
Promotion of Ru or Ni on Alumina Catalysts with a Basic Metal for CO2 Hydrogenation: Effect of the Type of Metal (Na, K, Ba)

 ,
,  ,
, 
 and
and 
Abstract
:
1. Introduction
2. Experimental
2.1. Catalyst Preparation
2.2. Catalytic Testing
2.3. Catalyst Characterization
3. Results and Discussion
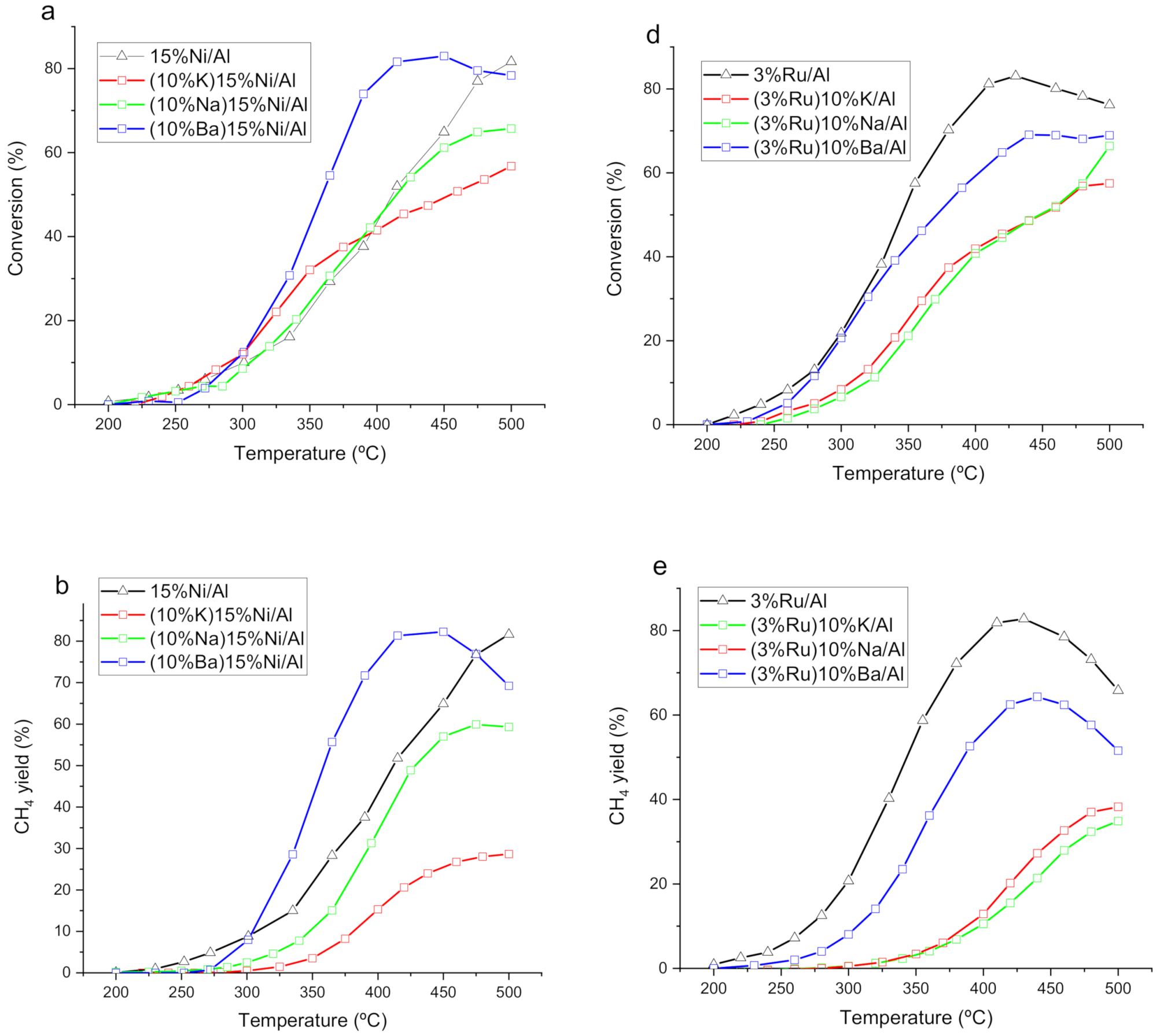
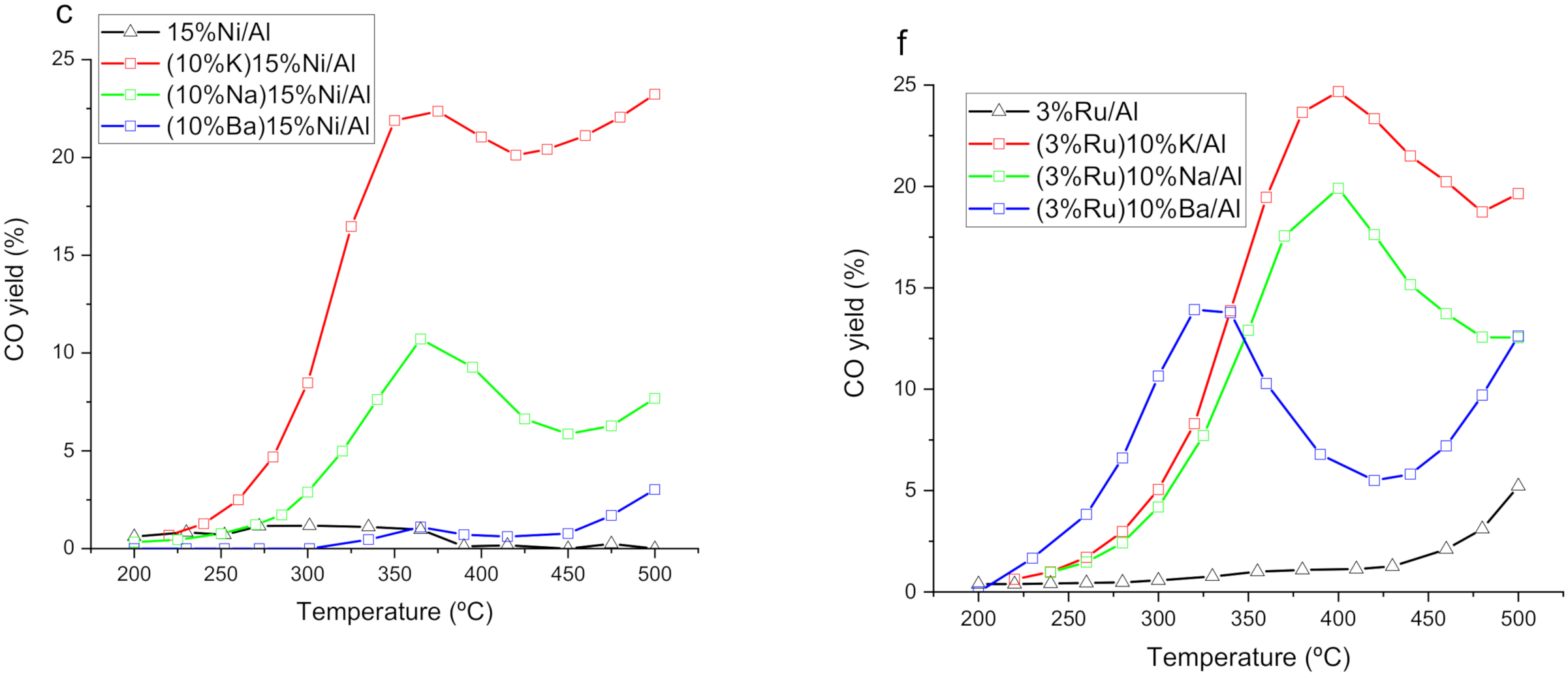
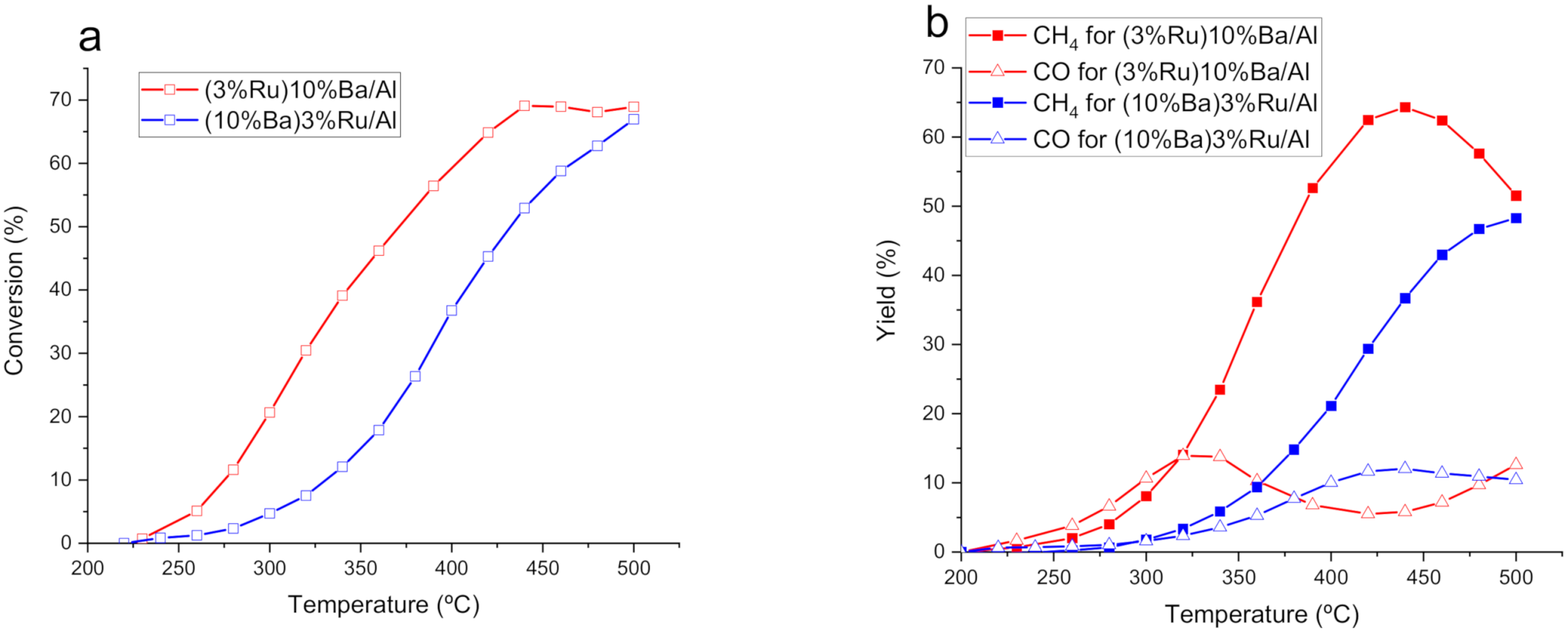
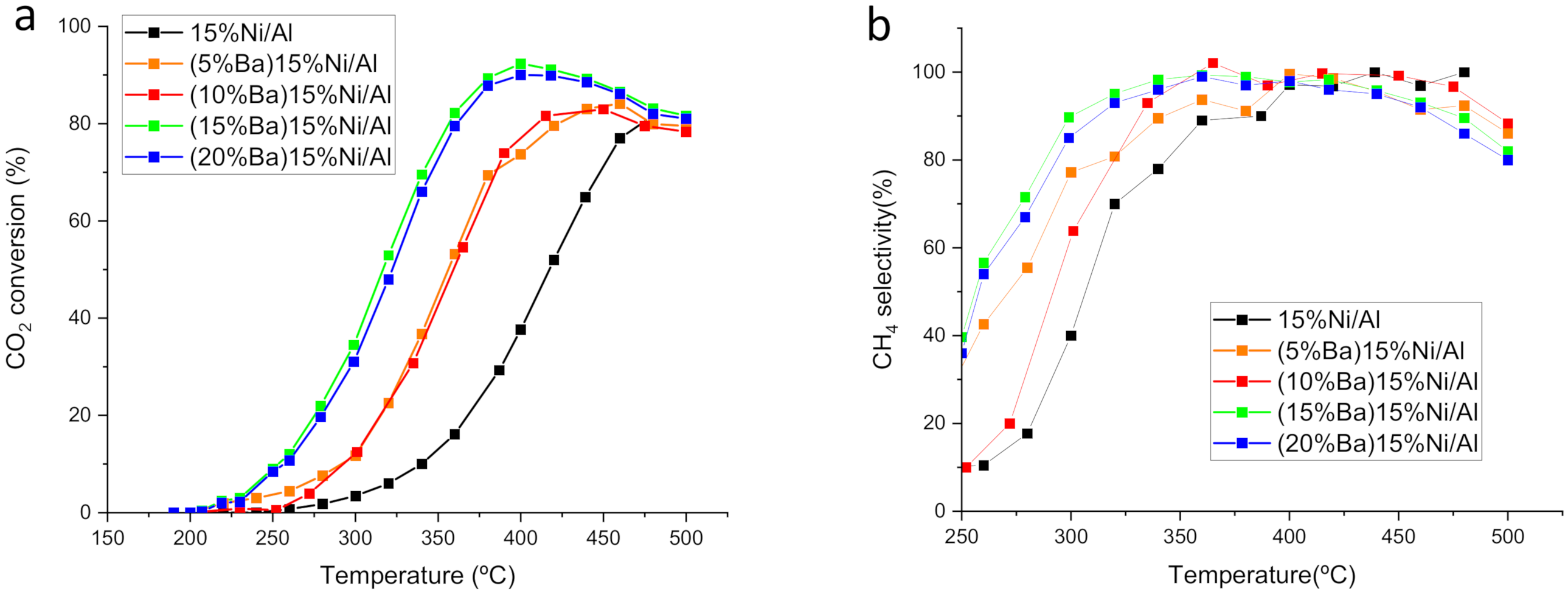
3.1. Catalytic Performance at Steady State
3.2. Basicity Assessed by Temperature-Programed Desorption of CO2 (CO2-TPD)
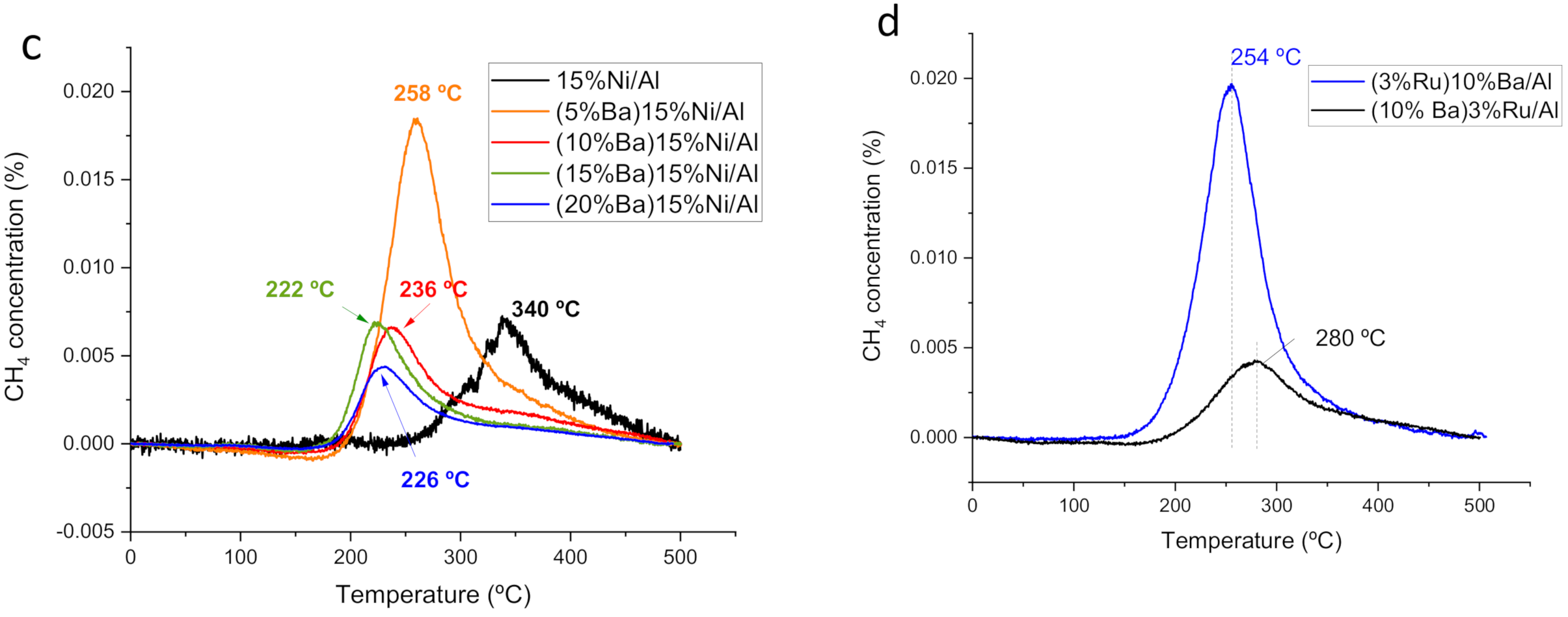
3.3. Temperature-Programed Surface Reaction of Adsorbed CO2 (TPSR)
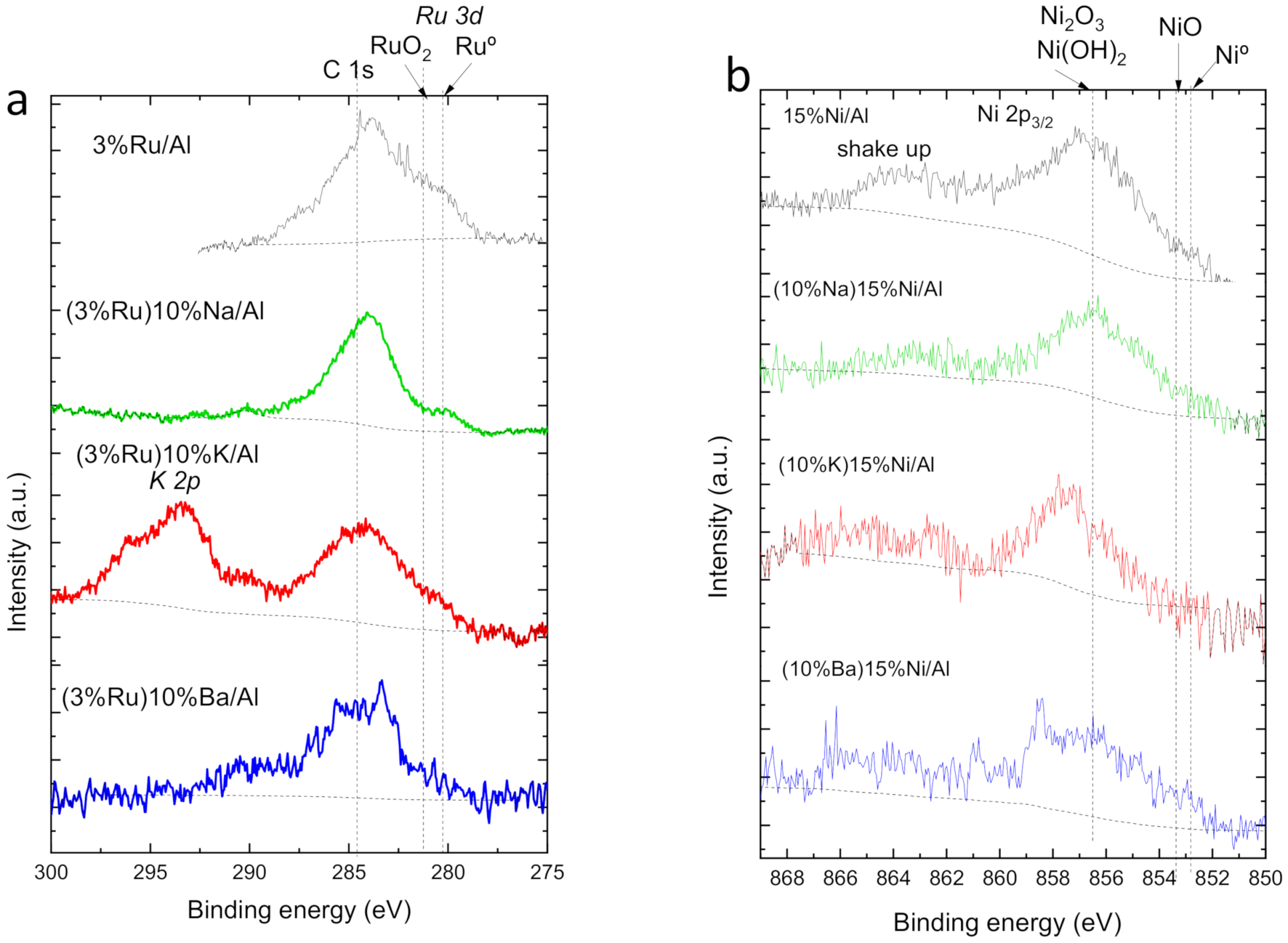
3.4. Catalysts Characterization by Instrumental Techniques
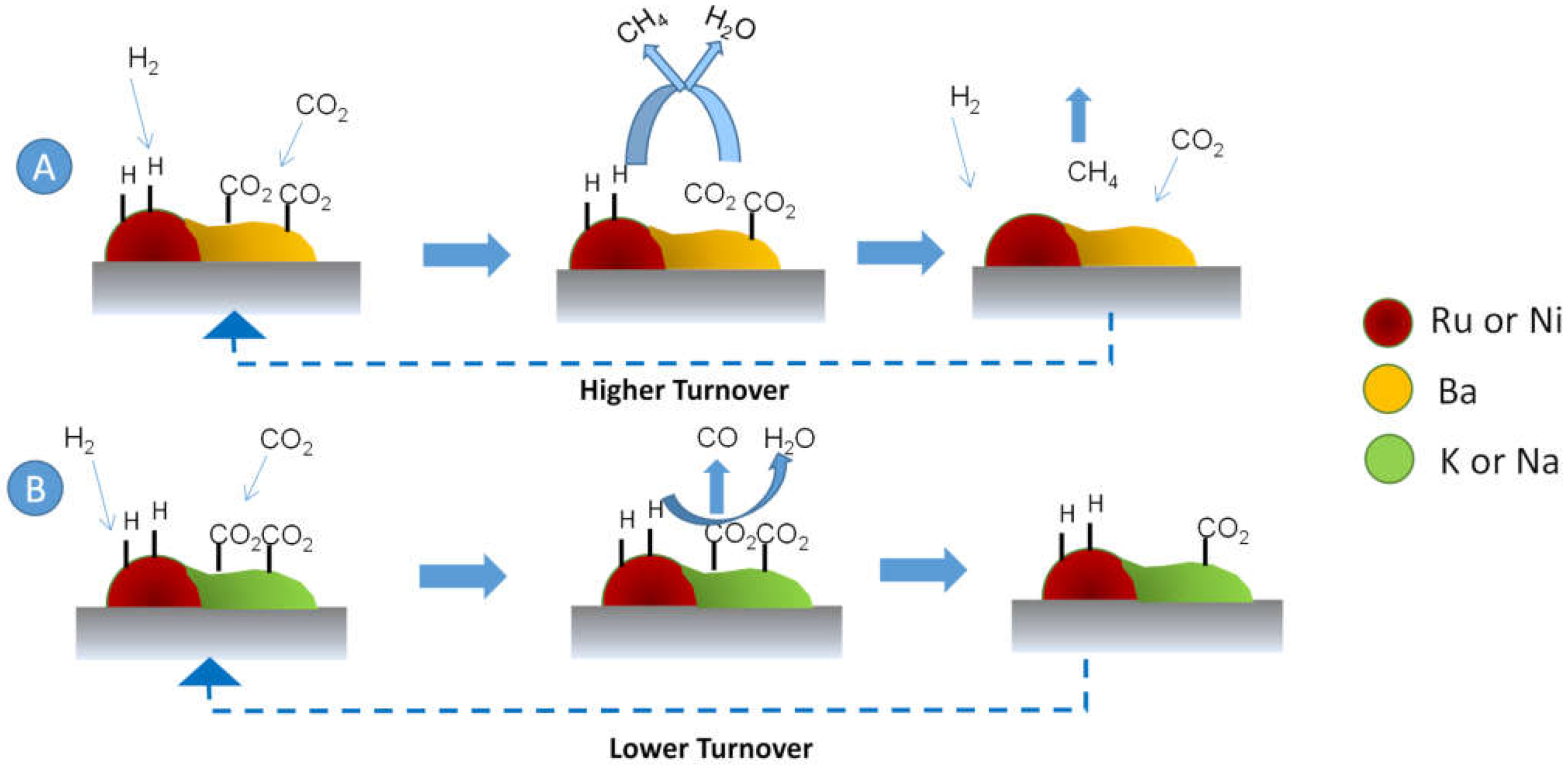
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Friedlingstein, P.; Andrew, R.M.; Rogelj, J.; Peters, G.P.; Canadell, J.G.; Knutti, R.; Luderer, G.; Raupach, M.R.; Schaeffer, M.; van Vuuren, D.P.; et al. Persistent growth of CO2 emissions and implications for reaching climate targets. Nat. Geosci. 2014, 7, 709–715. [Google Scholar] [CrossRef]
- 2030 Climate & Energy Framework. Available online: https://ec.europa.eu/clima/policies/strategies/2030_en (accessed on 1 December 2021).
- Kwak, J.H.; Kovarik, L.; Szanyi, J. Heterogeneous Catalysis on Atomically Dispersed Supported Metals: CO2 Reduction on Multifunctional Pd Catalysts. ACS Catal. 2013, 3, 2094–2100. [Google Scholar] [CrossRef]
- Burger, T.; Koschany, F.; Wenng, A.; Thomys, O.; Köhler, K.; Hinrichsen, O. Simultaneous activity and stability increase of co-precipitated Ni–Al CO2 methanation catalysts by synergistic effects of Fe and Mn promoters. Catal. Sci. Technol. 2018, 8, 5920–5932. [Google Scholar] [CrossRef]
- Pan, Q.; Peng, J.; Sun, T.; Wang, S.; Wang, S. Insight into the reaction route of CO2 methanation: Promotion effect of medium basic sites. Catal. Commun. 2014, 45, 74–78. [Google Scholar] [CrossRef]
