
Impact of Adsorption of Straight Chain Alcohol Molecules on the Optical Properties of Calcite (10.4) Surface

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
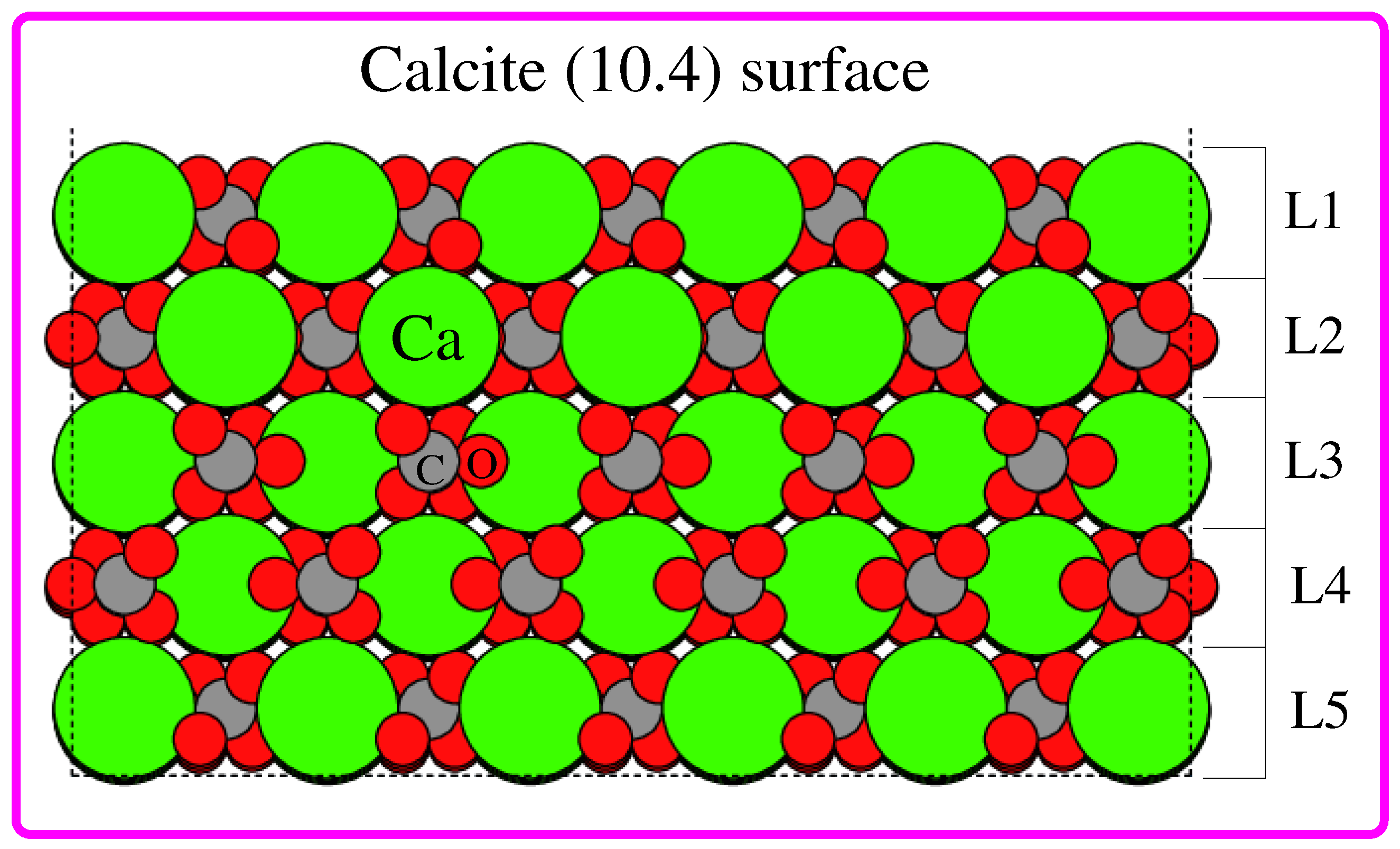
2. Computational Methodology
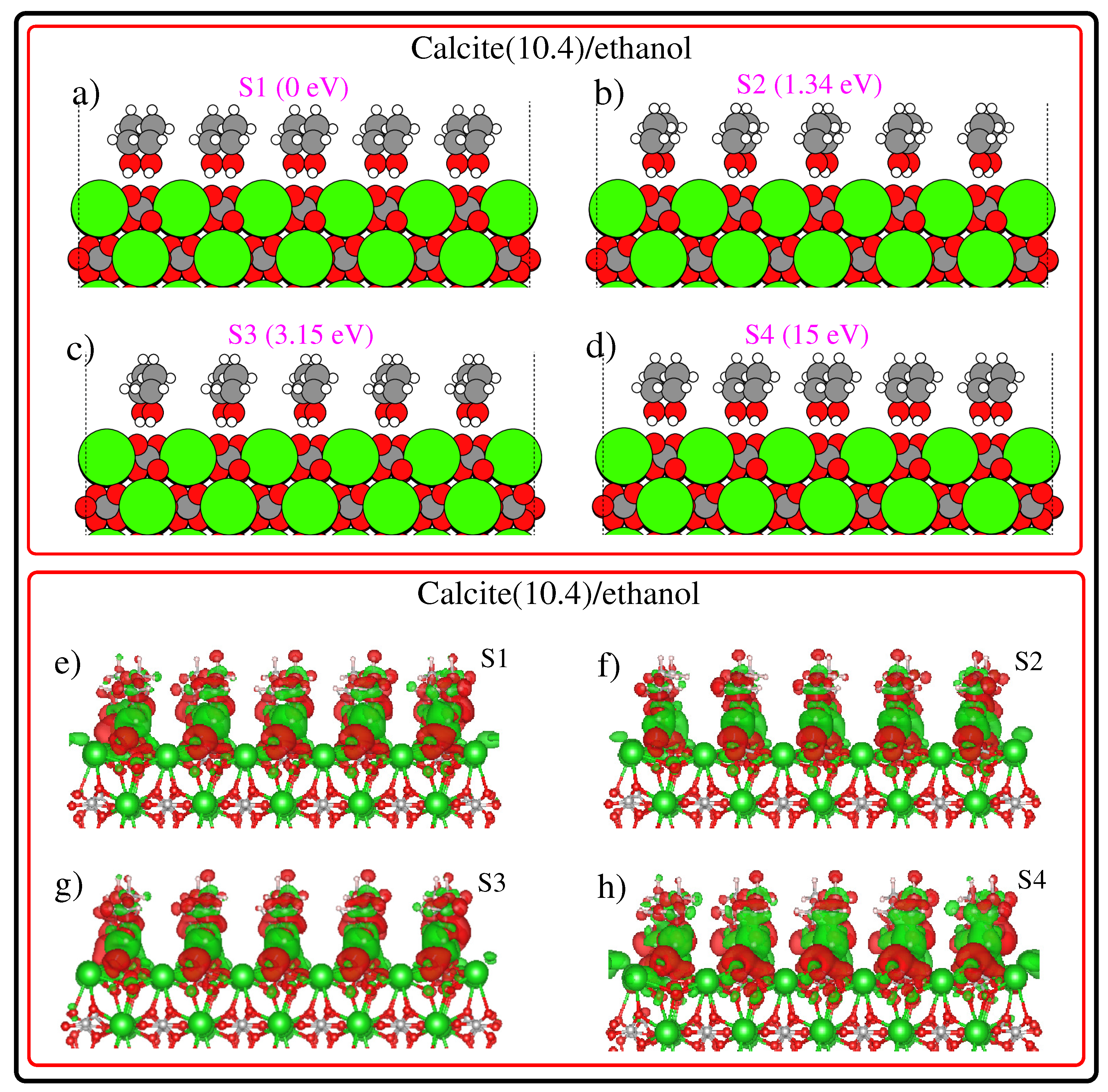
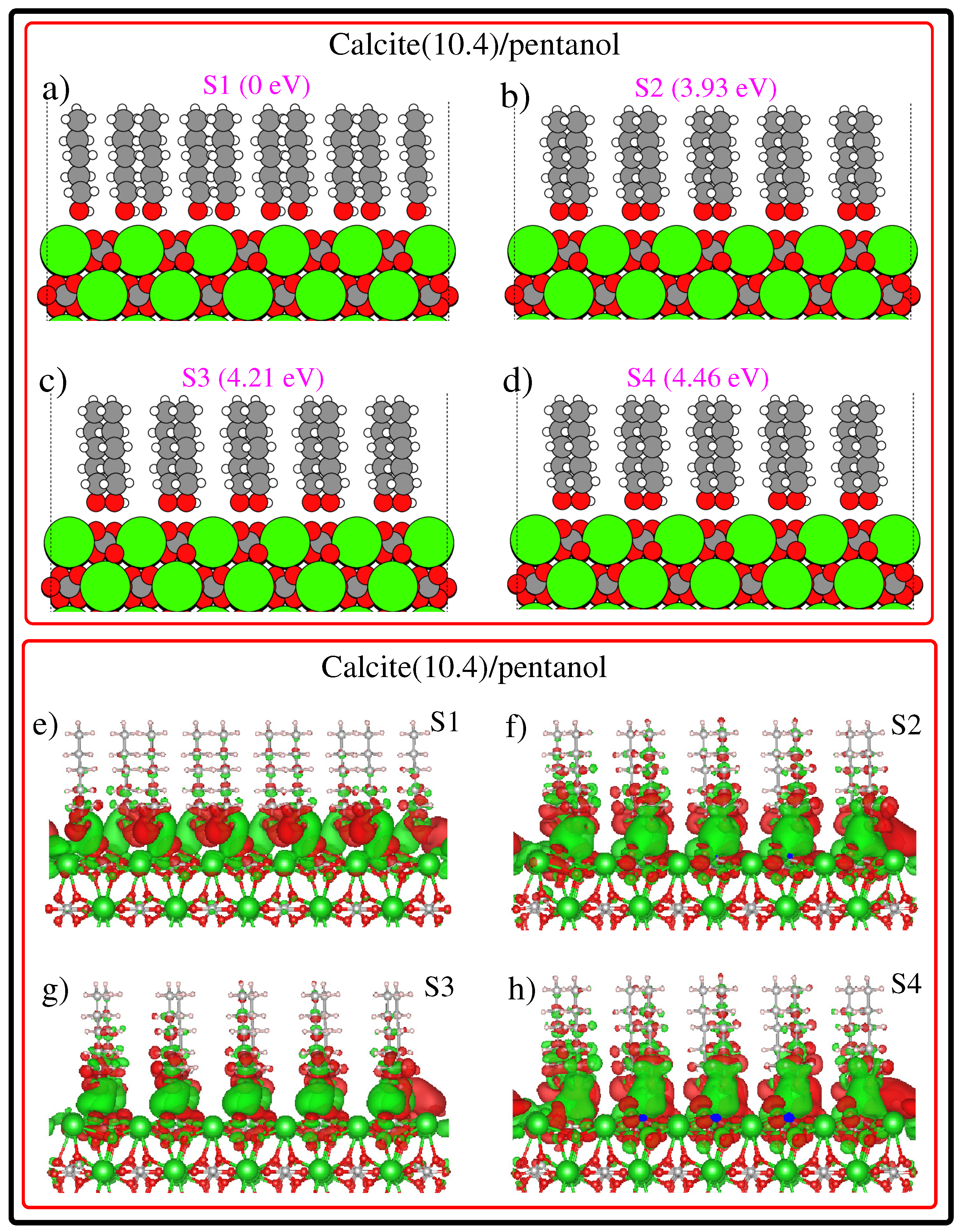
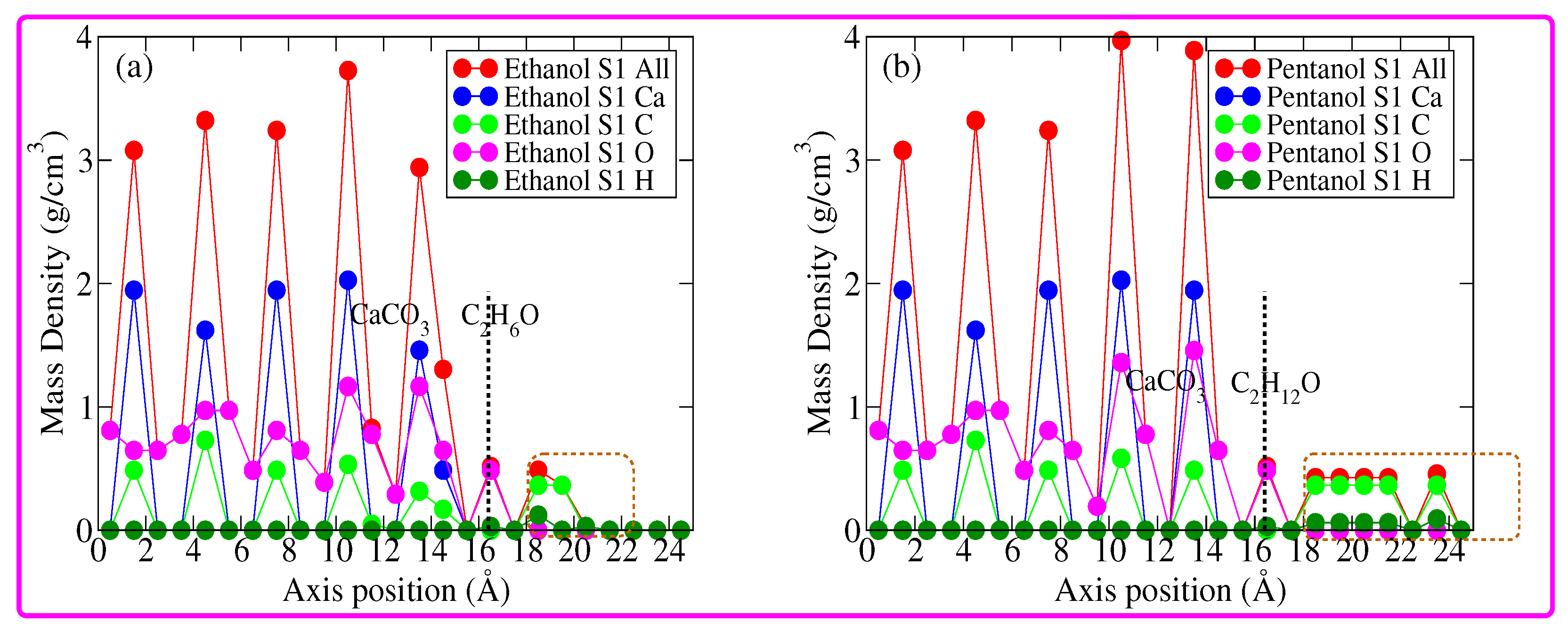
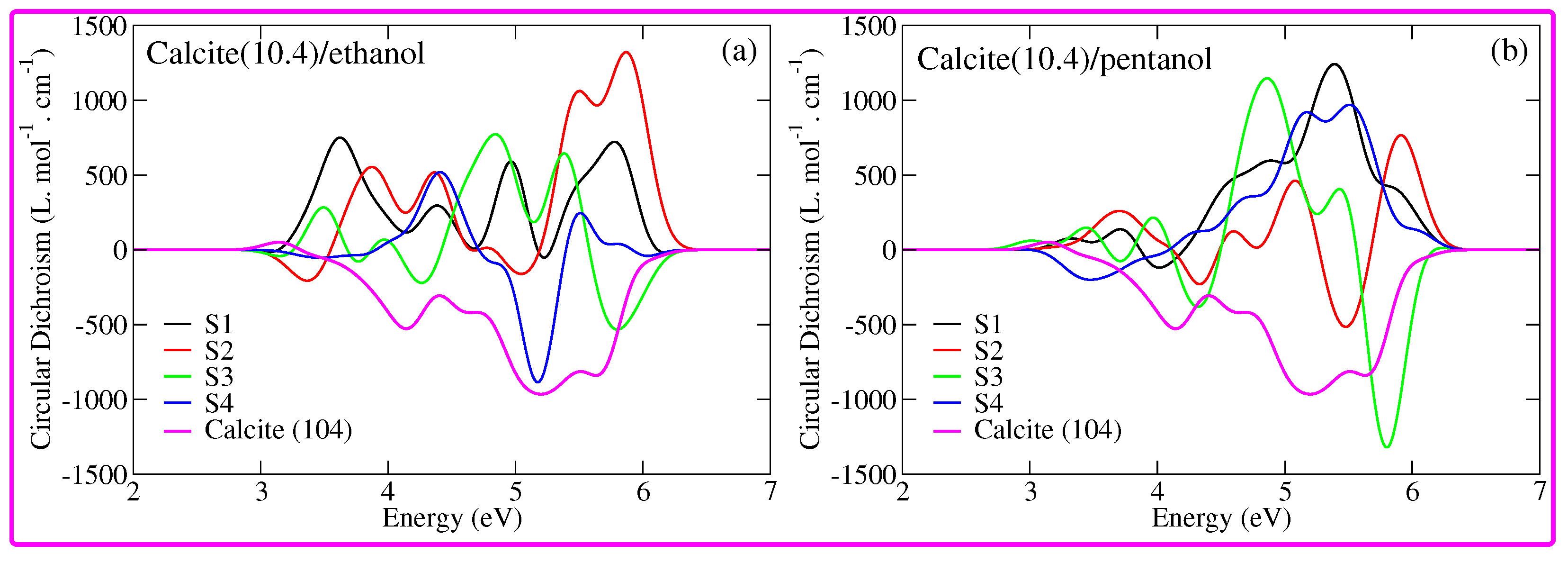
3. Results and Discussion
4. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Millero, F.J. Chemical Oceanography, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Morse, J.W.; Mackenzie, F.T. Geochemistry of Sedimentary Carbonates; Elsevier: Amsterdam, The Netherlands, 1990. [Google Scholar]
- Usher, C.R.; Michel, A.E.; Grassian, V.H. Reactions on Mineral Dust. Chem. Rev. 2003, 103, 4883–4940. [Google Scholar] [CrossRef] [PubMed]
- Talham, D.R. Biomineralization: Principles and Concepts in Bioinorganic Materials Chemistry Stephen Mann. Oxford University Press, New York, USA, 2001. Cryst. Growth Des. 2002, 2, 675. [Google Scholar] [CrossRef]
- Meldrum, F.C.; Cölfen, H. Controlling Mineral Morphologies and Structures in Biological and Synthetic Systems. Chem. Rev. 2008, 108, 4332–4432. [Google Scholar] [CrossRef] [PubMed]
- Sommerdijk, N.A.J.M.; With, G.d. Biomimetic CaCO3 Mineralization using Designer Molecules and Interfaces. Chem. Rev. 2008, 108, 4499–4550. [Google Scholar] [CrossRef]
- Reddy, M.S. Biomineralization of calcium carbonates and their engineered applications: A review. Front. Microbiol. 2013, 4, 314. [Google Scholar]
- Addadi, L.; Weiner, S. Control and Design Principles in Biological Mineralization. Angew. Chem. Int. Ed. Engl. 1992, 31, 153–169. [Google Scholar] [CrossRef]
