1. Introduction
The rapid growth of the world’s population and the industrialization era have led to unprecedented energy consumption, which has resulted in environmental issues, concerns about environmental sustainability, and economic obstacles. Fossil fuels are currently utilized as an energy source for more than 80% of the world’s paramount energy and are directly involved in greenhouse gas emissions [
1]. However, fossil fuels will not be able to fulfill the huge demand for energy in the future, due to their limited supply. Hence, there is a great need for the deployment of clean and renewable sources of energy. Renewable resources such as solar, hydraulic, biomass, wind, and geothermal energy have been researched and developed over the last few decades and are still under investigation. Among these, sunlight offers a prominent energy source, with an irradiation level of 1.8 × 10
14 kW at the Earth’s surface [
2], which can be converted into electricity and heat with a nominal impact on the environment [
3]. Photovoltaic solar cells are among the most effective applications which convert optical energy (sunlight) to electrical energy. Therefore, with the invention of solar photovoltaic devices in the 1950s, numerous techniques and device designs have been suggested and achieved, such as single-/multijunction III-V solar cells [
4,
5,
6], dye/quantum-dot-sensitized solar cells [
7,
8,
9], chalcopyrite solar cells [
10,
11], and crystalline/amorphous Si solar cells [
12]. In particular, photovoltaic (PV) panels based on silicon technology (crystalline silicon and polycrystalline silicon) are widely installed, owing to their eco-friendly and stable characteristics, superior PCE, and reliable supports in the semiconductor industry. Si-based solar cells have provided an outstanding efficiency of 25–26.7% in recent years [
13,
14,
15]. On the other hand, GaAs-based III-V compound semiconductor solar cells have attracted huge attention due to their direct bandgap properties that exhibit high absorption over the entire visible portion of the solar spectrum. Therefore, GaAs-based solar cells are regarded as a promising technology for a high PCE. A GaAs solar cell with a single-crystal structure and thin-film crystal devices achieved high PCEs of 27.8% and 29.1%, respectively [
16].
Despite the high PCEs, solar cells such as Si, GaAs, CIGS, and CdTe still exhibit poor spectral response in short-wavelength regions [
17,
18,
19,
20]. In addition, the surface recombination loss and surface Fresnel reflection are too high in these types of solar cells [
21,
22]. Fresnel reflection occurs due to the refractive index discrepancy between substrate–air interfaces. This discrepancy can be efficiently controlled by inserting an antireflective coating (ARC) between surface–air interfaces, which also offers a significant impact on enhancing the light utilization of the solar device [
23,
24,
25]. Furthermore, to minimize the surface Fresnel reflection by using ARC, we must also employ the entire solar spectrum efficiently to increase the PCE, especially in the ultraviolet (UV) region. However, in the UV region, high-energy photons are easily absorbed by the material near to the surface of the solar cell, and the recombination loss of the generated electron–hole pair near to the surface is also high [
26]. For this reason, a solar cell with the ordinary design cannot utilize the short-wavelength part of the solar spectrum effectively. This is a barrier to achieving a high PCE for any kind of solar cell. Fortunately, this issue can be solved by converting high-energy photons into lower-energy photons; this phenomenon is known as the luminescent downshifting (LDS) effect [
27,
28,
29,
30].
In 1990, Hovel et al. first demonstrated the advantages of utilizing the LDS effect for solar cell applications [
31]. Subsequently, various kinds of work have been reported using quantum dots (QDs) as an LDS layer incorporated into different kinds of solar cells, such as silicon [
32], GaAs [
33], CdTe [
20], organic [
34], and CIGS [
35] solar cells, to enhance the cell performance in the short-wavelength region, resulting in a higher PCE. The LDS materials absorb the light in the UV region and re-emit photons in the visible wavelength region, which can enter profoundly into the solar cell. Due to the fewer nonideal recombination centers in the bulk [
26,
36], more photons can be used to generate electron–hole pairs, which has a significant impact on increasing the external quantum efficiency (EQE) and generating a higher short-circuit current (J
sc). However, the open-circuit voltage (V
OC) and fill factor (FF) do not change significantly, and as a result, the electronic characteristics of the device resistance or the semiconducting material are not changed. Hence, due to the high current generation, the PCE can increase efficiently.
The host materials embedded with luminescent dye also have a high impact on maximizing the benefits of the LDS layer and reducing additional losses. A suitable host material should have a high transmittance and a low scattering effect in the high-response region of the cell. Many researchers have reported different kinds of host materials, such as polyvinyl acetate (PVA) [
37,
38], polymethyl methacrylate (PMMA) [
28,
39], poly(dimethylsiloxane) (PDMS) [
40,
41], CaF
2 [
42], Al
2O
3 [
31], and SiO
2 [
43].
A proper LDS material should contain some significant characteristics, such as (i) a wide absorption band, especially in the low-EQE region of the cell; (ii) a high absorption coefficient; (iii) a high luminescence quantum yield (LQE); (iv) a confined emission band that corresponds to the EQE peak of the cell; (v) a large Stokes shift, in order to decrease the losses due to reabsorption; (vi) high photostability; and (vii) low cost.
