T.; Kubo, G.; Sakurai, K.; Yamaguchi, K.; Nakanishi, J. Preparation of a series of photoresponsive polymersomes bearing photocleavable a 2-nitrobenzyl group at the hydrophobic/hydrophilic interfaces and their payload releasing behaviors. Polymers 2019, 11, 1254. [Google Scholar] [CrossRef] [PubMed]
- Oz, U.C.; Kucukturkmen, B.; Ozkose, U.U.; Gulyuz, S.; Bolat, Z.B.; Telci, D.; Sahin, F.; Yilmaz, O.; Bozkir, A. Design of Colloidally Stable and Non-Toxic Petox-Based Polymersomes for Cargo Molecule Encapsulation. ChemNanoMat 2019, 5, 766–775. [Google Scholar] [CrossRef]
- Habel, J.; Ogbonna, A.; Larsen, N.; Cherré, S.; Kynde, S.; Midtgaard, S.R.; Kinoshita, K.; Krabbe, S.; Jensen, G.V.; Hansen, J.S.; et al. Selecting analytical tools for characterization of polymersomes in aqueous solution. RSC Adv. 2015, 5, 79924–79946. [Google Scholar] [CrossRef]
- Tan, H.; Liu, Y.; Xie, J.; Gao, Y.; Li, Y.; Ma, L.; Zhang, L.; Tang, T.; Zhu, J. Light-triggered disassembly of photo-responsive gold nanovesicles for controlled drug release. Mater. Chem. Front. 2020, 4, 2805–2811. [Google Scholar] [CrossRef]
- Kuntsche, J.; Horst, J.C.; Bunjes, H. Cryogenic transmission electron microscopy (cryo-TEM) for studying the morphology of colloidal drug delivery systems. Int. J. Pharm. 2011, 417, 120–137. [Google Scholar] [CrossRef]
- Lichtman, J.W.; Conchello, J.A. Fluorescence microscopy. Nat. Methods 2005, 2, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Disalvo, G.M.; Robinson, A.R.; Aly, M.S.; Hoglund, E.R.; O’malley, S.M.; Griepenburg, J.C. Polymersome poration and rupture mediated by plasmonic nanoparticles in response to single-pulse irradiation. Polymers 2020, 12, 2381. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, G.; Lopresti, C.; Massignani, M.; Warren, N.J.; Madsen, J.; Forster, S.; Vasilev, C.; Hobbs, J.K.; Armes, S.P.; Chirasatitsin, S.; et al. Wet nanoscale imaging and testing of polymersomes. Small 2011, 7, 2010–2015. [Google Scholar] [CrossRef] [PubMed]
- Jaskiewicz, K.; Makowski, M.; Kappl, M.; Landfester, K.; Kroeger, A. Mechanical Properties of Poly(dimethylsiloxane)-block-poly(2-methyloxazoline) Polymersomes Probed by Atomic Force Microscopy. Langmuir 2012, 28, 12629–12636. [Google Scholar] [CrossRef]
- Iyisan, B.; Janke, A.; Reichenbach, P.; Eng, L.M.; Appelhans, D.; Voit, B. Immobilized Multifunctional Polymersomes on Solid Surfaces: Infrared Light-Induced Selective Photochemical Reactions, pH Responsive Behavior, and Probing Mechanical Properties under Liquid Phase. ACS Appl. Mater. Interfaces 2016, 8, 15788–15801. [Google Scholar] [CrossRef]
- Keeler, J. Understanding NRM Spectroscopy; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2010; ISBN 978-0-470-74608-0. [Google Scholar]
- Huang, Y.; Thanneeru, S.; Zhang, Q.; He, J. A new design of cleavable acetal-containing amphiphilic block copolymers triggered by light. J. Polym. Sci. Part A Polym. Chem. 2018, 56, 1815–1824. [Google Scholar] [CrossRef]
- Schlick, S.; Jeschke, G. Electron Spin Resonance Spectroscopy. Polym. Sci. A Compr. Ref. 2012, 2, 221–253. [Google Scholar] [CrossRef]
- Baumann, P.; Balasubramanian, V.; Onaca-Fischer, O.; Sienkiewicz, A.; Palivan, C.G. Light-responsive polymer nanoreactors: A source of reactive oxygen species on demand. Nanoscale 2013, 5, 217–224. [Google Scholar] [CrossRef]
- Wang, H.; Chu, P.K. Chapter 4-Surface Characterization of Biomaterials. In Characterization of Biomaterials; Elsevier: Amsterdam, The Netherlands, 2013; pp. 105–174. ISBN 9780124158009. [Google Scholar]
- Song, Y.; Chen, Y.; Li, P.; Dong, C.M. Photoresponsive Polypeptide-Glycosylated Dendron Amphiphiles: UV-Triggered Polymersomes, OVA Release, and in Vitro Enhanced Uptake and Immune Response. Biomacromolecules 2020, 21, 5345–5357. [Google Scholar] [CrossRef]
- Hu, X.; Liu, S.; Chen, X.; Mo, G.; Xie, Z.; Jing, X. Biodegradable amphiphilic block copolymers bearing protected hydroxyl groups: Synthesis and characterization. Biomacromolecules 2008, 9, 553–560. [Google Scholar] [CrossRef]
- Safar Sajadi, S.M.; Khoee, S. The simultaneous role of porphyrins’ H- and J- aggregates and host–guest chemistry on the fabrication of reversible Dextran-PMMA polymersome. Sci. Rep. 2021, 11, 2832. [Google Scholar] [CrossRef] [PubMed]
- Garni, M.; Thamboo, S.; Schoenenberger, C.A.; Palivan, C.G. Biopores/membrane proteins in synthetic polymer membranes. Biochim. Biophys. Acta-Biomembr. 2017, 1859, 619–638. [Google Scholar] [CrossRef] [PubMed]