- Wang, X.; Shen, M.; Song, L.; Su, Y.; Wang, J. Surface basicity on bulk modified phosphorus alumina through different synthesis methods. Phys. Chem. Chem. Phys. 2011, 13, 15589–15596. [Google Scholar] [CrossRef]
- Bustinza, A.; Frías, M.; Liu, Y.; García-Bordejé, E. Mono- and bimetallic metal catalysts based on Ni and Ru supported on alumina-coated monoliths for CO2 methanation. Catal. Sci. Technol. 2020, 10, 4061–4071. [Google Scholar] [CrossRef]
- Ilsemann, J.; Murshed, M.M.; Gesing, T.M.; Kopyscinski, J.; Bäumer, M. On the support dependency of the CO2 methanation–decoupling size and support effects. Catal. Sci. Technol. 2021, 11, 4098–4114. [Google Scholar] [CrossRef]
- Tada, S.; Shimizu, T.; Kameyama, H.; Haneda, T.; Kikuchi, R. Ni/CeO2 catalysts with high CO2 methanation activity and high CH4 selectivity at low temperatures. Int. J. Hudorgen Energy 2012, 37, 5527–5531. [Google Scholar] [CrossRef]
- Montanari, T.; Castoldi, L.; Lietti, L.; Busca, G. Basic catalysis and catalysis assisted by basicity: FT-IR and TPD characterization of potassium-doped alumina. Appl. Catal. Gen. 2011, 400, 61–69. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Vernoux, P.; Goula, G.; Caravaca, A. Electropositive Promotion by Alkalis or Alkaline Earths of Pt-Group Metals in Emissions Control Catalysis: A Status Report. Catalysts 2019, 9, 157. [Google Scholar] [CrossRef] [Green Version]
- Campbell, T.K.; Falconer, J.L. Carbon dioxide hydrogenation on potassium-promoted nickel catalysts. Appl. Catal. 1989, 50, 189–197. [Google Scholar] [CrossRef]
- Tsounis, C.; Wang, Y.; Arandiyan, H.; Wong, R.J.; Toe, C.Y.; Amal, R.; Scott, J. Tuning the Selectivity of LaNiO3 Perovskites for CO2 Hydrogenation through Potassium Substitution. Catalysts 2020, 10, 409. [Google Scholar] [CrossRef] [Green Version]
- Porta, A.; Matarrese, R.; Visconti, C.G.; Castoldi, L.; Lietti, L. Storage Material Effects on the Performance of Ru-Based CO2 Capture and Methanation Dual Functioning Materials. Ind. Eng. Chem. Res. 2021, 60, 6706–6718. [Google Scholar] [CrossRef]