There are essentially three types of LDS materials that have been reported so far: (i) rare-earth ions/complexes [
37,
38,
42,
44]; (ii) organic dyes [
28,
39,
45,
46,
47]; and (iii) QDs [
48,
49,
50,
51,
52]. Among these, QDs provide significant advantages over organic dyes and rare-earth complexes. QDs have a broad absorption band and a tunable emission band, a large Stokes shift, high photostability, and a high emission intensity [
52,
53,
54]. On the other hand, rare-earth complexes have a high LQE [
55,
56] but a relatively low absorption coefficient [
56]. Organic dyes have a small Stokes shift, a narrow absorption band [
57], and poor photostability [
31,
58]. Semiconductor-based QDs, such as CdS, CdSe/ZnS, CsPbBr
3, and CsPbI
3, have recently gained prime importance as the LDS materials of solar cells due to the great improvements to the PCEs of solar cells [
33,
41,
59], whereas they may be instable when exposed to air, heat, and moisture [
60,
61]. Therefore, many works have been reported that focus on improving the stability issues of QDs, perfecting them for practical application.
Thus, in this review, we will first discuss the physical and optical properties of QDs. Secondly, different approaches to the stability improvement and synthesis process of QDs will be presented. Thereafter, we will focus on the application of LDS layers in different kinds of solar cells. Finally, the challenges of the QD-based LDS layer for solar cells will be illustrated.
2. Physical and Optical Characteristics of QDs
QDs are three-dimensional confined nanocrystals, and their electronic and photonic characteristics could depend on their size [
62,
63,
64]. Due to the quantum confinement effect, the energy bandgap increases as the size of the QDs decreases. Hence, it is viable to obtain different emission colors by tuning the size of QDs. Colors such as violet, blue, and green are produced by smaller QDs, whereas yellow, orange, and red are produced by larger QDs. In view of their electronic wave functions and discrete electronic states, QDs are considered artificial atoms, since they are more akin to atoms than bulk materials [
65,
66]. Due to their small size, the electron of these nanoparticles is confined, and the energy levels are quantized according to Pauli’s exclusion principle when the nanocrystal radius is less than the exciton Bohr radius [
67,
68]. Their tunable emission, high thermal and optical stability, high quantum yield, and very short fluorescent lifetime make QDs promising candidates for application in field-effect transistors [
69], photodetectors [
70,
71], solar cells [
33,
72,
73], LEDs [
74,
75], biological fields [
76,
77], and visible-light communications [
78]. Cd-based QDs have been one of the most prominent commercially available QDs used in optoelectronic applications for many years. However, Cd
2+ ions are toxic in nature and have a negative impact on devices and tissue cells [
79,
80]. Consequently, scientists have concentrated their efforts on producing Cd-free light emitters that are as efficient, brilliant, and long-lasting as their environmentally harmful competitors.
Perovskite QDs (PQDs) are a class of direct-bandgap semiconductor materials that feature a high photoluminescence quantum yield (PLQY) [
81] and a high PL intensity and exhibit tunable narrow emission with symmetric PL peaks by altering the halide configuration [
82]. Due to these unique properties, PQDs are emerging as the next-generation materials in photonic applications. In addition, being highly chemically processable and possessing superior optical properties, PQDs make excellent models of colloidal nanocrystals (NCs). PQDs can be blended efficiently using cost-effective precursors at ambient temperature or at a lower temperature (<180 °C) and exhibit both high electron mobility and a large exciton diffusion length [
83,
84]. PQDs are 3D-structure-based materials that can be expressed chemically as AMX
3, where an organic cation (MA
+ or FA
+) or monovalent inorganic metal cations (Cs
+ or Rb
+) occupy the A-site; divalent metal cations (Ni
2+, Cu
2+, Eu
2+, Sn
2+, Pb
2+, etc.) occupy the M-site; and the halide anion (Br
−, Cl
−, or I
−) occupies the X-site [
85].