- Cölfen, H. Precipitation of carbonates: Recent progress in controlled production of complex shapes. Curr. Opin. Colloid Interface Sci. 2003, 8, 23–31. [Google Scholar] [CrossRef]
- Hlubina, P.; Urbańczyk, W. Dispersion of the group birefringence of a calcite crystal measured by white-light spectral interferometry. Meas. Sci. Technol. 2005, 16, 1267–1271. [Google Scholar] [CrossRef]
- de Leeuw, N.H.; Parker, S.C. Surface Structure and Morphology of Calcium Carbonate Polymorphs Calcite, Aragonite, and Vaterite: An Atomistic Approach. J. Phys. Chem. B 1998, 102, 2914–2922. [Google Scholar] [CrossRef]
- Marutschke, C.; Walters, D.; Cleveland, J.; Hermes, I.; Bechstein, R.; Kühnle, A. Three-dimensional hydration layer mapping on the (10.4) surface of calcite using amplitude modulation atomic force microscopy. Nanotechnology 2014, 25, 335703. [Google Scholar] [CrossRef]
- Hauke, C.M.; Rahe, P.; Nimmrich, M.; Schütte, J.; Kittelmann, M.; Stará, I.G.; Starý, I.; Rybáček, J.; Kühnle, A. Molecular Self-Assembly of Enantiopure Heptahelicene-2-Carboxylic Acid on Calcite (1014). J. Phys. Chem. C 2012, 116, 4637–4641. [Google Scholar] [CrossRef] [Green Version]
- de Leeuw, N.H.; Parker, S.C.; Rao, K.H. Modeling the Competitive Adsorption of Water and Methanoic Acid on Calcite and Fluorite Surfaces. Langmuir 1998, 14, 5900–5906. [Google Scholar] [CrossRef]
- Stöckelmann, E.; Hentschke, R. Adsorption Isotherms of Water Vapor on Calcite: A Molecular Dynamics—Monte Carlo Hybrid Simulation Using a Polarizable Water Model. Langmuir 1999, 15, 5141–5149. [Google Scholar] [CrossRef]
- Kerisit, S.; Parker, S.C.; Harding, J.H. Atomistic Simulation of the Dissociative Adsorption of Water on Calcite Surfaces. J. Phys. Chem. B 2003, 107, 7676–7682. [Google Scholar] [CrossRef]