- Tanner, P.; Baumann, P.; Enea, R.; Onaca, O.; Palivan, C.; Meier, W. Polymeric Vesicles: From Drug Carriers to Nanoreactors and Artificial Organelles. Acc. Chem. Res. 2011, 44, 1039–1049. [Google Scholar] [CrossRef]
- Chang, H.Y.; Sheng, Y.J.; Tsao, H.K. Structural and mechanical characteristics of polymersomes. Soft Matter 2014, 10, 6373–6381. [Google Scholar] [CrossRef]
- Discher, D.E.; Ahmed, F. Polymersomes. Annu. Rev. Biomed. Eng. 2006, 8, 323–341. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.J.; Pearce, A.K.; Foster, J.C.; O’Reilly, R.K. Probing and Tuning the Permeability of Polymersomes. ACS Cent. Sci. 2021, 7, 30–38. [Google Scholar] [CrossRef]
- Rodríguez-García, R.; Mell, M.; López-Montero, I.; Netzel, J.; Hellweg, T.; Monroy, F. Polymersomes: Smart vesicles of tunable rigidity and permeability. Soft Matter 2011, 7, 1532–1542. [Google Scholar] [CrossRef]
- Wang, X.; Yao, C.; Zhang, G.; Liu, S. Regulating vesicle bilayer permeability and selectivity via stimuli-triggered polymersome-to-PICsome transition. Nat. Commun. 2020, 11, 1524. [Google Scholar] [CrossRef]
- Messager, L.; Burns, J.R.; Kim, J.; Cecchin, D.; Hindley, J.; Pyne, A.L.B.; Gaitzsch, J.; Battaglia, G.; Howorka, S. Biomimetic Hybrid Nanocontainers with Selective Permeability. Angew. Chem. 2016, 128, 11272–11275. [Google Scholar] [CrossRef]
- Lomora, M.; Itel, F.; Dinu, I.A.; Palivan, C.G. Selective ion-permeable membranes by insertion of biopores into polymersomes. Phys. Chem. Chem. Phys. 2015, 17, 15538–15546. [Google Scholar] [CrossRef]
- Einfalt, T.; Goers, R.; Dinu, I.A.; Najer, A.; Spulber, M.; Onaca-Fischer, O.; Palivan, C.G. Stimuli-Triggered Activity of Nanoreactors by Biomimetic Engineering Polymer Membranes. Nano Lett. 2015, 15, 7596–7603. [Google Scholar] [CrossRef]
- Habel, J.; Hansen, M.; Kynde, S.; Larsen, N.; Midtgaard, S.R.; Jensen, G.V.; Bomholt, J.; Ogbonna, A.; Almdal, K.; Schulz, A.; et al. Aquaporin-Based Biomimetic Polymeric Membranes: Approaches and Challenges. Membranes 2015, 5, 307. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.E.; Abram, S.L.; Craciun, I.; Palivan, C.G. Biomolecule-polymer hybrid compartments: Combining the best of both worlds. Phys. Chem. Chem. Phys. 2020, 22, 11197–11218. [Google Scholar] [CrossRef]
- Feng, A.; Yuan, J. Smart nanocontainers: Progress on novel stimuli-responsive polymer vesicles. Macromol. Rapid Commun. 2014, 35, 767–779. [Google Scholar] [CrossRef]
- Thambi, T.; Lee, D.S. Stimuli-Responsive Polymersomes for Cancer Therapy; Elsevier: Amsterdam, The Netherlands, 2019; Volume 2, pp. 413–438. ISBN 9780081019955. [Google Scholar]
- Makhlouf, A.S.H.; Abu-Thabit, N.Y. Stimuli Responsive Polymeric Nanocarriers for Drug Delivery Applications, Volume 1: Types and Triggers; Woodhead Publishing: Sawston, UK, 2018; ISBN 9780081019979. [Google Scholar]
- Alibolandi, M.; Shahriari, M.; Ramezani, M. Chapter 16—Smart Polymersomes as Intelligent Nanomedicines in Cancer Treatment. In Polymeric Nanoparticles as a Promising Tool for Anti-Cancer Therapeutics; Academic Press: Cambridge, MA, USA, 2019; pp. 343–371. [Google Scholar]
- Li, F.; Qin, Y.; Lee, J.; Liao, H.; Wang, N.; Davis, T.P.; Qiao, R.; Ling, D. Stimuli-responsive nano-assemblies for remotely controlled drug delivery. J. Control. Release 2020, 322, 566–592. [Google Scholar] [CrossRef]
- Che, H.; van Hest, J.C.M. Stimuli-responsive polymersomes and nanoreactors. J. Mater. Chem. B 2016, 4, 4632–4647. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.F.; Lo, Y.L.; Huang, Y.C.; Yu, C.C.; Wu, Y.T.; Su, C.H.; Wang, L.F. Multi-stimuli-responsive dox released from magnetosome for tumor synergistic theranostics. Int. J. Nanomed. 2020, 15, 8623–8639. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Fei, Z.; Jin, L.; Zhou, P.; Li, C.; Liu, X.; Zhao, C. Dual-responsive polymersomes as anticancer drug carriers for the co-delivery of doxorubicin and paclitaxel. J. Mater. Chem. B 2021, 9, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Iyisan, B.; Kluge, J.; Formanek, P.; Voit, B.; Appelhans, D. Multifunctional and Dual-Responsive Polymersomes as Robust Nanocontainers: Design, Formation by Sequential Post-Conjugations, and pH-Controlled Drug Release. Chem. Mater. 2016, 28, 1513–1525. [Google Scholar] [CrossRef]
- Zhang, Y.; Uthaman, S.; Song, W.; Eom, K.H.; Jeon, S.H.; Huh, K.M.; Babu, A.; Park, I.K.; Kim, I. Multistimuli-Responsive Polymeric Vesicles for Accelerated Drug Release in Chemo-photothermal Therapy. ACS Biomater. Sci. Eng. 2020, 6, 5012–5023. [Google Scholar] [CrossRef]