- Beierlein, D.; Häussermann, D.; Pfeifer, M.; Schwarz, T.; Stöwe, K.; Traa, Y.; Klemm, E. Is the CO2 methanation on highly loaded Ni-Al2O3 catalysts really structure-sensitive? Appl. Catal. B Environ. 2019, 247, 200–219. [Google Scholar] [CrossRef]
- Zhang, Z.; Hu, X.; Wang, Y.; Hu, S.; Xiang, J.; Li, C.; Chen, G.; Liu, Q.; Wei, T.; Dong, D. Regulation the reaction intermediates in methanation reactions via modification of nickel catalysts with strong base. Fuel 2019, 237, 566–579. [Google Scholar] [CrossRef]
- Büchel, R.; Baiker, A.; Pratsinis, S.E. Effect of Ba and K addition and controlled spatial deposition of Rh in Rh/Al2O3 catalysts for CO2 hydrogenation. Appl. Catal. Gen. 2014, 477, 93–101. [Google Scholar] [CrossRef]
- He, L.; Lin, Q.; Liu, Y.; Huang, Y. Unique catalysis of Ni-Al hydrotalcite derived catalyst in CO2 methanation: Cooperative effect between Ni nanoparticles and a basic support. J. Energy Chem. 2014, 23, 587–592. [Google Scholar] [CrossRef]
- Vogt, C.; Wijten, J.; Madeira, C.L.; Kerkenaar, O.; Xu, K.; Holzinger, R.; Monai, M.; Weckhuysen, B.M. Alkali Promotion in the Formation of CH4 from CO2 and Renewably Produced H2 over Supported Ni Catalysts. ChemCatChem 2020, 12, 2792–2800. [Google Scholar] [CrossRef]
- Li, X.; Wang, Y.; Zhang, G.; Sun, W.; Bai, Y.; Zheng, L.; Han, X.; Wu, L. Influence of Mg-promoted Ni-based Catalyst Supported on Coconut Shell Carbon for CO2 Methanation. ChemistrySelect 2019, 4, 838–845. [Google Scholar] [CrossRef]
- WANG, X.; YANG, M.; ZHU, L.; ZHU, X.; WANG, S. CO2 methanation over Ni/Mg@MCM-41 prepared by in-situ synthesis method. J. Fuel Chem. Technol. 2020, 48, 456–465. [Google Scholar] [CrossRef]
- Bacariza, M.C.; Graça, I.; Bebiano, S.S.; Lopes, J.M.; Henriques, C. Magnesium as Promoter of CO2 Methanation on Ni-Based USY Zeolites. Energy Fuels 2017, 31, 9776–9789. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Xu, L.; Chen, M.; Lv, C.; Cui, Y.; Wen, X.; Wu, C.; Yang, B.; Miao, Z.; Hu, X.; et al. Facilely fabricating highly dispersed Ni-based catalysts supported on mesoporous MFI nanosponge for CO2 methanation. Microporous Mesoporous Mater. 2020, 302, 110250. [Google Scholar] [CrossRef]
