
Understanding the CH4 Conversion over Metal Dimers from First Principles


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Computational Methods
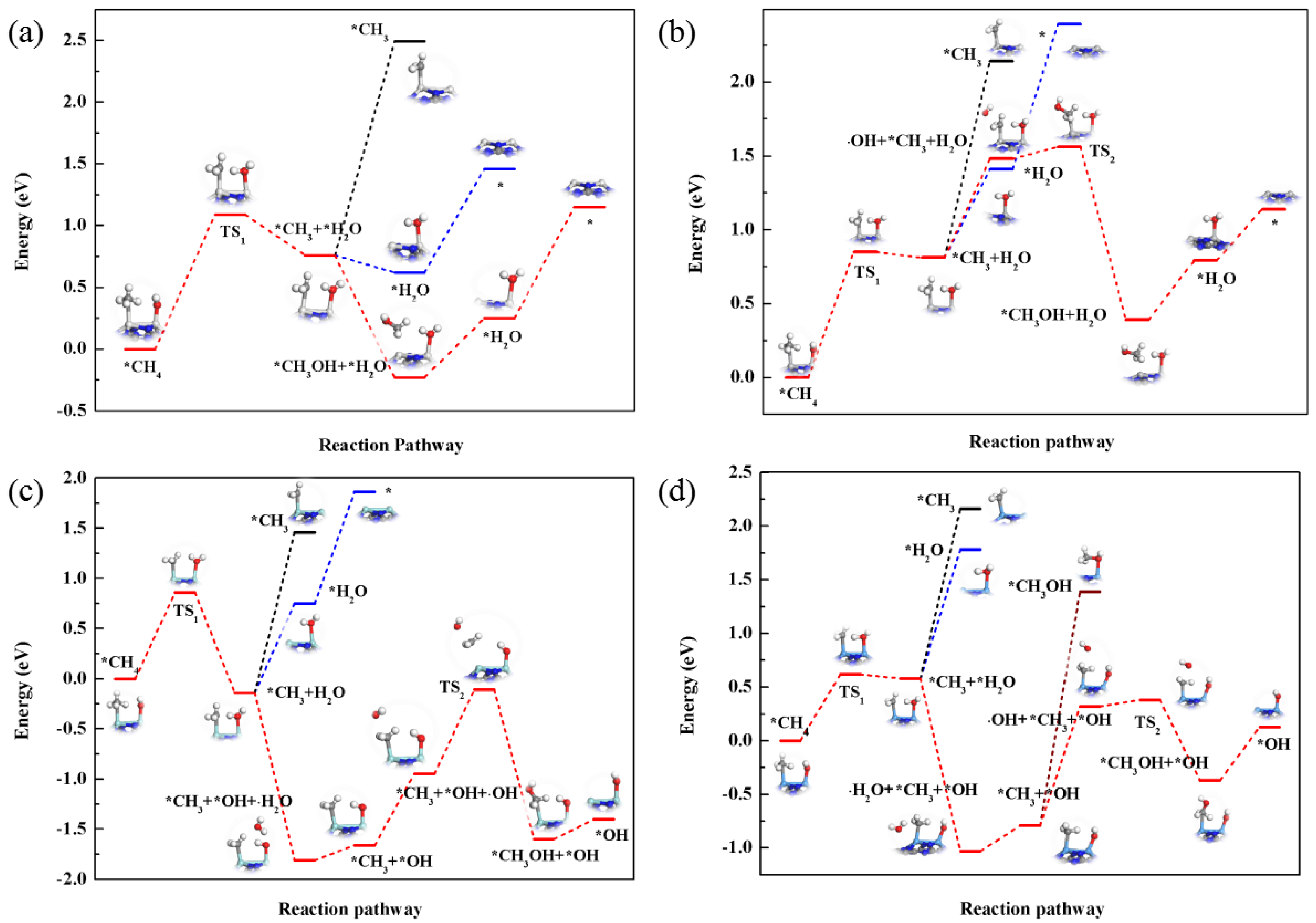
3. Results and Discussion
3.1. Geometric Structure and Stability of M2-Pc
3.2. Decomposition of H2O2 on M2-Pc
3.3. Catalytic Conversion of Methane on the M2-Pc
3.3.1. OH-Assisted Methane Conversion
3.3.2. O-Assisted Methane Conversion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, Z.; Grace, J.R.; Lim, J.C.; Zhang, L. Combustion of Low-Concentration Coal Bed Methane in a Fluidized Bed. Energy Fuels 2011, 25, 975–980. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; Palagin, D.; Ranocchiari, M.; Van Bokhoven, J.A. Selective Anaerobic Oxidation of Methane Enables Direct Synthesis of Methanol. Science 2017, 356, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Zhang, W.; Li, X.; Yang, J. A High Performance Catalyst for Methane Conversion to Methanol: Graphene Supported Single Atom Co. Chem. Commun. 2018, 54, 2284–2287. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.; Zhu, Q.; Wu, Z.; Ma, D. Methane Activation: The Past and Future. Energy Environ. Sci. 2014, 7, 2580–2591. [Google Scholar] [CrossRef]
- Narsimhan, K.; Iyoki, K.; Dinh, K.; Román-Leshkov, Y. Catalytic Oxidation of Methane into Methanol over Copper-Exchanged Zeolites with Oxygen at Low Temperature. ACS Cent. Sci. 2016, 2, 424–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Fang, G.; Li, G.; Ma, H.; Fan, H.; Yu, L.; Ma, C.; Wu, X.; Deng, D.; Wei, M.; et al. Direct, Nonoxidative Conversion of Methane to Ethylene, Aromatics, and Hydrogen. Science 2014, 344, 616–619. [Google Scholar] [CrossRef]
- Kwon, Y.; Kim, T.Y.; Kwon, G.; Yi, J.; Lee, H. Selective Activation of Methane on Single-Atom Catalyst of Rhodium Dispersed on Zirconia for Direct Conversion. J. Am. Chem. Soc. 2017, 139, 17694–17699. [Google Scholar] [CrossRef] [PubMed]
- Holmen, A. Direct Conversion of Methane to Fuels and Chemicals. Catal. Today 2009, 142, 2–8. [Google Scholar] [CrossRef]
- Jin, D.; Zhu, B.; Hou, Z.; Fei, J.; Lou, H.; Zheng, X. Dimethyl Ether Synthesis via Methanol and Syngas over Rare Earth Metals Modified Zeolite Y and Dual Cu–Mn–Zn Catalysts. Fuel 2007, 86, 2707–2713. [Google Scholar] [CrossRef]
- Wang, B.; Albarracín-Suazo, S.; Pagán-Torres, Y.; Nikolla, E. Advances in Methane Conversion Processes. Catal. Today 2017, 285, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.Z.; Yu, Z.L.; Ng, C.F.; Au, C.T. CO2/CH4 Reforming over Ni–La2O3/5A: An Investigation on Carbon Deposition and Reaction Steps. J. Catal. 2000, 194, 198–210. [Google Scholar] [CrossRef]