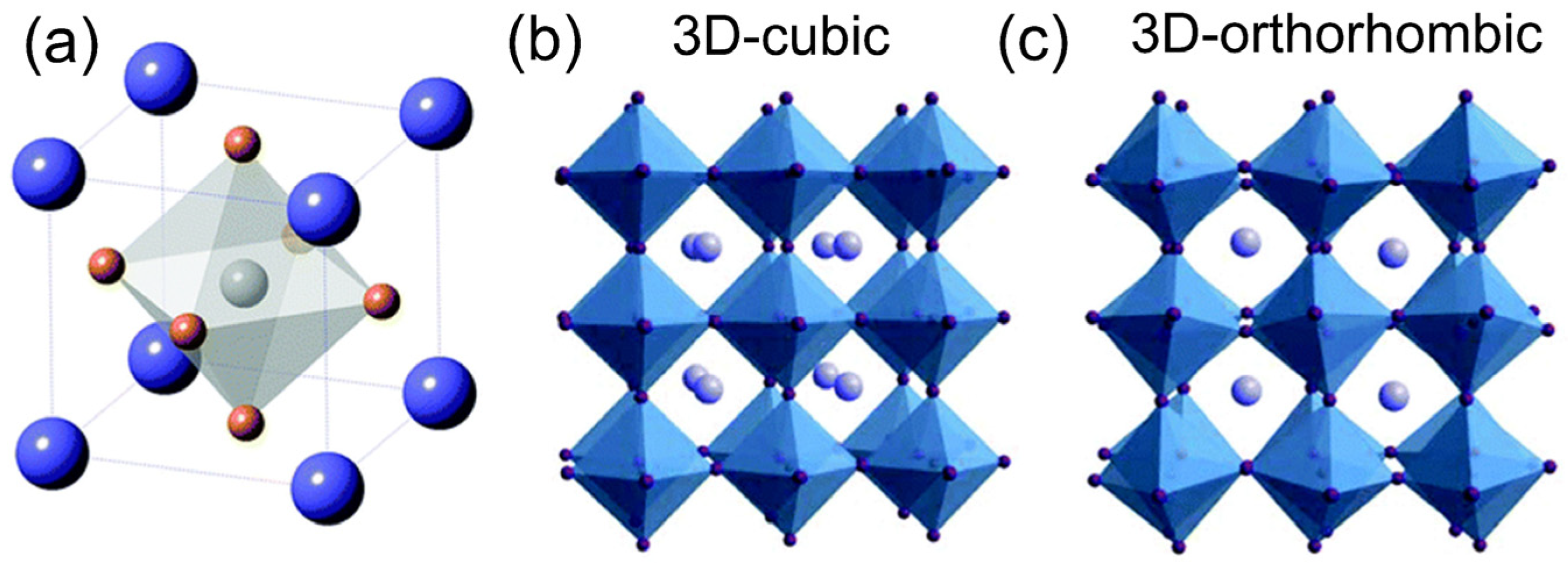
Figure 1a indicates the perovskite crystal structure, where the X anion occupies the center face of the cube and the A and M cations occupy the vertexes and the center of the cube, respectively.
Figure 1b depicts a regular octahedral structure that can be formed with this M cation (namely Pb
2+) and the six bounded X anions, which can indicate an optimal 3D cube-shaped perovskite structure in an octahedral PbX
6 arrangement. Nevertheless, when the octahedral PbX
6 arrangement deforms from the 3D cubic shape, the perovskite formation changes to a less symmetric orthorhombic shape (
Figure 1c).
Perovskites are categorized into all-inorganic and hybrid (or inorganic/organic) materials. CsPbX
3 belongs to the all-inorganic perovskite category, whereas MAPbX
3 and FAPbX
3 belong to the hybrid material category. Perovskite nanocrystals that are synthesized colloidally are typically categorized into QDs as 0D nanoparticles, nanowires (NWs), nanorods (NRs) as 1D nanoparticles, and nanoplatelets (NPLs) as a 2D nanocomposite. CsPbX
3 crystals are very colorful. For instance, CsPbI
3 crystals are black, CsPbBr
3 crystals are orange, and CsPbCl
3 crystals are pale yellow, while Cs
4PbX
6 (X = I, Cl, or Br) crystals have no color. In addition, CsPbX
3 shows photoconductivity properties and exhibits maximal spectrum sensitivity towards violet (CsPbCl
3), blue to green (CsPbBr
3), and red (CsPbI
3) [
86,
87]. Many researchers have reported that CsPbX
3 perovskite is a good light absorber (
Figure 2) due to its high absorption coefficient [
88,
89,
90,
91,
92]. Despite having excellent physical and chemical characteristics, perovskite materials have been used in optoelectronics for just a few decades. The capability to fabricate colloidal PQDs in solution allows the production of cost-efficient emitting materials that are also suitable for flexible devices [
93,
94,
95,
96].
3. Synthesis Method of QDs
Recently, the most common method used for the production of QDs has been hot injection (HI). To produce QDs by hot injection, a pre-prepared precursor solution is injected into another while it is surrounded by a protective gas at a specific temperature. Due to the rapid nucleation and growth dynamics, this synthesis method can be completed in just a few seconds, resulting in QDs with good monodispersity, controllable composition, and a high PLQY.
Figure 3a depicts the hot-injection synthesis process of CdSe QDs [
97]. The materials needed to synthesis CdSe QDs are oleic acid (OA), cadmium oxide (CdO), octadecene (ODE), selenium powder (Se), and trioctylphosphine (TOP). By dissolving the selenium powder in TOP and injecting the Se–TOP solution into a hot Cd-precursor that comprises CdO, ODE, and OA, CdSe QDs can be obtained, as shown in
Figure 3a. The inset of
Figure 3a indicates that the emission colors of the QD range from blue to yellow under UV radiation, depending on the QD growth time.
Using ligands to cap QDs is one of the most well-known and simple processes for achieving a high PLQY. It is notable that the PLQY and optical characteristics of QDs are strongly associated with the surface ligands. The ligands play an important role in regulating the surface defects and passivating the dangling bond on the exposed surface of QDs, which subsequently modulate the deep trap states [
98]. In this regard, Bansal et al. synthesized organic ligand-capped CdS QDs by the solvothermal synthesis method [
99]. Tri-n-octylphosphine oxide (TOPO) and octylphosphine (TPO) were used as organic ligands, and a standard solvent for the precursor. A coordinating solvent, such as TOPO, and a noncoordinating solvent, such as ODE, OA, diphenylphosphine (DPP), or TOP, were used for the synthesis of CdS QDs. DPP/TOP acted as a sulfur ligand [
98], and TOPO/OA acted as a cadmium ligand [
100]. It is also notable that a high PLQY was only attained when OA was coupled with DPP to decrease the defects on both Cd and S atoms. Similarly, a high PLQY was also realized when TOPO/OA (or TOPO) was coupled with DPP (or TOP).