- Rahaman, A.; Grassian, V.H.; Margulis, C.J. Dynamics of Water Adsorption onto a Calcite Surface as a Function of Relative Humidity. J. Phys. Chem. C 2008, 112, 2109–2115. [Google Scholar] [CrossRef]
- Sand, K.K.; Yang, M.; Makovicky, E.; Cooke, D.J.; Hassenkam, T.; Bechgaard, K.; Stipp, S.L.S. Binding of Ethanol on Calcite: The Role of the OH Bond and Its Relevance to Biomineralization. Langmuir 2010, 26, 15239–15247. [Google Scholar] [CrossRef]
- Cooke, D.J.; Gray, R.J.; Sand, K.K.; Stipp, S.L.S.; Elliott, J.A. Interaction of Ethanol and Water with the 1014 Surface of Calcite. Langmuir 2010, 26, 14520–14529. [Google Scholar] [CrossRef]
- Bovet, N.; Yang, M.; Javadi, M.S.; Stipp, S.L.S. Interaction of alcohols with the calcite surface. Phys. Chem. Chem. Phys. 2015, 17, 3490–3496. [Google Scholar] [CrossRef]
- Teng, H.H.; Chen, Y.; Pauli, E. Direction Specific Interactions of 1,4-Dicarboxylic Acid with Calcite Surfaces. J. Am. Soc. 2006, 128, 14482–14484. [Google Scholar] [CrossRef]
- Freeman, C.L.; Asteriadis, I.; Yang, M.; Harding, J.H. Interactions of Organic Molecules with Calcite and Magnesite Surfaces. J. Phys. Chem. C 2009, 113, 3666–3673. [Google Scholar] [CrossRef]
- Sánchez, V.M.; Miranda, C.R. Modeling Acid Oil Component Interactions with Carbonate Reservoirs: A First-Principles View on Low Salinity Recovery Mechanisms. J. Phys. Chem. C 2014, 118, 19180–19187. [Google Scholar] [CrossRef]
- Sonnenberg, L.; Luo, Y.; Schlaad, H.; Seitz, M.; Cölfen, H.; Gaub, H.E. Quantitative Single Molecule Measurements on the Interaction Forces of Poly(l-glutamic acid) with Calcite Crystals. J. Am. Soc. 2007, 129, 15364–15371. [Google Scholar] [CrossRef] [PubMed]
- Freeman, C.L.; Harding, J.H.; Quigley, D.; Rodger, P.M. Simulations of Ovocleidin-17 Binding to Calcite Surfaces and Its Implications for Eggshell Formation. J. Phys. Chem. C 2011, 115, 8175–8183. [Google Scholar] [CrossRef]