- Aflori, M. Smart nanomaterials for biomedical applications—A review. Nanomaterials 2021, 11, 396. [Google Scholar] [CrossRef] [PubMed]
- Laskar, P.; Dey, J.; Ghosh, S.K. Spontaneously formed redox- and pH-sensitive polymersomes by mPEG based cytocompatible random copolymers. J. Colloid Interface Sci. 2017, 501, 22–33. [Google Scholar] [CrossRef]
- Deng, Z.; Hu, J.; Liu, S. Reactive Oxygen, Nitrogen, and Sulfur Species (RONSS)-Responsive Polymersomes for Triggered Drug Release. Macromol. Rapid Commun. 2017, 38, 1600685. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Li, X.; Huang, X.; Pan, J.; Wang, Y.; Zhou, S. Development of a pH-responsive polymersome inducing endoplasmic reticulum stress and autophagy blockade. Sci. Adv. 2020, 6, eabb8725. [Google Scholar] [CrossRef]
- Kanamala, M.; Wilson, W.R.; Yang, M.; Palmer, B.D.; Wu, Z. Mechanisms and biomaterials in pH-responsive tumour targeted drug delivery: A review. Biomaterials 2016, 85, 152–167. [Google Scholar] [CrossRef]
- Wang, L.; Liu, G.; Wang, X.; Hu, J.; Zhang, G.; Liu, S. Acid-Disintegratable Polymersomes of pH-Responsive Amphiphilic Diblock Copolymers for Intracellular Drug Delivery. Macromolecules 2015, 48, 7262–7272. [Google Scholar] [CrossRef]
- Gannimani, R.; Walvekar, P.; Naidu, V.R.; Aminabhavi, T.M.; Govender, T. Acetal containing polymers as pH-responsive nano-drug delivery systems. J. Control. Release 2020, 328, 736–761. [Google Scholar] [CrossRef]
- Welzen, P.L.W.; Martinez Ciriano, S.W.; Cao, S.; Mason, A.F.; Welzen-Pijpers, I.A.B.; van Hest, J.C.M. Reversibly self-assembled pH-responsive PEG-p(CL-g-TMC) polymersomes. J. Polym. Sci. 2021, 59, 1241–1252. [Google Scholar] [CrossRef]
- Persi, E.; Duran-Frigola, M.; Damaghi, M.; Roush, W.R.; Aloy, P.; Cleveland, J.L.; Gillies, R.J.; Ruppin, E. Systems analysis of intracellular pH vulnerabilities for cancer therapy. Nat. Commun. 2018, 9, 2997. [Google Scholar] [CrossRef]
- Piasentin, N.; Milotti, E.; Chignola, R. The control of acidity in tumor cells: A biophysical model. Sci. Rep. 2020, 10, 13613. [Google Scholar] [CrossRef]
- Koltai, T. The Ph paradigm in cancer. Eur. J. Clin. Nutr. 2020, 74, 14–19. [Google Scholar] [CrossRef]
- Das, S.S.; Bharadwaj, P.; Bilal, M.; Barani, M.; Rahdar, A.; Taboada, P.; Bungau, S.; Kyzas, G.Z. Stimuli-responsive polymeric nanocarriers for drug delivery, imaging, and theragnosis. Polymers 2020, 12, 1397. [Google Scholar] [CrossRef]
- Wang, X.; Xuan, Z.; Zhu, X.; Sun, H.; Li, J.; Xie, Z. Near-infrared photoresponsive drug delivery nanosystems for cancer photo-chemotherapy. J. Nanobiotechnol. 2020, 18, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wang, G. NIR light-responsive nanocarriers for controlled release. J. Photochem. Photobiol. C Photochem. Rev. 2021, 47, 100420. [Google Scholar] [CrossRef]
- Oerlemans, C.; Bult, W.; Bos, M.; Storm, G.; Nijsen, J.F.W.; Hennink, W.E. Polymeric micelles in anticancer therapy: Targeting, imaging and triggered release. Pharm. Res. 2010, 27, 2569–2589. [Google Scholar] [CrossRef]
- Jhaveri, A.; Deshpande, P.; Torchilin, V. Stimuli-sensitive nanopreparations for combination cancer therapy. J. Control. Release 2014, 190, 352–370. [Google Scholar] [CrossRef]
- Huang, Y.; Dong, R.; Zhu, X.; Yan, D. Photo-responsive polymeric micelles. Soft Matter 2014, 10, 6121–6138. [Google Scholar] [CrossRef]
- Weinstain, R.; Slanina, T.; Kand, D.; Klán, P. Visible-to-NIR-Light Activated Release: From Small Molecules to Nanomaterials. Chem. Rev. 2020, 120, 13135–13272. [Google Scholar] [CrossRef] [PubMed]
- Speight, J.G. Redox Transformations. In Reaction Mechanisms in Environmental Engineering; Butterworth-Heinemann: Oxford, UK, 2018; pp. 231–267. ISBN 978-0-12-804422-3. [Google Scholar]
- Ciminelli, C.; Granucci, G.; Persico, M. The Photoisomerization Mechanism of Azobenzene: A Semiclassical Simulation of Nonadiabatic Dynamics. Chem.–A Eur. J. 2004, 10, 2327–2341. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, J. Photoresponsive molecular switches for biotechnology. J. Photochem. Photobiol. C Photochem. Rev. 2012, 13, 299–309. [Google Scholar] [CrossRef]
- Zhao, H.; Sterner, E.S.; Coughlin, E.B.; Theato, P. o-Nitrobenzyl Alcohol Derivatives: Opportunities in Polymer and Materials Science. Macromolecules 2012, 45, 1723–1736. [Google Scholar] [CrossRef]
