
Amphipathic Solvent-Assisted Synthetic Strategy for Random Lamellae of the Clinoptilolites with Flower-like Morphology and Thinner Nanosheet for Adsorption and Separation of CO2 and CH4


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Synthesized CP
2.3. Random Lamellae of Synthesized CP
2.4. Characterizations
2.5. Adsorption Measurements
2.5.1. Adsorption Volume Method
2.5.2. Multi-Constituent Adsorption Breakthrough Method
2.5.3. Adsorption-Desorption Cycles Measurement Method
3. Results and Discussion
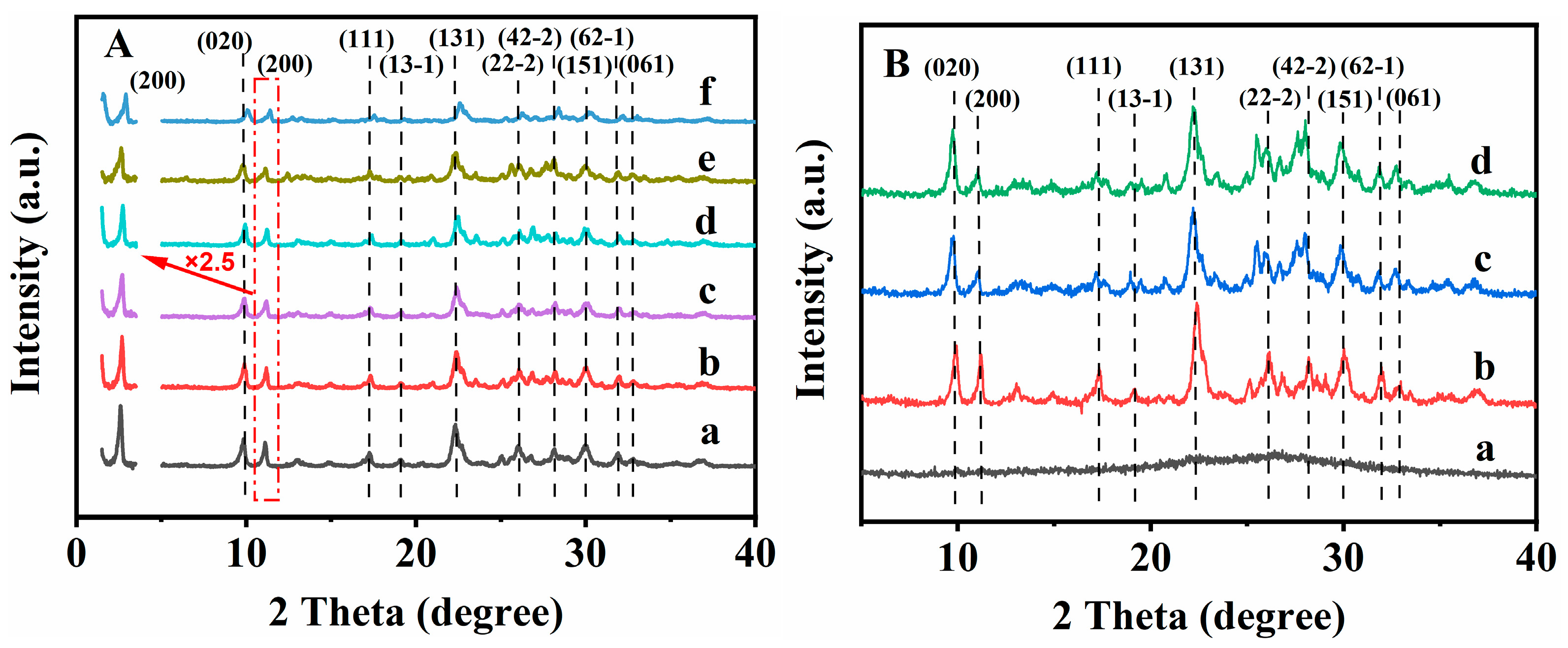
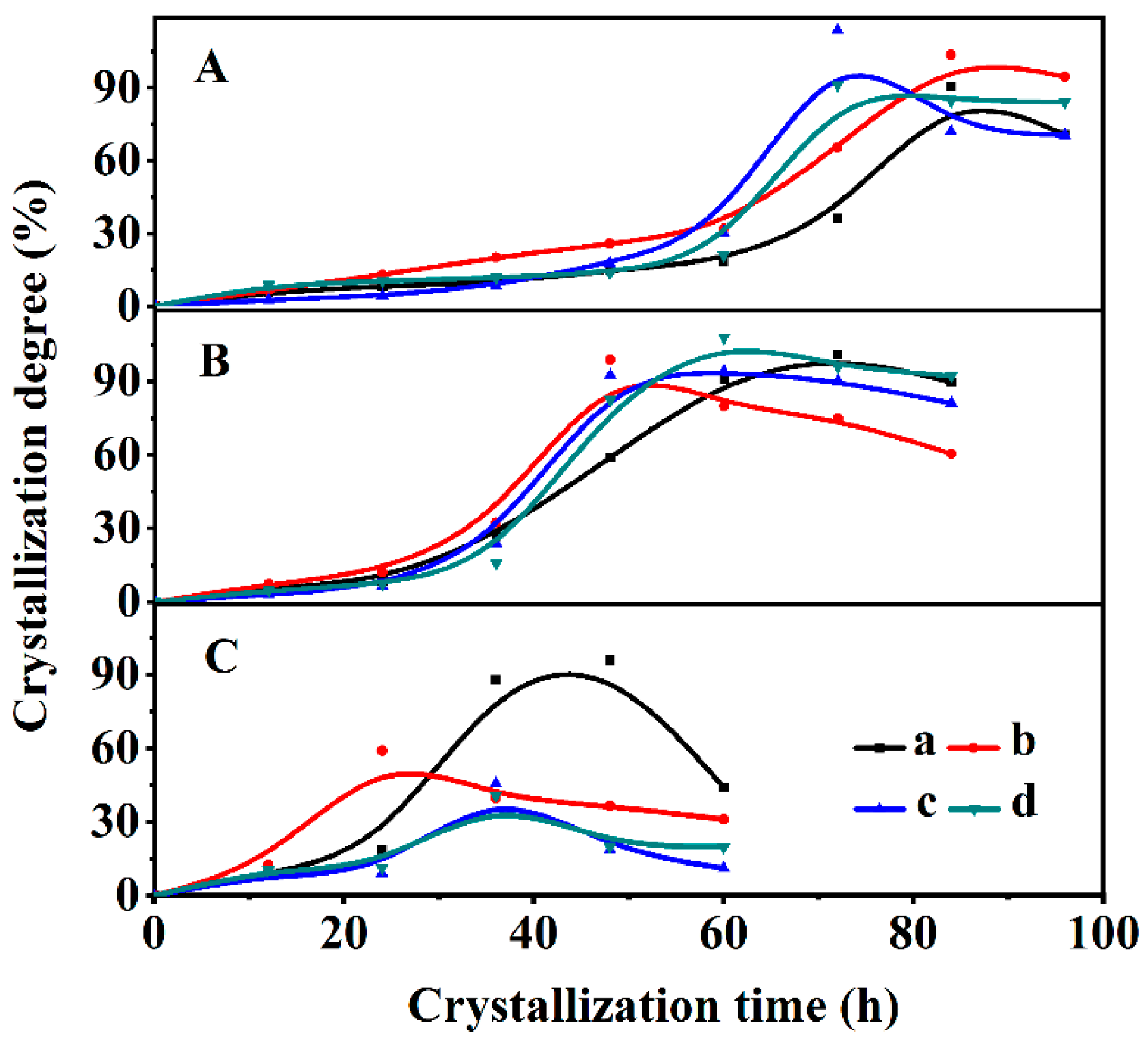