- Yang, M.; Stipp, S.L.S.; Harding, J. Biological Control on Calcite Crystallization by Polysaccharides. Cryst. Growth Des. 2008, 8, 4066–4074. [Google Scholar] [CrossRef]
- Rode, S.; Oyabu, N.; Kobayashi, K.; Yamada, H.; Kühnle, A. True Atomic-Resolution Imaging of (1014) Calcite in Aqueous Solution by Frequency Modulation Atomic Force Microscopy. Langmuir 2009, 25, 2850–2853. [Google Scholar] [CrossRef] [Green Version]
- Schütte, J.; Rahe, P.; Tröger, L.; Rode, S.; Bechstein, R.; Reichling, M.; Kühnle, A. Clear Signature of the (2 × 1) Reconstruction of Calcite (1014). Langmuir 2010, 26, 8295–8300. [Google Scholar] [CrossRef]
- Fenter, P.; Geissbühler, P.; DiMasi, E.; Srajer, G.; Sorensen, L.; Sturchio, N. Surface speciation of calcite observed in situ by high-resolution X-ray reflectivity. Geochim. Cosmochim. Acta 2000, 64, 1221–1228. [Google Scholar] [CrossRef]
- Kerisit, S.; Marmier, A.; Parker, S.C. Ab Initio Surface Phase Diagram of the 1014 Calcite Surface. J. Phys. Chem. B 2005, 109, 18211–18213. [Google Scholar] [CrossRef]
- Raina, G.; Gauldie, R.W.; Sharma, S.K.; Helsley, C.E. A study of the calcite cleavage plane using the atomic force microscope. Ferroelectr. Sect. 1994, 17, 65–72. [Google Scholar] [CrossRef]
- Blanchard, D.L.; Baer, D. The interactions of Co, Mn and water with calcite surfaces. Surf. Sci. 1992, 276, 27–39. [Google Scholar] [CrossRef]
- Didymus, J.M.; Oliver, P.; Mann, S.; DeVries, A.L.; Hauschka, P.V.; Westbroek, P. Influence of low-molecular-weight and macromolecular organic additives on the morphology of calcium carbonate. J. Chem. Soc. Faraday Trans. 1993, 89, 2891–2900. [Google Scholar] [CrossRef]