QDs synthesis in an aqueous solution is another effortless process. Nevertheless, this kind of water-soluble CdSe QD exhibits wide absorption and PL spectra but a poor PLQY. Through the hydrothermal process, it is possible to improve such poor optical properties of CdSe QDs. To prepare the precursor of CdSe QDs for the hydrothermal process, Cd ions and Se ions were mixed together with N-acetyl-L-cysteine (NAC) as a ligand [
101]. The precursor solution was then treated to prepare the CdSe QD solution at a specific temperature using an oil bath for a particular period of time inside an autoclave and cooled in the ambient environment. CdSe/ZnS QDs were produced under a specific temperature of heating by constantly mixing the ZnS precursor solution into the CdSe QD solution.
Colloidal PQDs have also attracted huge attention in recent years due to their superior optoelectronic characteristics. They can be synthesized to achieve various goals such as a high PLQY, a cost-effective production, and a tunable bandgap. Colloidal PQDs have been synthesized by a variety of methods in recent decades [
102,
103,
104,
105,
106]. HI process can also be used to synthesize colloidal PQDs. In 2015, Protesescu et al. first developed thoroughly inorganic CsPbX
3 (X = Br, Cl, and I, or mixed halide) employing a cost-effective commercial precursor [
107].
Figure 3b indicates the schematic picture of the synthesis procedure of CsPbBr
3 PQDs using the HI method [
83]. The Cs precursor solution contained CsCO
3, ODE, and OA. The CsPbBr
3 QDs were obtained by injecting the Cs precursor solution into a heated mixture of PbBr
2, ODE, OA, and oleylamine (OAm).
The HI process requires a specific injection temperature, a protective inert gas, and a high reaction temperature. Therefore, it is essential to develop a simple and easy-to-use technique to synthesize PQDs. The ligand-assisted reprecipitation (LARP) and supersaturated recrystallization (SR) techniques at room temperature have been reported in recent years. Based on the polarity of the reaction solvent system, two categories can be distinguished: nonpolar solvent systems and polar solvent systems. The reprecipitation technique is an easy procedure that employs solvent mixing to simultaneously produce polymer dots and organic nanoparticles. The usage of capping ligands on the surface of the nanoparticles has received a lot of attention recently, and it is becoming a more significant approach for regulating their size and configuration [
108,
109]. Zhang et al. proposed a LARP procedure to synthesize color-tunable and brightly luminescent colloidal CH
3NH
3PbX
3 (X = Br, I, and Cl) QDs, which provided a PLQY of up to 70% at ambient atmosphere and low excitation fluencies [
110].
Figure 4a schematically illustrates the fabrication of PQDs using the LARP process. The precursor solution for the synthesis of CH
3NH
3PbBr
3 QDs was prepared by mixing PbBr
2, CH
3NH
3Br, n-octylamine, and OA into a standard solvent, DMF, which has an excellent capability for dissolving inorganic salts and small molecules. The precursor solution was then gradually added into a vigorously stirred poor solvent, toluene, to eventually produce a yellow-green colloidal solution, indicating the aggregation process of the precursors into nanoparticles. Subsequently, small-sized nanoparticles were obtained by centrifuging the colloidal solution at a high rpm to remove large particles. Other longer chains of alkylamines, such as hexylamine, hexadecylamine, and dodecylamine, are also capable of controlling crystallization and producing colloidal QD solutions. However, it was found that n-octylamine had the potentiality to adjust the QD size by regulating the crystallization kinetics, while the aggregation of QDs was suppressed by OA, which assured the stability of the colloidal solution. Li et al. developed a room-temperature SR method (RT-SR) to fabricate CsPbX
3 within a few seconds, without any inert gas or injection operation, as shown in
Figure 4b [
111]. A combination of ion sources containing CsX and PbX
2 (X = Br, I) was mixed in DMF or dimethyl sulfoxide (DMSO) with the surface ligands OAm and OA to prepare a precursor solution. Subsequently, a supersaturated state was achieved due to the fast recrystallization after adding the precursor solution into toluene under vigorous stirring. Finally, after the centrifugation of the resultant solution, a stable toluene dispersion of PQDs with ligand protection was obtained (
Figure 4b). Moreover, OAm and OA provided an advantage for tuning the crystal size and the ability to disperse them into various nonpolar solvents.
Another work has been reported by Song et al. that focused on the synthesis of inorganic CsPbBr
3 QDs using synergistic short ligands in toluene under room-temperature conditions without employing an inert gas as a protector [
112]. Three synergistic short ligands, namely octanoic acid (OTAc), didodecyldimethylammonium bromide (DDAB), and tetraoctylammonium bromide (TOAB) (
Figure 5a), provide excellent electrical transportation capabilities, a high ink stability and superior emissive properties, and a capability for dissolving PbBr
2, respectively.
Figure 5b describes the synthesis process of the PQDs.