- Hansen, M.J.; Velema, W.A.; Lerch, M.M.; Szymanski, W.; Feringa, B.L. Wavelength-selective cleavage of photoprotecting groups: Strategies and applications in dynamic systems. Chem. Soc. Rev. 2015, 44, 3358–3377. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.K. Photocleavable linkers: Design and applications in nanotechnology. Photonanotechnol. Ther. Imaging 2020, 243–275. [Google Scholar] [CrossRef]
- Song, J.; Fang, Z.; Wang, C.; Zhou, J.; Duan, B.; Pu, L.; Duan, H. Photolabile plasmonic vesicles assembled from amphiphilic gold nanoparticles for remote-controlled traceable drug delivery. Nanoscale 2013, 5, 5816–5824. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, G.; Hu, J.; Zhang, G.; Liu, S. Concurrent block copolymer polymersome stabilization and bilayer permeabilization by stimuli-regulated “traceless” crosslinking. Angew. Chem. Int. Ed. Engl. 2014, 53, 3138–3142. [Google Scholar] [CrossRef]
- Wang, X.; Hu, J.; Liu, G.; Tian, J.; Wang, H.; Gong, M.; Liu, S. Reversibly Switching Bilayer Permeability and Release Modules of Photochromic Polymersomes Stabilized by Cooperative Noncovalent Interactions. J. Am. Chem. Soc. 2015, 137, 15262–15275. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, R.; Yang, H.; Bao, C.; Fan, J.; Wang, C.; Lin, Q.; Zhu, L. Light-responsive polymersomes with a charge-switch for targeted drug delivery. J. Mater. Chem. B 2020, 8, 727–735. [Google Scholar] [CrossRef]
- Kim, B.S.; Naito, M.; Kamegawa, R.; Kim, H.J.; Iizuka, R.; Funatsu, T.; Ueno, S.; Ichiki, T.; Kishimura, A.; Miyata, K. Photo-reactive oligodeoxynucleotide-embedded nanovesicles (PROsomes) with switchable stability for efficient cellular uptake and gene knockdown. Chem. Commun. 2020, 56, 9477–9480. [Google Scholar] [CrossRef]
- Mallory, M.; Gogineni, E.; Jones, G.C.; Greer, L.; Simone, C.B. Therapeutic hyperthermia: The old, the new, and the upcoming. Crit. Rev. Oncol. Hematol. 2016, 97, 56–64. [Google Scholar] [CrossRef]
- Li, J.; Zhang, W.; Ji, W.; Wang, J.; Wang, N.; Wu, W.; Wu, Q.; Hou, X.; Hu, W.; Li, L. Near infrared photothermal conversion materials: Mechanism, preparation, and photothermal cancer therapy applications. J. Mater. Chem. B 2021, 9, 7909–7926. [Google Scholar] [CrossRef]
- Eskiizmir, G.; Ermertcan, A.T.; Yapici, K. Nanomaterials: Promising structures for the management of oral cancer. In Nanostructures for Oral Medicine; Elsevier: Amsterdam, The Netherlands, 2017; pp. 511–544. ISBN 9780323477215. [Google Scholar]
- Han, H.S.; Choi, K.Y. Advances in nanomaterial-mediated photothermal cancer therapies: Toward clinical applications. Biomedicines 2021, 9, 305. [Google Scholar] [CrossRef]
- Li, Z.; Chen, Y.; Yang, Y.; Yu, Y.; Zhang, Y.; Zhu, D.; Yu, X.; Ouyang, X.; Xie, Z.; Zhao, Y.; et al. Recent Advances in Nanomaterials-Based Chemo-Photothermal Combination Therapy for Improving Cancer Treatment. Front. Bioeng. Biotechnol. 2019, 7, 293. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Cheng, L.; Liu, A.; Yin, J.; Kuang, M.; Duan, H. Plasmonic vesicles of amphiphilic gold nanocrystals: Self-assembly and external-stimuli-triggered destruction. J. Am. Chem. Soc. 2011, 133, 10760–10763. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Chen, K.; Xue, G.; Cai, J.; Zou, G.; Li, Y.; Zhang, Q. Near-infrared light induced fusion and fission of azobenzene-containing polymer vesicles. RSC Adv. 2013, 3, 23997–24000. [Google Scholar] [CrossRef]
- Benov, L. Photodynamic Therapy: Current Status and Future Directions. Med. Princ. Pract. 2014, 24, 14. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, S.; Knap, B.; Przystupski, D.; Saczko, J.; Kędzierska, E.; Knap-Czop, K.; Kotlińska, J.; Michel, O.; Kotowski, K.; Kulbacka, J. Photodynamic therapy–mechanisms, photosensitizers and combinations. Biomed. Pharmacother. 2018, 106, 1098–1107. [Google Scholar] [CrossRef]
- Pervaiz, S.; Olivo, M. Art and science of photodynamic therapy. Clin. Exp. Pharmacol. Physiol. 2006, 33, 551–556. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Amin Doustvandi, M.; Mohammadnejad, F.; Kamari, F.; Gjerstorff, M.F.; Baradaran, B.; Hamblin, M.R. Photodynamic therapy for cancer: Role of natural products. Photodiagn. Photodyn. Ther. 2019, 26, 395–404. [Google Scholar] [CrossRef]
- Hamblin, M.R.; Luke Newman, E. New trends in photobiology. On the mechanism of the tumour-localising effect in photodynamic therapy. J. Photochem. Photobiol. B Biol. 1994, 23, 3–8. [Google Scholar] [CrossRef]