- Tang, S.; Ji, L.; Lin, J.; Zeng, H.C.; Tan, K.L.; Li, K. CO2 Reforming of Methane to Synthesis Gas over Sol–Gel-made Ni/γ-Al2O3 Catalysts from Organometallic Precursors. J. Catal. 2000, 194, 424–430. [Google Scholar] [CrossRef]
- McFarland, E. Unconventional Chemistry for Unconventional Natural Gas. Science 2012, 338, 340–342. [Google Scholar] [CrossRef]
- Da Silva, M.J. Synthesis of Methanol from Methane: Challenges and Advances on the Multi-Step (Syngas) and One-Step Routes (DMTM). Fuel Process. Technol. 2016, 145, 42–61. [Google Scholar] [CrossRef]
- Schroder, D.; Fiedler, A.; Hrusak, J.; Schwarz, H. Experimental and Theoretical Studies toward a Characterization of Conceivable Intermediates Involved in the Gas-Phase Oxidation of Methane by Bare FeO+. Generation of Four Distinguishable [Fe,C,H4,O]+ Isomers. J. Am. Chem. Soc. 1992, 114, 1215–1222. [Google Scholar] [CrossRef]
- Chen, Y.-M.; Clemmer, D.E.; Armentrout, P.B. Conversion of CH4 to CH3OH: Reactions of CoO+ with CH4 and D2, Co+ with CH3OD and D2O, and Co+(CH3OD) with Xe. J. Am. Chem. Soc. 1994, 116, 7815–7826. [Google Scholar] [CrossRef]
- Lieberman, R.L.; Rosenzweig, A.C. Crystal Structure of a Membrane-Bound Metalloenzyme that Catalyses the Biological Oxidation of Methane. Nature 2005, 434, 177–182. [Google Scholar] [CrossRef]
- Hakemian, A.S.; Rosenzweig, A.C. The Biochemistry of Methane Oxidation. Annu. Rev. Biochem. 2007, 76, 223–241. [Google Scholar] [CrossRef]
- Rosenzweig, A.C.; Frederick, C.A.; Lippard, S.J.; Nordlund, P. Crystal Structure of A Bacterial Non-Haem Iron Hydroxylase that Catalyses the Biological Oxidation of Methane. Nature 1993, 366, 537–543. [Google Scholar] [CrossRef]
- Lipscomb, J.D. Biochemistry of the Soluble Methane Monooxygenase. Annu. Rev. Microbiol. 1994, 48, 371–399. [Google Scholar] [CrossRef]
- Colby, J.; Stirling, D.I.; Dalton, H. The Soluble Methane Mono-Oxygenase of Methylococcus Capsulatus (Bath). Its Ability to Oxygenate n-Alkanes, n-Alkenes, Ethers, and Alicyclic, Aromatic and Heterocyclic Compounds. Biochem. J. 1977, 165, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramanian, R.; Smith, S.M.; Rawat, S.; Yatsunyk, L.A.; Stemmler, T.L.; Rosenzweig, A.C. Oxidation of Methane by a Biological Dicopper Centre. Nature 2010, 465, 115–119. [Google Scholar] [CrossRef] [Green Version]