- Petala, A.; Panagiotopoulou, P. Methanation of CO2 over alkali-promoted Ru/TiO2 catalysts: I. Effect of alkali additives on catalytic activity and selectivity. Appl. Catal. B Environ. 2018, 224, 919–927. [Google Scholar] [CrossRef]
- Xu, L.; Wang, F.; Chen, M.; Yang, H.; Nie, D.; Qi, L.; Lian, X. Alkaline-promoted Ni based ordered mesoporous catalysts with enhanced low-temperature catalytic activity toward CO2 methanation. RSC Adv. 2017, 7, 18199–18210. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Lu, G. The effect of impregnation strategy on structural characters and CO2 methanation properties over MgO modified Ni/SiO2 catalysts. Catal. Commun. 2014, 54, 55–60. [Google Scholar] [CrossRef]
- Takano, H.; Shinomiya, H.; Izumiya, K.; Kumagai, N.; Habazaki, H.; Hashimoto, K. CO2 methanation of Ni catalysts supported on tetragonal ZrO2 doped with Ca2+ and Ni2+ ions. Int. J. Hydrog. Energy 2015, 40, 8347–8355. [Google Scholar] [CrossRef]
- Tan, J.; Wang, J.; Zhang, Z.; Ma, Z.; Wang, L.; Liu, Y. Highly dispersed and stable Ni nanoparticles confined by MgO on ZrO2 for CO2 methanation. Appl. Surf. Sci. 2019, 481, 1538–1548. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, S.; Zhao, G.; Yang, H.; Yuan, M.; An, X.; Zhou, H.; Qiao, Y.; Tian, Y. CO2 methanation over ordered mesoporous NiRu-doped CaO-Al2O3 nanocomposites with enhanced catalytic performance. Int. J. Hydrog. Energy 2018, 43, 239–250. [Google Scholar] [CrossRef]
- Xu, L.; Yang, H.; Chen, M.; Wang, F.; Nie, D.; Qi, L.; Lian, X.; Chen, H.; Wu, M. CO2 methanation over Ca doped ordered mesoporous Ni-Al composite oxide catalysts: The promoting effect of basic modifier. J. CO2 Util. 2017, 21, 200–210. [Google Scholar] [CrossRef]
- Everett, O.E.; Zonetti, P.C.; Alves, O.C.; de Avillez, R.R.; Appel, L.G. The role of oxygen vacancies in the CO2 methanation employing Ni/ZrO2 doped with Ca. Int. J. Hydrog. Energy 2020, 45, 6352–6359. [Google Scholar] [CrossRef]