In most situations, the nanocrystallization of lead halide PQDs takes place by dispersing lead halides into standard solvents and then injecting them into poor solvents. Nevertheless, the PQDs generated using this method generally have a nonuniform spherical shape compared to the inorganic cesium (Cs)–lead halide analogs, which have a uniform cubic arrangement [
113,
114,
115]. In addition, the PLQY of these PQDs is up to 80% in the solution form; however, this decreases to 40% in the thin-film form, due to the severe quenching effect [
114]. To address this issue, Dai et al. developed PQDs using the spray-synthesis method by integrating conventional PQD synthesis with the spray-pyrolysis technique [
116]. According to this work, both the solution and thin-film PQDs exhibited a uniform cubic structure with a stable PLQY of 100%. A standard solvent DMF was used to dissolve the organic halides, such as methylammonium bromide (CH
3NH
3Br, MABr), and lead bromide (PbBr
2) with the organic ligands OA and n-octylamine, and this was sprayed into a poor solvent (toluene), as shown in
Figure 6a. Unlike the conventional process, the micro-sized droplets in the spray enabled a significantly higher contact surface area and enhanced the mixing between the precursor solvent and the poor solvent, allowing for superior-quality PQD crystallization in the poor solvent. By centrifuging the resulting solution, the precipitate was eliminated and then put back into the hexane suspension to fabricate a spin-coated thin film.
Figure 6b indicates the PL emission spectra of spray-synthesized QDs (S) and conventional drop-synthesized QDs (D) in toluene, where both QDs showed an emission peak around 511 nm. Nevertheless, solution D exhibited a shoulder peak at 470 nm due to the smaller size of the quantum dots in the solution.
Figure 6c depicts the stability performance of the PLQY for solutions D and S, demonstrating that solution D was significantly less stable than solution S, with a high QY deterioration over 30 days. On the other hand, the PLQY of solution S was about 100%, but due to the presence of excess OA, it was burdensome to fabricate a uniform film. The casted film with excess OA experienced severe quenching, resulting in an exceptionally low PLQY of ~1–2%. In contrast, the film spin-coated with the precipitate of solution S in hexane revealed a very high PLQY of 100% and only 9% PLQY deterioration over 60 days. In conclusion, halide PQDs fabricated through spray synthesis exhibit excellent optical properties in terms of stability and emission when cast into thin films, suggesting that they are well-suited for photon conversion for optoelectronics devices.
The presence of Pb in lead halide PQDs results in toxicity issues, impeding their commercialization, even though they have a high PLQY. Researchers have reported various approaches for synthesizing Pb-free PQDs, such as substituting Pb
2+ with Sn
2+ [
117] or partially substituting the B-site cation with various metal ions [
118], which could maintain a standard PLQY and increase the thermal stability of α-CsPbI
3 and orthorhombic CsPbBr
3 at ambient environment. Thus, the concurrent exchange of the anion and cation in PQDs can improve the thermal stability as well as provide a high PLQY. Following this trend, Singh et al. developed a favorable approach by simultaneously exchanging the cation and anion with cobalt and chloride ions in the host CH
3NH
3Pb
1–xCo
xBr
3–2xCl
2x (x = 0 to 0.5) PQDs, followed by PMMA encapsulation for PQD stabilization [
119]. The results indicated no crucial impact on the crystal structure after the partial substitution of Pb
2+ and Br
− with Co
2+ and Cl
−, respectively. Moreover, after increasing the concentration of the Co
2+ and Cl
− ion source, a blue shift in the PL was observed. This could be associated with the lattice contraction, along with the local anion clustering mechanism. It was also observed that after the partial substitution with Co
2+ and Cl
−, the bandgap of the PQDs changed from 2.29 to 2.41 eV, which was attributed to the electronegativity difference between the Pb
2+ and Co
2+ cations, as well as the Br
− and Cl
− anions. The host PQDs showed a PLQY of 95% after the substitution of the Co
2+ and Cl
− ions.
The use of surface ligands such as oleylamine (OLA) and OA during the synthesis stage is important for passivating the PQD structure, controlling the size of the PQDs, and stabilizing the PQDs in colloidal form. Additionally, encapsulation approaches have been utilized to boost both the moisture and the UV resistance of PQDs. Rather than incorporating carboxylic acids (such as OA) and alkylamines (such as OLA) in the perovskite precursor, Li et al. fabricated perovskite quantum dot papers (PQDPs) by casting cellulose nanocrystals (CNCs) as long-chain binding ligands, which contain a large number of -HSO
3− and -O
− groups (
Figure 7) [
120]. These two anions behave as capping ligands that restrict the growth of perovskite crystals and enable the forming of well-dispersed PQDPs. Moreover, during the preparation of the precursor solution (DMF + MAX + PbX
2 + CNCs, MA = CH
3NH
3+) of the PQDs, the -HSO
3− and -O
− groups of the CNCs were coordinated to the empty orbital of the PbX
2, and the PQDs were entangled between the long chains of the CNCs to avoid the dangling capping behavior of the OA and OLA and to increase the stability. The PQDPs exhibited a very stable PL intensity under continuous UV radiation for 60 days.
Table 1 demonstrates a comparison between Cd-based QDs and PQDs in terms of the improvement in the PLQY in recent years.