- Narsimhan, K.; Michaelis, V.; Mathies, G.; Gunther, M.; Griffin, W.R.; Román-Leshkov, Y. Methane to Acetic Acid over Cu-Exchanged Zeolites: Mechanistic Insights from a Site-Specific Carbonylation Reaction. J. Am. Chem. Soc. 2015, 137, 1825–1832. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Zhuang, J.; Nie, L.; Zhang, J.; Zhang, Y.; Gu, N.; Wang, T.; Feng, J.; Yang, D.; Perrett, S.; et al. Intrinsic Peroxidase-like Activity of Ferromagnetic Nanoparticles. Nat. Nanotechnol. 2007, 2, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Zhang, L.; Ma, K.; Riegman, M.; Chen, F.; Ingold, I.; Conrad, M.; Turker, M.Z.; Gao, M.; Jiang, X.; et al. Ultrasmall Nanoparticles Induce Ferroptosis in Nutrient-Deprived Cancer Cells and Suppress Tumour Growth. Nat. Nanotechnol. 2016, 11, 977–985. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wang, X.; Wang, Q.; Lou, Z.; Li, S.; Zhu, Y.; Qin, L.; Wei, H. Nanomaterials with Enzyme-like Characteristics (Nanozymes): Next-Generation Artificial Enzymes (II). Chem. Soc. Rev. 2019, 48, 1004–1076. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-Atom Catalysis of CO Oxidation Using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Gawande, M.B.; Ariga, K.; Yamauchi, Y. Single-Atom Catalysts. Small 2021, 17, 2101584. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Li, J.; Zhang, T. Heterogeneous Single-Atom Catalysis. Nat. Rev. Chem. 2018, 2, 65–81. [Google Scholar] [CrossRef]
- Deng, T.; Zheng, W.; Zhang, W. Increasing the Range of Non-Noble-Metal Single-Atom Catalysts. Chin. J. Catal. 2017, 38, 1489–1497. [Google Scholar] [CrossRef]
- Chen, Z.; Yan, J.-M.; Jiang, Q. Single or Double: Which Is the Altar of Atomic Catalysts for Nitrogen Reduction Reaction? Small Methods 2018, 3, 1800291. [Google Scholar] [CrossRef]
- Li, F.; Liu, X.; Chen, Z. 1 + 1′ > 2: Heteronuclear Bi-atom Catalyst Outperforms Its Homonuclear Counterparts for CO Oxidation. Small Methods 2019, 3, 1800480. [Google Scholar] [CrossRef]
- Yan, H.; Lin, Y.; Wu, H.; Zhang, W.; Sun, Z.; Cheng, H.; Liu, W.; Wang, C.; Li, J.; Huang, X.; et al. Bottom-up Precise Synthesis of Stable Platinum Dimers on Graphene. Nat. Commun. 2017, 8, 1070. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, Y.; Zeng, X.C.; Chen, Z. Exploration of High-Performance Single-Atom Catalysts on Support M1/FeOx for CO Oxidation via Computational Study. ACS Catal. 2015, 5, 544–552. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, J.; Li, F.; Chen, Z. Copper Dimer Supported on C2N-Layer as an Efficient Electrocatalyst for CO2 Reduction Reaction: A Computational Study. J. Phys. Chem. C 2018, 122, 19712–19721. [Google Scholar] [CrossRef]