- Do, J.Y.; Park, N.-K.; Seo, M.W.; Lee, D.; Ryu, H.-J.; Kang, M. Effective thermocatalytic carbon dioxide methanation on Ca-inserted NiTiO3 perovskite. Fuel 2020, 271, 117624. [Google Scholar] [CrossRef]
- Tsiotsias, A.I.; Charisiou, N.D.; Yentekakis, I.V.; Goula, M.A. The Role of Alkali and Alkaline Earth Metals in the CO2 Methanation Reaction and the Combined Capture and Methanation of CO2. Catalysts 2020, 10, 812. [Google Scholar] [CrossRef]
- Cimino, S.; Boccia, F.; Lisi, L. Effect of alkali promoters (Li, Na, K) on the performance of Ru/Al2O3 catalysts for CO2 capture and hydrogenation to methane. J. CO2 Util. 2020, 37, 195–203. [Google Scholar] [CrossRef]
- Liang, C.; Ye, Z.; Dong, D.; Zhang, S.; Liu, Q.; Chen, G.; Li, C.; Wang, Y.; Hu, X. Methanation of CO2: Impacts of modifying nickel catalysts with variable-valence additives on reaction mechanism. Fuel 2019, 254, 115654. [Google Scholar] [CrossRef]
- Liang, C.; Hu, X.; Wei, T.; Jia, P.; Zhang, Z.; Dong, D.; Zhang, S.; Liu, Q.; Hu, G. Methanation of CO2 over Ni/Al2O3 modified with alkaline earth metals: Impacts of oxygen vacancies on catalytic activity. Int. J. Hydrog. Energy 2019, 44, 8197–8213. [Google Scholar] [CrossRef]
- Guo, M.; Lu, G. The difference of roles of alkaline-earth metal oxides on silica-supported nickel catalysts for CO2 methanation. RSC Adv. 2014, 4, 58171–58177. [Google Scholar] [CrossRef]
- Liu, K.; Xu, X.; Xu, J.; Fang, X.; Liu, L.; Wang, X. The distributions of alkaline earth metal oxides and their promotional effects on Ni/CeO2 for CO2 methanation. J. CO2 Util. 2020, 38, 113–124. [Google Scholar] [CrossRef]
- Roldán, L.; Marco, Y.; García-Bordejé, E. Origin of the Excellent Performance of Ru on Nitrogen-Doped Carbon Nanofibers for CO2 Hydrogenation to CH4. ChemSusChem 2017, 10, 1139–1144. [Google Scholar] [CrossRef] [Green Version]
- Levenspiel Chemical Reaction Engineering; John Wiley and Sons: New York, NY, USA, 1962; p. 578.