- Mokkath, J.H. Water-calcite (104) surface interactions using first-principles simulations. J. Phys. Chem. Solids 2022, 161, 110394. [Google Scholar] [CrossRef]
- Rahe, P.; Schütte, J.; Kühnle, A. NC-AFM contrast formation on the calcite (104) surface. J. Phys. Condens. Matter 2012, 24, 084006. [Google Scholar] [CrossRef] [PubMed]
- Smidstrup, S.; Markussen, T.; Vancraeyveld, P.; Wellendorff, J.; Schneider, J.; Gunst, T.; Verstichel, B.; Stradi, D.; Khomyakov, P.A.; Vej-Hansen, U.G.; et al. QuantumATK: An integrated platform of electronic and atomic-scale modelling tools. J. Phys. Condens. Matter 2020, 32, 015901. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method forab initioorder-Nmaterials simulation. J. Phys. Condens. 2002, 14, 2745–2779. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Barone, V.; Casarin, M.; Forrer, D.; Pavone, M.; Sambi, M.; Vittadini, A. Role and effective treatment of dispersive forces in materials: Polyethylene and graphite crystals as test cases. J. Comput. Chem. 2009, 30, 934–939. [Google Scholar] [CrossRef]
- Okhrimenko, D.V.; Nissenbaum, J.; Andersson, M.P.; Olsson, M.H.M.; Stipp, S.L.S. Energies of the Adsorption of Functional Groups to Calcium Carbonate Polymorphs: The Importance of –OH and –COOH Groups. Langmuir 2013, 29, 11062–11073. [Google Scholar] [CrossRef]
- Ataman, E.; Andersson, M.P.; Ceccato, M.; Bovet, N.; Stipp, S.L.S. Functional Group Adsorption on Calcite: II. Nitrogen and Sulfur Containing Organic Molecules. J. Phys. Chem. C 2016, 120, 16597–16607. [Google Scholar] [CrossRef]
- Li, H.; Vovusha, H.; Sharma, S.; Singh, N.; Schwingenschlögl, U. Mechanism of wettability alteration of the calcite 104 surface. Phys. Chem. Chem. Phys. 2020, 22, 15365–15372. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, C.; Grimme, S. A simplified time-dependent density functional theory approach for electronic ultraviolet and circular dichroism spectra of very large molecules. Comput. Theor. Chem. 2014, 1040–1041, 45–53. [Google Scholar] [CrossRef]