- Abel, M.; Clair, S.; Ourdjini, O.; Mossoyan, M.; Porte, L. Single Layer of Polymeric Fe-Phthalocyanine: An Organometallic Sheet on Metal and Thin Insulating Film. J. Am. Chem. Soc. 2011, 133, 1203–1205. [Google Scholar] [CrossRef]
- Matsushita, O.; Derkacheva, V.M.; Muranaka, A.; Shimizu, S.; Uchiyama, M.; Luk’yanets, E.A.; Kobayashi, N. Rectangular-Shaped Expanded Phthalocyanines with Two Central Metal Atoms. J. Am. Chem. Soc. 2012, 134, 3411–3418. [Google Scholar] [CrossRef]
- Gueorguiev, G.K.; Pacheco, J.M.; Stafström, S.; Hultman, L. Silicon–metal Clusters: Nano-templates for Cluster Assembled Materials. Thin Solid Films 2006, 515, 1192–1196. [Google Scholar] [CrossRef]
- Dos Santos, P.B.; Rivelino, R.; de Brito Mota, F.; Gueorguiev, G.K.; Kakanakova-Georgieva, A. Dopant Species with Al-Si and N-Si Bonding in The MOCVD of AlN Implementing Trimethylaluminum, Ammonia and Silane. J. Phys. D Appl. Phys. 2015, 48, 295104. [Google Scholar]
- Bučko, T.; Hafner, J.; Lebègue, S.; Àngyán, J.G. Improved Description of the Structure of Molecular and Layered Crystals: Ab Initio DFT Calculations with van der Waals Corrections. J. Phys. Chem. A 2010, 114, 11814–11824. [Google Scholar] [CrossRef]
- Blöch, P. Projector Augmented-Wave Method. Phy. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.; Pack, J. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
- Henkelman, G.; Jónsson, H. Improved Tangent Estimate in the Nudged Elastic Band Method for Finding Minimum Energy Paths and Saddle Points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef] [Green Version]
- Sangiovanni, D.G.; Gueorguiev, G.K.; Kakanakova-Georgieva, A. Ab initio Molecular Dynamics of Atomic-Scale Surface Rreactions: Insights into Metal Organic Chemical Vapor Deposition of AlN on Graphene. Phys. Chem. Chem. Phys. 2018, 20, 17751–17761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos-Martin, J.M.; Blanco-Brieva, G.; Fierro, J.L.G. Hydrogen Peroxide Synthesis: An Outlook beyond the Anthraquinone Process. Angew. Chem. Int. Ed. 2006, 45, 6962–6984. [Google Scholar] [CrossRef]
- Sajith, P.K.; Staykov, A.; Yoshida, M.; Shiota, Y.; Yoshizawa, K. Theoretical Study of the Direct Conversion of Methane to Methanol Using H2O2 as an Oxidant on Pd and Au/Pd Surfaces. J. Phys. Chem. C 2020, 124, 13231–13239. [Google Scholar] [CrossRef]
- Hammond, C.; Forde, M.M.; Ab Rahim, M.H.; Thetford, A.; He, Q.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Dummer, N.F.; Murphy, D.M.; et al. Direct Catalytic Conversion of Methane to Methanol in an Aqueous Medium by Using Copper-Promoted Fe-ZSM-5. Angew. Chem. Int. Ed. 2012, 51, 5129–5133. [Google Scholar] [CrossRef]