- Qidwai, A.; Anuu; Nabi, B.; Kotta, S.; Narang, J.K.; Baboota, S.; Ali, J. Role of nanocarriers in photodynamic therapy. Photodiagn. Photodyn. Ther. 2020, 30, 101782. [Google Scholar] [CrossRef]
- Hsu, C.Y.; Nieh, M.P.; Lai, P.S. Facile self-assembly of porphyrin-embedded polymeric vesicles for theranostic applications. Chem. Commun. 2012, 48, 9343–9345. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Xiao, L.; Anraku, Y.; Mi, P.; Liu, X.; Cabral, H.; Inoue, A.; Nomoto, T.; Kishimura, A.; Nishiyama, N.; et al. Polyion complex vesicles for photoinduced intracellular delivery of amphiphilic photosensitizer. J. Am. Chem. Soc. 2014, 136, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wei, K.; Zuo, S.; Xu, Y.; Zha, Z.; Ke, W.; Chen, H.; Ge, Z. Light-Triggered Clustered Vesicles with Self-Supplied Oxygen and Tissue Penetrability for Photodynamic Therapy against Hypoxic Tumor. Adv. Funct. Mater. 2017, 27, 1702108. [Google Scholar] [CrossRef]
- Lu, X.; Liu, L. Asymmetric polyplex-nanocapsules loaded with photosentisizer for light-assisted gene transfer. J. Photochem. Photobiol. B Biol. 2017, 174, 269–275. [Google Scholar] [CrossRef]
- Qi, J.; Chen, C.; Ding, D.; Tang, B.Z. Aggregation-Induced Emission Luminogens: Union Is Strength, Gathering Illuminates Healthcare. Adv. Healthc. Mater. 2018, 7, 1800477. [Google Scholar] [CrossRef]
- Hong, Y.; Lam, J.W.Y.; Tang, B.Z. Aggregation-induced emission: Phenomenon, mechanism and applications. Chem. Commun. 2009, 4332–4353. [Google Scholar] [CrossRef]
- Chen, Y.; Lam, J.W.Y.; Kwok, R.T.K.; Liu, B.; Tang, B.Z. Aggregation-induced emission: Fundamental understanding and future developments. Mater. Horiz. 2019, 6, 428–433. [Google Scholar] [CrossRef]
- Wang, H.; Zhao, E.; Lam, J.W.Y.; Tang, B.Z. AIE luminogens: Emission brightened by aggregation. Mater. Today 2015, 18, 365–377. [Google Scholar] [CrossRef]
- Guan, J.; Wei, R.; Prlj, A.; Peng, J.; Lin, K.; Liu, J.; Han, H.; Corminboeuf, C.; Zhao, D.; Yu, Z.; et al. Direct Observation of Aggregation-Induced Emission Mechanism. Angew. Chem. 2020, 132, 15013–15019. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.; Li, H.; Song, N.; Wang, D.; Tang, B.Z. Supramolecular materials based on AIE luminogens (AIEgens): Construction and applications. Chem. Soc. Rev. 2020, 49, 1144–1172. [Google Scholar] [CrossRef]
- Zhou, T.; Hu, R.; Wang, L.; Qiu, Y.; Zhang, G.; Deng, Q.; Zhang, H.; Yin, P.; Situ, B.; Zhan, C.; et al. An AIE-Active Conjugated Polymer with High ROS-Generation Ability and Biocompatibility for Efficient Photodynamic Therapy of Bacterial Infections. Angew. Chem. Int. Ed. Engl. 2020, 59, 9952–9956. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Lu, H.; Jiang, Z.; Guan, Y.; Zou, J.; Wang, X.; Cheng, R.; Gao, H. Low-power white light triggered AIE polymer nanoparticles with high ROS quantum yield for mitochondria-targeted and image-guided photodynamic therapy. J. Mater. Chem. B 2017, 5, 6277–6281. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; He, W.; Tang, B.Z. Aggregate Materials beyond AIEgens. Acc. Mater. Res. 2021, 2, 1251–1260. [Google Scholar] [CrossRef]
- Cao, S.; Shao, J.; Wu, H.; Song, S.; De Martino, M.T.; Pijpers, I.A.B.; Friedrich, H.; Abdelmohsen, L.K.E.A.; Williams, D.S.; van Hest, J.C.M. Photoactivated nanomotors via aggregation induced emission for enhanced phototherapy. Nat. Commun. 2021, 12, 2077. [Google Scholar] [CrossRef]
- Pan, P.; Svirskis, D.; Rees, S.W.P.; Barker, D.; Waterhouse, G.I.N.; Wu, Z. Photosensitive drug delivery systems for cancer therapy: Mechanisms and applications. J. Control. Release 2021, 338, 446–461. [Google Scholar] [CrossRef]
- Wen, S.; Zhou, J.; Zheng, K.; Bednarkiewicz, A.; Liu, X.; Jin, D. Advances in highly doped upconversion nanoparticles. Nat. Commun. 2018, 9, 2415. [Google Scholar] [CrossRef]
- Bagheri, A.; Arandiyan, H.; Boyer, C.; Lim, M. Lanthanide-doped upconversion nanoparticles: Emerging intelligent light-activated drug delivery systems. Adv. Sci. 2016, 3, 1500437. [Google Scholar] [CrossRef]
- Zhang, L.; Jin, D.; Stenzel, M.H. Polymer-Functionalized Upconversion Nanoparticles for Light/Imaging-Guided Drug Delivery. Biomacromolecules 2021, 22, 3168–3201. [Google Scholar] [CrossRef]
- Gao, C.; Zheng, P.; Liu, Q.; Han, S.; Li, D.; Luo, S.; Temple, H.; Xing, C.; Wang, J.; Wei, Y.; et al. Recent advances of upconversion nanomaterials in the biological field. Nanomaterials 2021, 11, 2474. [Google Scholar] [CrossRef]