- Grimme, S. A simplified Tamm-Dancoff density functional approach for the electronic excitation spectra of very large molecules. J. Chem. Phys. 2013, 138, 244104. [Google Scholar] [CrossRef] [PubMed]
- de Wergifosse, M.; Seibert, J.; Grimme, S. Simplified time-dependent density functional theory (sTD-DFT) for molecular optical rotation. J. Chem. Phys. 2020, 153, 084116. [Google Scholar] [CrossRef] [PubMed]
- de Wergifosse, M.; Grimme, S. Nonlinear-response properties in a simplified time-dependent density functional theory (sTD-DFT) framework: Evaluation of excited-state absorption spectra. J. Chem. Phys. 2019, 150, 094112. [Google Scholar] [CrossRef]
- Bannwarth, C.; Seibert, J.; Grimme, S. Electronic Circular Dichroism of [16]Helicene With Simplified TD-DFT: Beyond the Single Structure Approach. Chirality 2016, 28, 365–369. [Google Scholar] [CrossRef]
- de Wergifosse, M.; Grimme, S. Perspective on Simplified Quantum Chemistry Methods for Excited States and Response Properties. J. Phys. Chem. A 2021, 125, 3841–3851. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef] [Green Version]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef] [Green Version]
- Heyd, J.; Scuseria, G.E. Assessment and validation of a screened Coulomb hybrid density functional. J. Chem. Phys. 2004, 120, 7274–7280. [Google Scholar] [CrossRef] [PubMed]
- Yersin, H.; Humbs, W.; Strasser, J. Low-lying electronic states of [Rh(bpy)3]3+, [Pt(bpy)2]2+, and [Ru(bpy)3]2+. A comparative study based on highly resolved and time-resolved spectra. Coord. Chem. 1997, 159, 325–358. [Google Scholar] [CrossRef]