- Ab Rahim, M.H.; Forde, M.M.; Jenkins, R.; Hammond, C.; He, Q.; Dimitratos, N.; Lopez-Sanchez, J.A.; Carley, A.F.; Taylor, S.H.; Willock, D.J.; et al. Oxidation of Methane to Methanol with Hydrogen Peroxide Using Supported Gold-Palladium Alloy Nanoparticles. Angew. Chem. Int. Ed. 2013, 52, 1280–1284. [Google Scholar] [CrossRef]
- Osadchii, D.Y.; Olivos-Suarez, A.L.; Szecsenyi, Á.; Li, G.; Nasalevich, M.A.; Dugulan, L.A.; Crespo, P.S.; Hensen, E.J.M.; Veber, S.L.; Fedin, M.V.; et al. Isolated Fe Sites in Metal Organic Frameworks Catalyze the Direct Conversion of Methane to Methanol. ACS Catal. 2018, 8, 5542–5548. [Google Scholar] [CrossRef] [Green Version]
- Xiao, P.; Wang, Y.; Nishitoba, T.; Kondo, J.N.; Yoko, T. Selective Oxidation of Methane to Methanol with H2O2 over An Fe-MFI Zeolite Catalyst Using Sulfolane Solvent. Chem. Commun. 2019, 55, 2896–2899. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.S.; Schumann, J.; Studt, F.; Abild-Pedersen, F.; Nørskov, J.K. Theoretical Investigation of Methane Oxidation on Pd(111) and Other Metallic Surfaces. J. Phys. Chem. C 2018, 122, 16023–16032. [Google Scholar] [CrossRef]
- Dinh, K.T.; Sullivan, M.M.; Serna, P.; Meyer, R.J.; Dincă, M.; Román-Leshkov, Y. Viewpoint on the Partial Oxidation of Methane to Methanol Using Cu- and Fe-Exchanged Zeolites. ACS Catal. 2018, 8, 8306–8313. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.; Latimer, A.; Yoo, J.S.; Studt, F.; Abild-Pedersen, F. Predicting Promoter-Induced Bond Activation on Solid Catalysts Using Elementary Bond Orders. J. Phys. Chem. Lett. 2015, 6, 3670–3674. [Google Scholar] [CrossRef]
- Yoo, J.S.; Khan, T.S.; Abild-Pedersen, F.; Nørskov, J.K.; Studt, F. On the Role of the Surface Oxygen Species during A–H (A = C, N, O) Bond Activation: A Density Functional Theory Study. Chem. Commun. 2015, 51, 2621–2624. [Google Scholar] [CrossRef]
- Li, J.; Staykov, A.; Ishihara, T.; Yoshizawa, K. Theoretical Study of the Decomposition and Hydrogenation of H2O2 on Pd and Au@Pd Surfaces: Understanding toward High Selectivity of H2O2 Synthesis. J. Phys. Chem. C 2011, 115, 7392–7398. [Google Scholar] [CrossRef]
- Serra-Maia, R.; Michel, F.M.; Kang, Y.; Stach, E.A. Decomposition of Hydrogen Peroxide Catalyzed by AuPd Nanocatalysts during Methane Oxidation to Methanol. ACS Catal. 2020, 10, 5115–5123. [Google Scholar] [CrossRef]
- Latimer, A.A.; Aljama, H.; Kakekhani, A.; Yoo, J.S.; Kulkarni, A.; Tsai, C.; Garcia-Melchor, M.; Abild-Pedersen, F.; Nørskov, J.K. Mechanistic Insights into Heterogeneous Methane Activation. Phys. Chem. Chem. Phys. 2017, 19, 3575–3581. [Google Scholar] [CrossRef]