4. Strategies of Stability Improvement for PQDs
Despite its excellent physical and chemical properties, the perovskite material still has a lower stability compared with Cd-based QDs [
122]. Furthermore, the low stability of hybrid organic–inorganic perovskite nanomaterials due to their organic component and their peripheral devices is a key issue for optoelectronic applications [
123,
124,
125]. On comparison with organic–inorganic perovskite hybrid analogs, all-inorganic perovskite QDs such as CsPbI
3, CsPbCl
3, and CsPbBr
3 have better thermal stability in the atmospheric environment [
126]. Among them, CsPbI
3 shows a comparatively confined bandgap (1.73 eV) at the cubic phase (α-CsPbI
3), as well as better chemical and compositional stability [
92,
127,
128]. However, a quick degradation occurs from its cubic phase (black phase) to a non-perovskite yellow phase (δ-CsPbI
3, bandgap 2.82 eV) at room temperature [
92,
113,
127,
128,
129,
130], which then requires a very high temperature (above 310 °C) to change into the black phase from the yellow phase [
127,
128,
130,
131,
132,
133,
134,
135]. In this scenario, it is also necessary to note that the CsPbI
3 black phase at room temperature is a cubic α-phase or orthorhombic γ-phase (bandgap 1.75 eV) [
136,
137,
138,
139]. Generally, both the α- and γ-phases have an optimal 3D perovskite structure in an octahedral PbX
6 arrangement, whereas the non-perovskite yellow phase (δ-CsPbI
3) has lost the 3D configuration [
140]. Furthermore, both the α- and γ-phases show similar optoelectronics characteristics [
137,
139].
Thus, it is significantly difficult to employ CsPbI
3 in a solar cell and a stable black phase at ambient temperature [
141,
142]. CsPbBr
3 could be a substitute to overcome the black phase instability, but its large bandgap (2.25 eV) restricts photon harvesting, which then reduces the PCE of solar cells [
143,
144]. Therefore, many attempts have been reported to improve the phase stability of CsPbI
3 in recent years. The most-often utilized processes are (1) 2D nanocrystal engineering, (2) doping with an alloying compound, and (3) solvent-additive techniques.
4.1. 2D Nanocrystal Engineering
CsPbI
3 nanocrystals can improve the stability of CsPbI
3 perovskite due to their immense surface energy and strong microstrain [
107]. Nevertheless, the phase diagrams of size-dependent nanocrystals state that the size of the nanocrystals and the cubic phase stability of CsPbI
3 are inversely correlated. Hence, α-CsPbI
3 exhibits better stability as size of the nanocrystals decreases. Based on this hypothesis, Protesescu et al. demonstrated that cubic CsPbI
3 4–15 nm in size provided comparatively better stability when retained at room temperature for 30 days [
107], while cubic CsPbI
3 100–200 nm in size strongly degraded into the yellow phase. Dutta et al. [
145] reported that the phase stability of CsPbI
3 also depends on the reaction temperature. This study then mentioned that colloidal and thermally stable CsPbI
3 NCs can be produced via a reaction with high temperatures. The high temperature (~160 °C) allowed the alkylammonium ions to passivate the surface briskly and hinder the phase deterioration of the NCs. The attained NCs provided higher stability in the film both under an ambient environment and upon prolonged annealing at high temperatures.
Recently, Mir et al. [
140] reported Mn-doped CsPbI
3 NCs which showed better black phase stability for nearly a month compared to non-doped nanocrystals. The results revealed that the thermal and colloidal stability of the CsPbI
3 black phase was improved due to the utilization of the postsynthesis Mn-doping procedure in an ambient environment via surface passivation. It could also be noticed that after the addition of the Mn dopant under ambient conditions into the CsPbI
3 NCs, no structural changes occurred from the black phase (in both the α-CsPbI
3 and γ-CsPbI
3), and the transformation into the non-perovskite yellow phase (δ-CsPbI
3) was prevented. Furthermore, these findings highlight the importance of addressing the fundamental chemistry of such NCs, which might assist scientists in improving the stability of nanocrystal-based inorganic perovskite solar cells.