- Pasarín, I.S.; Yang, M.; Bovet, N.; Glyvradal, M.; Nielsen, M.M.; Bohr, J.; Feidenhans’l, R.; Stipp, S.L.S. Molecular Ordering of Ethanol at the Calcite Surface. Langmuir 2012, 28, 2545–2550. [Google Scholar] [CrossRef] [PubMed]
- Haddad, J.; Steinrück, H.-G.; Hlaing, H.; Kewalramani, S.; Pontoni, D.; Reichert, H.; Murphy, B.M.; Festersen, S.; Runge, B.; Magnussen, O.M.; et al. Order and Melting in Self-Assembled Alkanol Monolayers on Amorphous SiO2. J. Phys. Chem. 2015, 119, 17648–17654. [Google Scholar] [CrossRef]
- Russell, T. X-ray and neutron reflectivity for the investigation of polymers. Mater. Sci. Rep. 1990, 5, 171–271. [Google Scholar] [CrossRef]
- Weber, W.; Lengeler, B. Diffuse scattering of hard x rays from rough surfaces. Phys. Rev. B 1992, 46, 7953–7956. [Google Scholar] [CrossRef]
- Garoff, S.; Sirota, E.B.; Sinha, S.K.; Stanley, H.B. The effects of substrate roughness on ultrathin water films. J. Chem. 1989, 90, 7505–7515. [Google Scholar] [CrossRef]
- Braslau, A.; Deutsch, M.; Pershan, P.S.; Weiss, A.H.; Als-Nielsen, J.; Bohr, J. Surface Roughness of Water Measured by X-ray Reflectivity. Phys. Rev. Lett. 1985, 54, 114–117. [Google Scholar] [CrossRef] [Green Version]
- Stipp, S.L.; Hochella, M.F. Structure and bonding environments at the calcite surface as observed with X-ray photoelectron spectroscopy (XPS) and low energy electron diffraction (LEED). Geochim. Cosmochim. Acta 1991, 55, 1723–1736. [Google Scholar] [CrossRef]
- Kypr, J.; Kejnovská, I.; Renčiuk, D.; Vorlíčková, M. Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res. 2009, 37, 1713–1725. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Absolute structure determination: Pushing the limits. Acta Cryst. Sect. B 2016, 72, 659–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, T.J.; Gómez, D.E. Interaction of localized surface plasmons with chiral molecules. Phys. Rev. B 2014, 90, 235424. [Google Scholar] [CrossRef]