- Wang, X.; Hong, Y.; Shi, H.; Szanyi, J. Kinetic modeling and transient DRIFTS–MS studies of CO2 methanation over Ru/Al2O3 catalysts. Catal. CO2 Convers. Process. Fuels Small Mol. 2016, 343, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Bermejo-López, A.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R. Ni loading effects on dual function materials for capture and in-situ conversion of CO2 to CH4 using CaO or Na2CO3. J. CO2 Util. 2019, 34, 576–587. [Google Scholar] [CrossRef]
- Wang, Y.; Winter, L.R.; Chen, J.G.; Yan, B. CO2 hydrogenation over heterogeneous catalysts at atmospheric pressure: From electronic properties to product selectivity. Green Chem. 2021, 23, 249–267. [Google Scholar] [CrossRef]
- Mutschler, R.; Moioli, E.; Luo, W.; Gallandat, N.; Züttel, A. CO2 hydrogenation reaction over pristine Fe, Co, Ni, Cu and Al2O3 supported Ru: Comparison and determination of the activation energies. J. Catal. 2018, 366, 139–149. [Google Scholar] [CrossRef]
- Weatherbee, G.D.; Bartholomew, C.H. Hydrogenation of CO2 on group VIII metals: IV. Specific activities and selectivities of silica-supported Co, Fe, and Ru. J. Catal. 1984, 87, 352–362. [Google Scholar] [CrossRef]
- Falbo, L.; Martinelli, M.; Visconti, C.G.; Lietti, L.; Bassano, C.; Deiana, P. Kinetics of CO2 methanation on a Ru-based catalyst at process conditions relevant for Power-to-Gas applications. Appl. Catal. B Environ. 2018, 225, 354–363. [Google Scholar] [CrossRef]
- Pan, Y.; Liu, C.; Ge, Q. Adsorption and Protonation of CO2 on Partially Hydroxylated γ-Al2O3 Surfaces: A Density Functional Theory Study. Langmuir 2008, 24, 12410–12419. [Google Scholar] [CrossRef] [PubMed]
- Proaño, L.; Tello, E.; Arellano-Trevino, M.A.; Wang, S.; Farrauto, R.J.; Cobo, M. In-situ DRIFTS study of two-step CO2 capture and catalytic methanation over Ru,“Na2O”/Al2O3 Dual Functional Material. Appl. Surf. Sci. 2019, 479, 25–30. [Google Scholar] [CrossRef]