4.2. Doping with Alloying Compound
In order to increase the value of the Goldschmidt tolerance factor (t), the stabilization process of the CsPbI
3 black phase can be carried out at ambient atmosphere by doping or partially alloying its B-site with metallic cations, which have a smaller radius [
146]. As reported by previous works, the typical metallic cations used are Eu
3+, Bi
3+, Sb
3+, Sn
3+, Ge
3+, and Mn
3+ [
147]. Hu et al. first proposed B-site doping using Bi
3+ in CsPbI
3 [
129]. The 4 mol% Bi
3+-integrated α-CsPb
0.96Bi
0.04I was produced by employing a one-step deposition technique and substituting Pb
2+ (radius = 1.19 Å) with a Bi
3+ cation (radius = 1.03 Å), resulting in a t-value increase from 0.81 to 0.84. The active layer of the solar cell with α-CsPb
0.96Bi
0.04I showed a high PCE of 13.21% and maintained 68% of the initial PCE for 168 h under atmospheric conditions. Moreover, it can be observed from
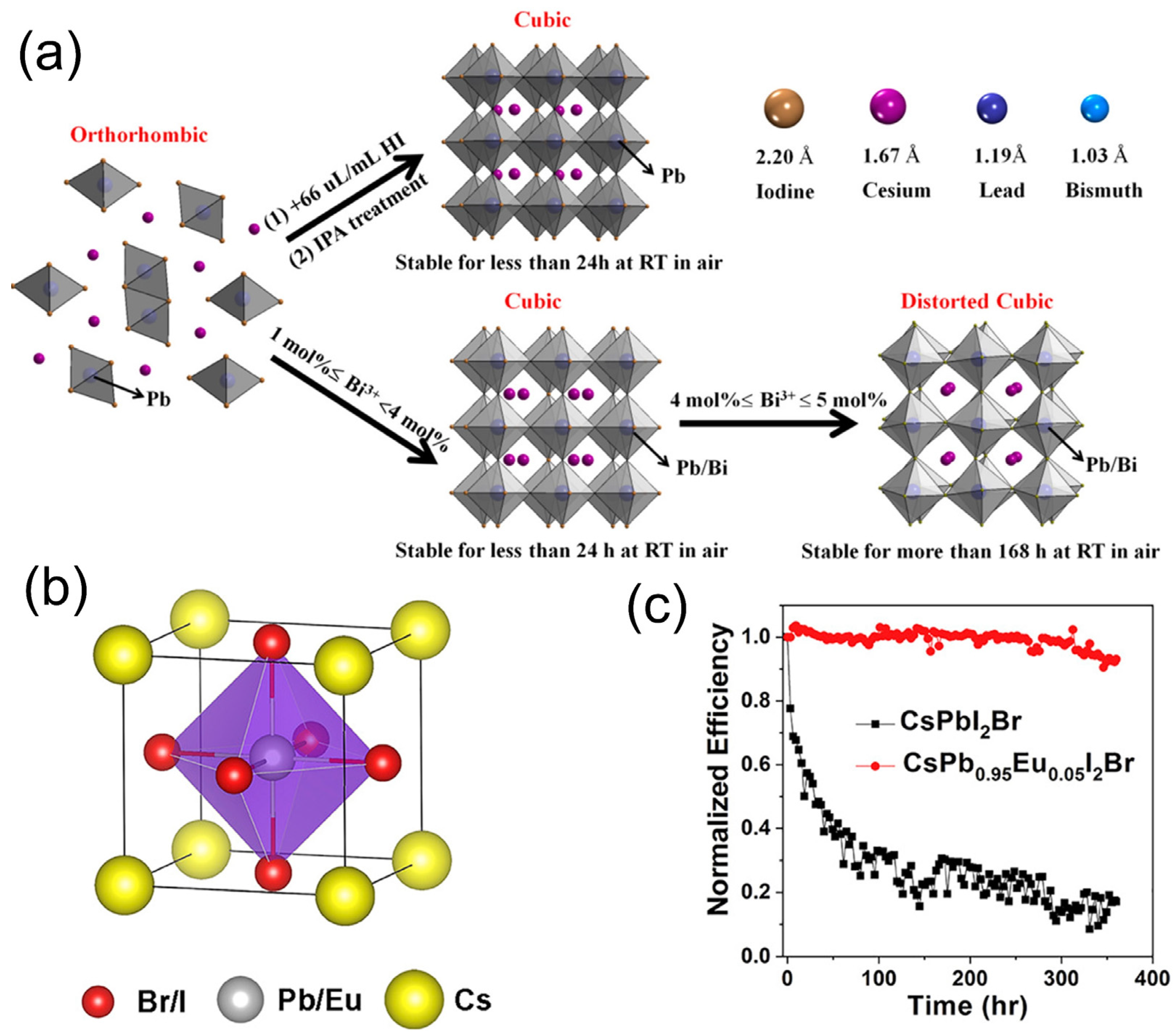
Figure 8a that the stabilization techniques for the α-CsPbI3 phase are identical, whether hydroiodic acid (HI) or Bi
3+ is utilized. The combination of Br and CsPbI
3 to create CsPbI
2Br has been shown to have high ambient stability and an appropriate bandgap (1.92 eV) for tandem solar cells [
148]. However, in order to improve the stability further, Xiang et al. [
149] recently proposed a CsPbI
2Br perovskite solar cell doped with Eu
3+ dopant (
Figure 8b). This work shows a stabilized black phase and a significant improvement in the cell performance. In fact, after 370 h of uninterrupted white-light illumination at 100 mW/cm
2, the device exhibited 93% of the initial PCE, as shown in
Figure 8c.
In 2017, another work developed by Akkerman et al. [
150] improved the cubic phase stability of CsPbI
3. In this work, an Mn
2+ dopant combined with CsPbI
3 was used to enhance the phase stability of the CsPbI
3 film as well as its NCs. Nevertheless, it was also emphasized that due to the comparatively smaller radius of the Mn
2+ cation (0.70 Å) with respect to Pb
2+ (1.19 Å), only a small amount of Mn
2+ could be used for the CsPbI
3 NC phase separation. Due to the relatively close radii of Sn
2+ (1.18 Å) and Pb
2+ (1.19 Å), Sn
2+ can be utilized to modify the t-value of CsPbI
3. Nonetheless, due to the fast oxidation of Sn
2+ in an atmospheric environment, it is necessary to incorporate it with another compound. Following this concept, Liang et al. [
151] employed Br
− to synthesize a CsPb
1−xSnI
3−xBr
x mixed-halide perovskite, which provided superior phase stabilization.