- Noguez, C.; Garzón, I.L. Optically active metal nanoparticles. Chem. Soc. Rev. 2009, 38, 757–771. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Moriyama, H. Encyclopedia of Polymer Science and Technology; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Castiglioni, E.; Biscarini, P.; Abbate, S. Experimental aspects of solid state circular dichroism. Chirality 2009, 21, E28–E36. [Google Scholar] [CrossRef]
- Dang, Y.; Liu, X.; Sun, Y.; Song, J.; Hu, W.; Tao, X. Bulk Chiral Halide Perovskite Single Crystals for Active Circular Dichroism and Circularly Polarized Luminescence. J. Phys. Chem. Lett. 2020, 11, 1689–1696. [Google Scholar] [CrossRef]
- Makkonen, E.; Rossi, T.P.; Larsen, A.H.; Lopez-Acevedo, O.; Rinke, P.; Kuisma, M.; Chen, X. Real-time time-dependent density functional theory implementation of electronic circular dichroism applied to nanoscale metal–organic clusters. J. Chem. Phys. 2021, 154, 114102. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mokkath, J.H. Impact of Adsorption of Straight Chain Alcohol Molecules on the Optical Properties of Calcite (10.4) Surface. Nanomaterials 2022, 12, 1460. https://doi.org/10.3390/nano12091460
Mokkath JH. Impact of Adsorption of Straight Chain Alcohol Molecules on the Optical Properties of Calcite (10.4) Surface. Nanomaterials. 2022; 12(9):1460. https://doi.org/10.3390/nano12091460
Chicago/Turabian StyleMokkath, Junais Habeeb. 2022. "Impact of Adsorption of Straight Chain Alcohol Molecules on the Optical Properties of Calcite (10.4) Surface" Nanomaterials 12, no. 9: 1460. https://doi.org/10.3390/nano12091460