- Wang, Y.; Song, S.; Zhang, S.; Zhang, H. Stimuli-responsive nanotheranostics based on lanthanide-doped upconversion nanoparticles for cancer imaging and therapy: Current advances and future challenges. Nano Today 2019, 25, 38–67. [Google Scholar] [CrossRef]
- Wang, C.; Cheng, L.; Liu, Z. Upconversion Nanoparticles for Photodynamic Therapy and Other Cancer Therapeutics. Theranostics 2013, 3, 317. [Google Scholar] [CrossRef] [PubMed]
- Hou, B.; Zheng, B.; Yang, W.; Dong, C.; Wang, H.; Chang, J. Construction of near infrared light triggered nanodumbbell for cancer photodynamic therapy. J. Colloid Interface Sci. 2017, 494, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhou, L.; Guan, Y.; Su, Y.; Dong, C.M. Multi-responsive polypeptidosome: Characterization, morphology transformation, and triggered drug delivery. Macromol. Rapid Commun. 2014, 35, 1673–1678. [Google Scholar] [CrossRef]
- Zhang, X.; Zhuo, R. Dual UV- and pH-Responsive Supramolecular Vesicles Mediated by Host–Guest Interactions for Drug Controlled Release. Macromol. Chem. Phys. 2016, 217, 1934–1940. [Google Scholar] [CrossRef]
- Yao, C.; Wang, X.; Liu, G.; Hu, J.; Liu, S. Distinct Morphological Transitions of Photoreactive and Thermoresponsive Vesicles for Controlled Release and Nanoreactors. Macromolecules 2016, 49, 8282–8295. [Google Scholar] [CrossRef]
- Sun, Z.; Liu, G.; Hu, J.; Liu, S. Photo- and Reduction-Responsive Polymersomes for Programmed Release of Small and Macromolecular Payloads. Biomacromolecules 2018, 19, 2071–2081. [Google Scholar] [CrossRef]
- Tong, Z.; Zhou, J.; Huang, R.; Zhou, J.; Zhang, R.; Zhuo, W.; Jiang, G. Dual-responsive supramolecular self-assembly of inclusion complex of an azobenzene-ended poly(ε-caprolactone) with a water-soluble pillar[6]arene and its application in controlled drug release. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 2477–2482. [Google Scholar] [CrossRef]
- Weng, C.; Chen, H.; Xu, T.; Li, Z.; Liu, X.; Ding, M.; Zhang, Q.; Tan, H.; Fu, Q. Photo-responsive Self-Reducible Polymers: Overcoming the Spatiotemporal Barriers for Hypersensitivity. ACS Mater. Lett. 2020, 2, 602–609. [Google Scholar] [CrossRef]
- Tsai, M.F.; Lo, Y.L.; Soorni, Y.; Su, C.H.; Sivasoorian, S.S.; Yang, J.Y.; Wang, L.F. Near-Infrared Light-Triggered Drug Release from Ultraviolet- And Redox-Responsive Polymersome Encapsulated with Core-Shell Upconversion Nanoparticles for Cancer Therapy. ACS Appl. Bio Mater. 2021, 4, 3264–3275. [Google Scholar] [CrossRef]
- Liao, J.F.; Li, W.T.; Peng, J.R.; Yang, Q.; Li, H.; Wei, Y.Q.; Zhang, X.N.; Qian, Z.Y. Combined cancer photothermal-chemotherapy based on doxorubicin/gold nanorod-loaded polymersomes. Theranostics 2015, 5, 345–356. [Google Scholar] [CrossRef]
- He, H.; Ji, S.; He, Y.; Zhu, A.; Zou, Y.; Deng, Y.; Ke, H.; Yang, H.; Zhao, Y.; Guo, Z.; et al. Photoconversion-Tunable Fluorophore Vesicles for Wavelength-Dependent Photoinduced Cancer Therapy. Adv. Mater. 2017, 29, 1606690. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Fan, F.; Huang, C.; Zhang, Z.; Qin, Y.; Lu, L.; Wang, H.; Jin, X.; Zhao, H.; Yang, H.; et al. Bubble-generating polymersomes loaded with both indocyanine green and doxorubicin for effective chemotherapy combined with photothermal therapy. Acta Biomater. 2018, 75, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Liu, G.; Zhang, G.; Hu, J.; Liu, S. Engineering Cross-Linkable Plasmonic Vesicles for Synergistic Chemo-Photothermal Therapy Using Orthogonal Light Irradiation. Macromolecules 2018, 51, 8530–8538. [Google Scholar] [CrossRef]
- Tang, L.; Yang, Z.; Zhou, Z.; Ma, Y.; Kiesewetter, D.O.; Wang, Z.; Fan, W.; Zhu, S.; Zhang, M.; Tian, R.; et al. A logic-gated modular nanovesicle enables programmable drug release for on-demand chemotherapy. Theranostics 2019, 9, 1358–1368. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cui, W.; Qu, X.; Wu, H.; Qu, L.; Zhang, X.; Mäkilä, E.; Salonen, J.; Zhu, Y.; Yang, Z.; et al. Photothermal-responsive nanosized hybrid polymersome as versatile therapeutics codelivery nanovehicle for effective tumor suppression. Proc. Natl. Acad. Sci. USA 2019, 116, 7744–7749. [Google Scholar] [CrossRef]
- Saravanakumar, G.; Park, H.; Kim, J.; Park, D.; Lim, J.; Lee, J.; Kim, W.J. Polymersomes with singlet oxygen-labile poly(β-aminoacrylate) membrane for NIR light-controlled combined chemo-phototherapy. J. Control. Release 2020, 327, 627–640. [Google Scholar] [CrossRef]
- Tang, Q.; Hu, P.; Peng, H.; Zhang, N.; Zheng, Q.; He, Y. Near-Infrared Laser-Triggered, Self-Immolative Smart Polymersomes for in vivo Cancer Therapy. Int. J. Nanomed. 2020, 15, 137–149. [Google Scholar] [CrossRef]