Recently, in 2020, Zhao et al. [
138] reported that tortuous 3D γ-CsPbI
3 films can be easily prepared under a low temperature (60 °C) without any additive and show similar optical properties to α-CsPbI
3. However, this metastable tortuous 3D γ-CsPbI
3 has a high tendency to convert into the non-perovskite yellow phase at ambient temperature. Ca
2+-doped γ-CsPbI
3 (CsPb
1−xCa
xI
3, x = 0–2%) has been found to provide a higher stability at room temperature.
Figure 9 reveals that the formation of γ-CsPbI
3 at 60 °C provides more stability than the formation of α-CsPbI
3 (>320 °C), due to its lower cohesive energy. On the other hand, the formation of tortuous 3D γ-CsPbI
3 doped with Ca
2+ has better thermal stability than non-doped γ-CsPbI
3, due to its lower cohesive energy and appropriate Goldschmidt tolerance factor (t).
Considering everything that has been said so far, it can be concluded that doping or alloying CsPbI3 with a small cation is a useful strategy to enhance the phase stability of the CsPbI3 layer. Nonetheless, further investigation is still required. Co-doping approaches might be an attractive route to pursue in further research.
4.3. Solvent-Additive Technique
In 2015, Eperon et al. first reported that the phase transformation of a CsPbI
3 perovskite can occur at ambient atmosphere [
127]. In fact, to form a black phase, it was necessary to rise the temperature above 335 °C. However, it could alter to the orthorhombic yellow phase when brought to an atmospheric environment. Nevertheless, a stabilized black phase with a 2.9% PCE could be produced at room temperature with the addition of a small amount of HI into the precursor solution (CsI and PbI at a ratio of 1:1 mixed in DMF) of CsPbI
3. The purpose of adding HI was to produce microstrain in the crystal lattice, which caused the α-CsPbI
3 to stabilize at room temperature. The entire process was performed in an inert environment at a temperature of about 100 °C, which was comparatively much lower than the initial temperature (above 310 °C) required to generate the black phase.
In 2016, Luo et al. [
128] demonstrated a sequential solvent process using HI and isopropyl alcohol (IPA) to fabricate a stable CsPbI
3 at room temperature with a humidity of less than 30%. According to this work, the first 66 µL/mL of HI was mixed into the Cs
4PbI
6 precursor solution at a low temperature for optimization. After being mixed with HI, the film exhibited a yellow-brown color. Second, the film was annealed for 5 min at 100 °C after adding a hot IPA solution in atmospheric conditions. Eventually, the film converted to a dark brown α-CsPbI
3 and provided a stability of up to 72 h, as shown in
Figure 10.
Recently, Zhao et al. proposed that the addition of a minute volume of H
2O into the precursor solution of γ-CsPbI3 could provide thermodynamic stability to the γ-CsPbI
3 films via a proton transfer reaction [
139]. The theoretical and experimental results revealed that at surface areas greater than 8600 m
2/mol, the γ-CsPbI
3 film with lower surface free energy had better thermal stability compared to δ-CsPbI
3. To increase the black-phase stability at ambient atmosphere, treatment with large-radius cations has been a found to be an alternative. For example, Li et al. [
152] proposed that the addition of a large-radius phenylethylammonium (PEA
+) cation into the CsPbI
3 perovskite profoundly enhanced the phase stability of the film. In fact, the resulting Cs
xPEA
1−xPbI
3 perovskite exhibited a PCE of 5.7% and α-phase stability at ambient temperature up to 250 °C in atmospheric conditions. Furthermore, Zhang et al. demonstrated that the incorporation of a small quantity of ethylenediamine cations (EDA
2+) in the α-CsPbI
3 perovskite showed superior black phase stability for a month at room temperature and more than 150 h at 100 °C, as well as a reproducible efficiency of 11.8% [
153]. In 2016, Liao et al. introduced a 2D cesium lead iodide perovskite BA
2CsPb
2I
7 (where BA denotes CH
3(CH
2)
3NH
3) with a combination of 3D CsPbI
3 and a bulky ammonium cation. The synthesized BA
2CsPb
2I
7 perovskite showed high stability under heating at 85 °C for 3 days and a relative humidity of 30% for one month [
154]. Afterward, Wang et al. [
155] used a single-step film deposition process to stabilize the α -CsPbI
3 film in an ambient environment to incorporate a small amount of sulfobetaine zwitterions (~1.5 wt.%). By interacting with ions and colloids in the CsPbI
3 precursor solution, the zwitterion molecules impeded the crystallization of the CsPbI
3 perovskite. Under one-sun illumination, solar cells containing these zwitterion-stabilized perovskite films had a stable efficiency of 11.4%.
Figure 11 describes the stabilization mechanism of the CsPbI
3 α-phase using zwitterions.
The research reported thus far shows that incorporating additives into the perovskite manufacturing process not only improves the crystallization, but also stabilizes the material in its 3D crystal phase.


 ,
, 























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}