- Gruene, P.; Belova, A.G.; Yegulalp, T.M.; Farrauto, R.J.; Castaldi, M.J. Dispersed Calcium Oxide as a Reversible and Efficient CO2−Sorbent at Intermediate Temperatures. Ind. Eng. Chem. Res. 2011, 50, 4042–4049. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y.; Zhang, J.; Liu, R.; Yang, J.; Yang, B.; Xu, H.; Ma, Y. High pressure X-ray diffraction study of sodium oxide (Na2O): Observations of amorphization and equation of state measurements to 15.9 GPa. J. Alloys Compd. 2020, 823, 153793. [Google Scholar] [CrossRef]
- Hartmann, P.; Bender, C.L.; Sann, J.; Dürr, A.K.; Jansen, M.; Janek, J.; Adelhelm, P. A comprehensive study on the cell chemistry of the sodium superoxide (NaO2) battery. Phys. Chem. Chem. Phys. 2013, 15, 11661–11672. [Google Scholar] [CrossRef]
- Zhang, S.; Gai, S.; He, F.; Dai, Y.; Gao, P.; Li, L.; Chen, Y.; Yang, P. Uniform Ni/SiO2@Au magnetic hollow microspheres: Rational design and excellent catalytic performance in 4-nitrophenol reduction. Nanoscale 2014, 6, 7025–7032. [Google Scholar] [CrossRef]
- Li, J.; Li, P.; Li, J.; Tian, Z.; Yu, F. Highly-Dispersed Ni-NiO Nanoparticles Anchored on an SiO2 Support for an Enhanced CO Methanation Performance. Catalysts 2019, 9, 506. [Google Scholar] [CrossRef] [Green Version]
- Foelske, A.; Barbieri, O.; Hahn, M.; Kötz, R. An X-Ray Photoelectron Spectroscopy Study of Hydrous Ruthenium Oxide Powders with Various Water Contents for Supercapacitors. Electrochem. Solid-State Lett. 2006, 9, A268. [Google Scholar] [CrossRef]
- Wang, G.; Hsieh, C.-S.; Tsai, D.-S.; Chen, R.-S.; Huang, Y.-S. Area-selective growth of ruthenium dioxide nanorods on LiNbO3(100) and Zn/Si substrates. J. Mater. Chem. 2004, 14, 3503–3508. [Google Scholar] [CrossRef]
- Over, H.; Seitsonen, A.P.; Lundgren, E.; Smedh, M.; Andersen, J.N. On the origin of the Ru-3d5/2 satellite feature from RuO2(110). Surf. Sci. 2002, 504, L196–L200. [Google Scholar] [CrossRef]
- Cuéllar-Cruz, M.; Schneider, D.K.; Stojanoff, V.; Islas, S.R.; Sánchez-Puig, N.; Arreguín-Espinosa, R.; Delgado, J.M.; Moreno, A. Formation of Crystalline Silica–Carbonate Biomorphs of Alkaline Earth Metals (Ca, Ba, Sr) from Ambient to Low Temperatures: Chemical Implications during the Primitive Earth’s Life. Cryst. Growth Des. 2020, 20, 1186–1195. [Google Scholar] [CrossRef]