- Hu, J.; Luo, H.; Qu, Q.; Liao, X.; Huang, C.; Chen, J.; Cai, Z.; Bao, Y.; Chen, G.; Li, B.; et al. Cell Membrane-Inspired Polymeric Vesicles for Combined Photothermal and Photodynamic Prostate Cancer Therapy. ACS Appl. Mater. Interfaces 2020, 12, 42511–42520. [Google Scholar] [CrossRef]
- He, Y.; Guo, S.; Zhang, Y.; Liu, Y.; Ju, H. Near-Infrared Photo-controlled Permeability of a Biomimetic Polymersome with Sustained Drug Release and Efficient Tumor Therapy. ACS Appl. Mater. Interfaces 2021, 13, 14951–14963. [Google Scholar] [CrossRef]
- Wang, L.; Yuan, Y.; Lin, S.; Huang, J.; Dai, J.; Jiang, Q.; Cheng, D.; Shuai, X. Photothermo-chemotherapy of cancer employing drug leakage-free gold nanoshells. Biomaterials 2016, 78, 40–49. [Google Scholar] [CrossRef]
- Zhu, K.; Liu, G.; Hu, J.; Liu, S. Near-Infrared Light-Activated Photochemical Internalization of Reduction-Responsive Polyprodrug Vesicles for Synergistic Photodynamic Therapy and Chemotherapy. Biomacromolecules 2017, 18, 2571–2582. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Ju, Y.; Lin, Y.; Bai, X.; Hu, W.; Wang, Y.; Luo, H.; Tong, Z. Reactive Oxygen Species Synergistic pH/H2O2-Responsive Poly(L-lactic acid)-block-poly(sodium 4-styrenesulfonate)/Citrate-Fe(III)@ZIF-8 Hybrid Nanocomposites for Controlled Drug Release. ACS Appl. Bio Mater. 2019, 2, 3648–3658. [Google Scholar] [CrossRef]
- Mena-Giraldo, P.; Orozco, J. Polymeric micro/nanocarriers and motors for cargo transport and phototriggered delivery. Polymers 2021, 13, 3920. [Google Scholar] [CrossRef] [PubMed]
- Mena-Giraldo, P.; Orozco, J. Photosensitive Polymeric Janus Micromotor for Enzymatic Activity Protection and Enhanced Substrate Degradation. ACS Appl. Mater. Interfaces 2022, 14, 5897–5907, acsami.1c14663. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Bhat, R.; Saini, D.; Ramachandran, A. Theragnostic nanomotors: Successes and upcoming challenges. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2021, 13, e1736. [Google Scholar] [CrossRef]
- Yao, C.; Li, Y.; Wang, Z.; Song, C.; Hu, X.; Liu, S. Cytosolic NQO1 Enzyme-Activated Near-Infrared Fluorescence Imaging and Photodynamic Therapy with Polymeric Vesicles. ACS Nano 2020, 14, 1919–1935. [Google Scholar] [CrossRef]
- Shi, Y.; van der Meel, R.; Chen, X.; Lammers, T. The EPR effect and beyond: Strategies to improve tumor targeting and cancer nanomedicine treatment efficacy. Theranostics 2020, 10, 7921. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amphiphilic Copolymer Type | Polymersome Building Blocks | Ref. |
|---|---|---|
| Block copolymer | PS-b-PAA | [77] |
| PEE37-b-PEO40 | [78] | |
| PNBA-b-PDMA | [80] | |
| PEG-P(CLgTMC) | [81] | |
| Graft copolymer | PHOU-g-PEG | [84] |
| PEG/EAB–PPPs | [85] | |
| PGA-g-(PCL-b-PEO) | [86] | |
| Random copolymer | NALGA-NADA | [90] |
| PPAPE | [91] | |
| Alternate copolymer | P(MF-alt-VBP) | [101] |
| Dendronized copolymer | PS-dendr-(NH2)8 | [108] |
| PEG-bis-MPA | [109,110] | |
| Gradient copolymer | MPOx | [122] |
| P(AA-grad-TFEMA) | [123] |
| Polymersome Building Blocks | Light-Activable Moieties | Light Stimulus Wavelength (nm) | Assembly Method | Therapeutic Cargo | Size (nm) | Cell Line/Biological Model | Ref. |
|---|---|---|---|---|---|---|---|
| PEG-AuNPs-PNBA | NBA | 365 | Film rehydratation | DOX | 210 a | MDA-MB-435 | [247] |
| PEO45-b-PNBOC30 | ONB | 365 | Nanoprecipitacion | DOX | - | - | [248] |
| PEO-b-PSPA | SP | 365/530 | Nanoprecipitation | 5-dFu | 450 a | - | [249] |
| PAMAM | Azide | 365 | Nanoprecipitation | OVA | 121 a | RAW264.7 | [198] |
| PDMA-b-PNBA | ONB | 365 | Emulsion | DOX | - | - | [80] |
| C12NB | ONB | 365 | Nanoprecipitation | DOX | 80–150 b | HeLa | [250] |
| PRO and PEG-P(Asp-AP) | PRO | 312 | Vortex mixing | AsNEAT2 | 80 a | A549 | [251] |
| Polymersome Building Blocks | Light-Activable Moieties | Light Stimulus Wavelength (nm) | Assembly Method | Therapeutic Cargo | Size (nm) | Ref. |
|---|---|---|---|---|---|---|
| PEG-AuNRs-PMMA | AuNRs | 785 | Film rehydratation | AuNRs | 200 b | [257] |
| PNIPAM-b-PAZO | AZO | 365 | Nanoprecipitation | MN | 5 µm b | [258] |
| PBD35-b-PEO20 | AuNPs | 532 nm | Gel-assisted rehydration | AuNPs | 71 a | [189] |
| Polymersome Building Blocks | Light-Activable Moieties | Light Stimulus Wavelength (nm) | Assembly Method | Therapeutic Cargo | Size (nm) | Cell Line/Biological Model | Ref. |
|---|---|---|---|---|---|---|---|