- Latimer, A.A.; Kulkarni, A.R.; Aljama, H.; Montoya, J.H.; Yoo, J.S.; Tsai, C.; Abild-Pedersen, F.; Studt, F.; Nørskov, J.K. Understanding Trends in C–H Bond Activation in Heterogeneous Catalysis. Nat. Mater. 2017, 16, 225–229. [Google Scholar] [CrossRef]
- Fernan, S.; Leonardo, B. Density-Functional Theory Models of Fe(IV)O Reactivity in metal-organic Frameworks: SelfInteraction Error, Spin Delocalisation and The Role of Hybrid Exchange. Phys. Chem. Chem. Phys. 2020, 22, 12821–12830. [Google Scholar]
- Yu, L.; Li, F.; Zhao, J.; Chen, Z. Revisiting Catalytic Performance of Supported Metal Dimers for Oxygen Reduction Reaction via Magnetic Coupling from First Principles. Adv. Powder Mater. 2022, 1, 100031. [Google Scholar] [CrossRef]
- Latimer, A.A.; Kakekhani, A.; Kulkarni, A.; Nørskov, J.K. Direct Methane to Methanol: The Selectivity-Conversion Limit and Design Strategies. ACS Catal. 2018, 8, 6894–6907. [Google Scholar] [CrossRef]
- Da Silva, J.C.S.; Pennifold, R.C.R.; Harvey, J.N.; Rocha, W.R.A. Radical Rebound Mechanism for the Methane Oxidation Reaction Promoted by the Dicopper Center of A pMMO Enzyme: A Computational Perspective. Dalton Trans. 2016, 45, 2492–2504. [Google Scholar] [CrossRef] [Green Version]
- Chin, Y.H.; Buda, C.; Neurock, M.; Iglesia, E. Consequences of Metal-Oxide Interconversion for C–H Bond Activation during CH4 Reactions on Pd Catalysts. J. Am. Chem. Soc. 2013, 135, 15425–15442. [Google Scholar] [CrossRef]
- Bernd, E.; Francesco, B.; Michiel, C.M.G.; Evert, J.B. Methane-to-Methanol Oxidation by the Hydrated Iron(IV) Oxo Species in Aqueous Solution: A Combined DFT and Car-Parrinello Molecular Dynamics Study. J. Am. Chem. Soc. 2004, 126, 4355–4365. [Google Scholar]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Phys. Chem. C 2003, 118, 8207–8215. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, H.; Han, B.; Li, F.; Zhao, J.; Chen, Z. Understanding the CH4 Conversion over Metal Dimers from First Principles. Nanomaterials 2022, 12, 1518. https://doi.org/10.3390/nano12091518
Meng H, Han B, Li F, Zhao J, Chen Z. Understanding the CH4 Conversion over Metal Dimers from First Principles. Nanomaterials. 2022; 12(9):1518. https://doi.org/10.3390/nano12091518
Chicago/Turabian StyleMeng, Haihong, Bing Han, Fengyu Li, Jingxiang Zhao, and Zhongfang Chen. 2022. "Understanding the CH4 Conversion over Metal Dimers from First Principles" Nanomaterials 12, no. 9: 1518. https://doi.org/10.3390/nano12091518