- Wang, R.; Li, Y.; Shi, R.; Yang, M. Effect of metal–support interaction on the catalytic performance of Ni/Al2O3 for selective hydrogenation of isoprene. J. Mol. Catal. Chem. 2011, 344, 122–127. [Google Scholar] [CrossRef]
- Mahata, N.; Cunha, A.F.; Órfão, J.J.M.; Figueiredo, J.L. Hydrogenation of nitrobenzene over nickel nanoparticles stabilized by filamentous carbon. Appl. Catal. Gen. 2008, 351, 204–209. [Google Scholar] [CrossRef]
- Hoffer, B.W.; Dick van Langeveld, A.; Janssens, J.-P.; Bonné, R.L.C.; Lok, C.M.; Moulijn, J.A. Stability of highly dispersed Ni/Al2O3 catalysts: Effects of pretreatment. J. Catal. 2000, 192, 432–440. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak Maximum Position | M/Al (at) | Al/O (at) | |
|---|---|---|---|
| 3%Ru/Al | 281.2 | 0.119 | 0.56 |
| (3%Ru)10%Ba/Al | 282.1 | 0.046 | 0.51 |
| (3%Ru)10%K/Al | 281.0 | 0.035 | 0.48 |
| (3%Ru)10%Na/Al | 280.3 | 0.068 | 0.37 |
| 15%Ni/Al | 856.8 | 0.061 | 0.57 |
| (10%Ba)15%Ni/Al | 858.5 | 0.084 | 0.39 |
| (10%K)15%Ni/Al | 857.7 | 0.025 | 0.55 |
| (10%Na)15%Ni/Al | 856.3 | 0.038 | 0.48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Bordejé, E.; Dongil, A.B.; Conesa, J.M.; Guerrero-Ruiz, A.; Rodríguez-Ramos, I. Promotion of Ru or Ni on Alumina Catalysts with a Basic Metal for CO2 Hydrogenation: Effect of the Type of Metal (Na, K, Ba). Nanomaterials 2022, 12, 1052. https://doi.org/10.3390/nano12071052
García-Bordejé E, Dongil AB, Conesa JM, Guerrero-Ruiz A, Rodríguez-Ramos I. Promotion of Ru or Ni on Alumina Catalysts with a Basic Metal for CO2 Hydrogenation: Effect of the Type of Metal (Na, K, Ba). Nanomaterials. 2022; 12(7):1052. https://doi.org/10.3390/nano12071052
Chicago/Turabian StyleGarcía-Bordejé, Enrique, Ana Belén Dongil, José M. Conesa, Antonio Guerrero-Ruiz, and Inmaculada Rodríguez-Ramos. 2022. "Promotion of Ru or Ni on Alumina Catalysts with a Basic Metal for CO2 Hydrogenation: Effect of the Type of Metal (Na, K, Ba)" Nanomaterials 12, no. 7: 1052. https://doi.org/10.3390/nano12071052