| PPLA | Porphyrin | 420 | Oil-in-water solvent evaporation | Porphyrin | 52 | HeLa | [265] |
| PMOXA–PDMS–PMOXA | Rose Bengal | 543 | Film rehydration | Rose Bengal | Rg: 105 a Rh: 110 a | HeLa | [196] |
| PEG45-(PAsp)75 homo-P(Asp-AP)82 | AlPcS2a | 400–700 | Vortex mixing | AlPcS2a | 106 a | A549 | [266] |
| CC-PAMAM Thioketal moiety-PEG | Ce6 | 660 | Nanoprecipitation | Ce6 | 187.5 a 155.2 b | BxPC-3 BALB/c nude mice | [267] |
| PEG-PCL-PEI | PheoA | 670 | Nanoprecipitation | PheoA | 200–280 a 200 b | HeLa | [268] |
| PEG-P(CLgTMC) | BODIPY | 606 | Direct hydratation | BODIPY | - | A549, HeLa, HepG2, and 3T3 A549 tumor-bearing nude mice | [81] |
| Stimuli | Polymersomes Building Blocks | Light-Activable Moieties | Light Stimulus Wavelength (nm) | Assembly Method | Therapeutic Cargo | Size (nm) | Cell Line/Biological Model | Ref. |
|---|---|---|---|---|---|---|---|---|
| Light Redox | PNBC-b-PEO | ONB Thiol | 365 | Film rehydratation | DOX | 83 a | HeLa | [287] |
| Light pH | β-CD-AZO-ACE⊂ α-CD | AZO Acetal moiety | 365 | Nanoprecipitation | DOX | 40 a 88.5 b | - | [288] |
| Light Thermo | PNIPAM31-b-PNBOCA53 | ONB PNIPAM | 365 808 | Nanoprecipitation | DOX | - | - | [289] |
| Light Redox | PEO45-b-PCSSMA22 | Coumarin Disulfide linkage | 430 | Nanoprecipitation | DOX TR-dextran | - | - | [290] |
| Light pH | PCL-Azo + WP6 | AZO WP6 | 365 435 | Nanoprecipitation | DOX | HepG2 | [291] | |
| Light Redox | ONB-DTT-CDI-MPEG | ONB Disulfide linkage | 365 | Solvent exchange | DOX | Rg:35.5 a Rh:34.8 a 90 a | MCF-7 | [292] |
| Light Redox pH | PEO45-b-P(NCMA0.55- co-DPA0.45)29; PEO45-b-PNCMA17-b-PDPA21; PEO45-b-P(DCMA0.45-co-PDPA0.55)33 | ONB Disulfide likage Carbamate Tertiary amine | 365 | Nanoprecipitation | Gem DOX 5-FU Calcein | 540–770 a | - | [207] |
| Light Redox | PCL-ONB-SS-PMAA | UCNP ONB Disulfide linkage | 980 | Double emulsion | DOX | - | A549, CR-5802, HEL-299 A549 tumor bearing mice | [293] |
| Therapies | Polymersome Building Blocks | Light-Activable Moieties | Light Stimuli Wavelength (nm) | Assembly Method | Therapeutic Cargo | Size (nm) | Cell Line/Biological Model | Ref. |
|---|---|---|---|---|---|---|---|---|
| Chemotherapy PTT | mPEG-PCL | AuNRs | 808 | Double emulsion | DOX | 208 a 175 b | C26 Mice bearing C26 tumors | [294] |
| PTT PDT | PEG45-PCL60-PNIPAM33 | BODIPY | 660 785 | Ultrasonication | - | 127.3 a 72.5 b | 4T1 Mice bearing 4T1 tumors | [295] |
| Chemotherapy PTT | PCL8000-PEG8000-PCL8000 | ICG | 808 | Film rehydration | DOX | 208.1 a | 4T1-Luc | [296] |
| Chemotherapy PTT | PEO-b-PMALA PNBOC-b-PMALA | ONB | 410 | Nanoprecipitation | DOX PTX AuNPs | 194 a,b | HepG2 | [297] |
| Chemotherapy PTT | PPS-PEG | CR780 dye | 808 | Film rehydration | DOX | 100 a | U87MG Mice bearing U87MG tumors | [298] |
| Chemotherapy PTT | PEG-b-PLA/DOPC | AuNRs | 808 | Double emulsion | DOX Docetaxel Rapamycin Afatinib | 286 b | SKBR-3/AR HER2-positive breast cancer mouse model | [299] |
| Chemotherapy PTT PDT | mPEG-b-PBAC-b-mPEG | IR-780 PBAC | 808 | Nanoprecipitacion | DOX | 204 b | 4T1, CT26 4T1 tumor bearing mice | [300] |
| Chemotherapy PDT | PPS20-b-PEG12 | ZnPc | 660 | Solvent dispersion | DOX | 150 a | A375 Nude mice bearing A375 | [301] |
| PTT PDT | PEG-PCL | ICG AuNRs | 785 | Ultrasonication | - | 238 b | PC3 Nude mice bearing PC3 | [302] |
| Chemotherapy PDT | b-(AZO-grafted Dex)-b((MMA)-r-(β-CDAc)-r-(PorAc)) | AZO Porphyrin | 365 | Nanoprecipitation | Quercetin 5-FU | 200 b | - | [200] |
| Chemotherapy PDT | P(OEGMA-co-EoS)-b-PNBOC | UCNPs ONB | 980 | Nanoprecipitation | AQ4N | 234 a 209 b | HepG2 | [303] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández Becerra, E.; Quinchia, J.; Castro, C.; Orozco, J. Light-Triggered Polymersome-Based Anticancer Therapeutics Delivery. Nanomaterials 2022, 12, 836. https://doi.org/10.3390/nano12050836
Hernández Becerra E, Quinchia J, Castro C, Orozco J. Light-Triggered Polymersome-Based Anticancer Therapeutics Delivery. Nanomaterials. 2022; 12(5):836. https://doi.org/10.3390/nano12050836
Chicago/Turabian StyleHernández Becerra, Elisa, Jennifer Quinchia, Cristina Castro, and Jahir Orozco. 2022. "Light-Triggered Polymersome-Based Anticancer Therapeutics Delivery" Nanomaterials 12, no. 5: 836. https://doi.org/10.3390/nano12050836
APA StyleHernández Becerra, E., Quinchia, J., Castro, C., & Orozco, J. (2022). Light-Triggered Polymersome-Based Anticancer Therapeutics Delivery. Nanomaterials, 12(5), 836. https://doi.org/10.3390/nano12050836