3.1. Structural Characterizations of the CPs and Their Crystallization Kinetics
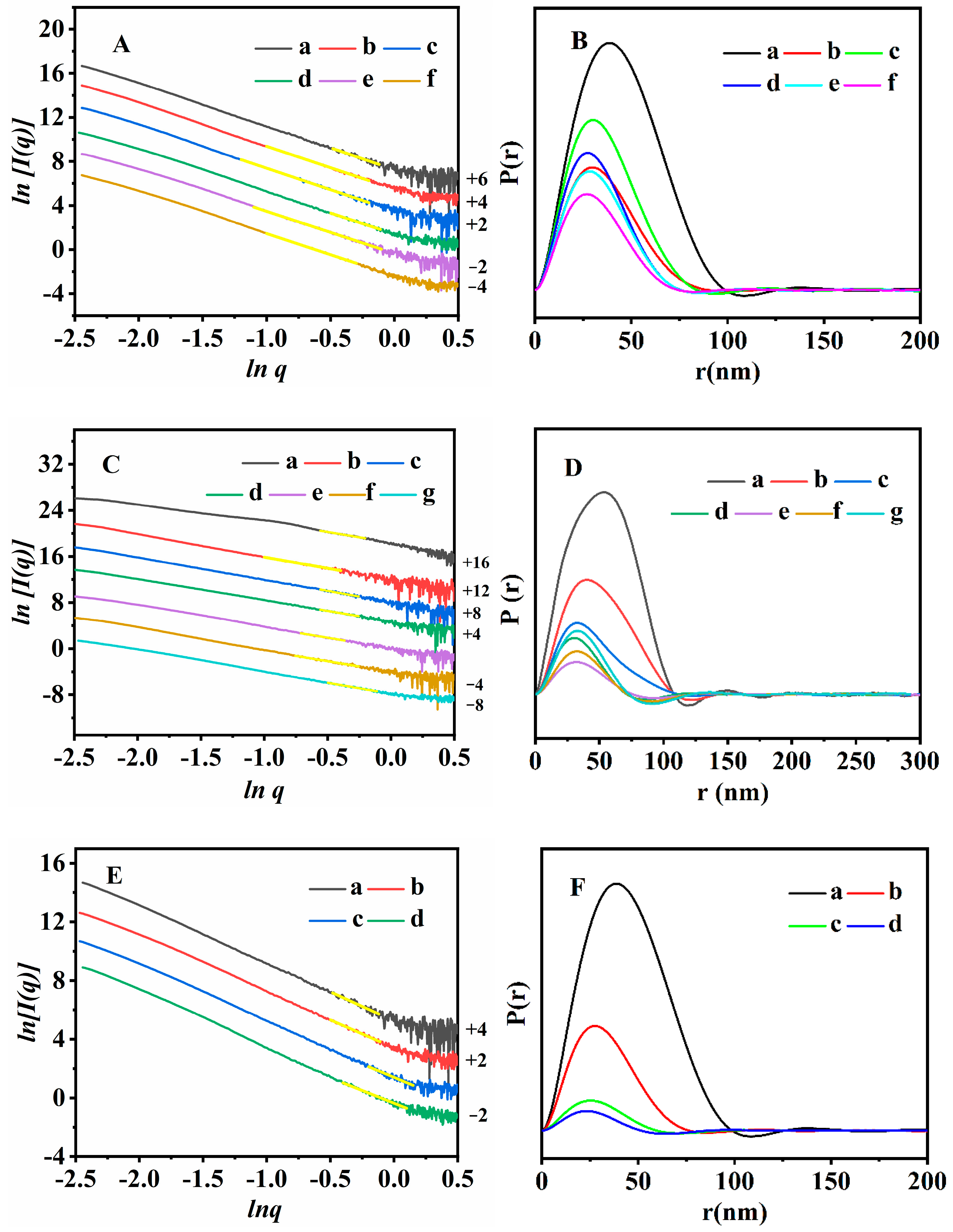
3.2. Structure and Morphology Characterizations of the Synthesized CPs
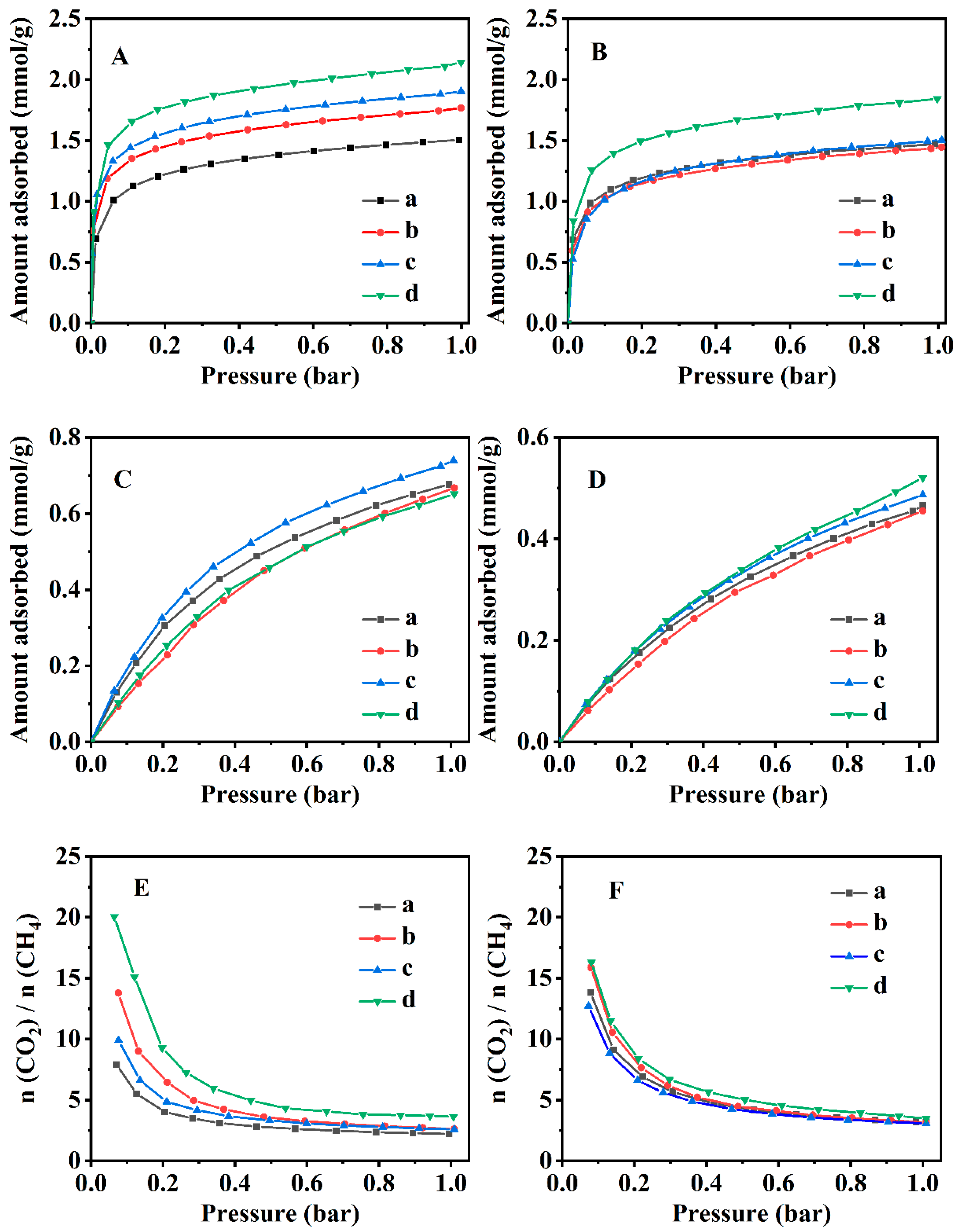
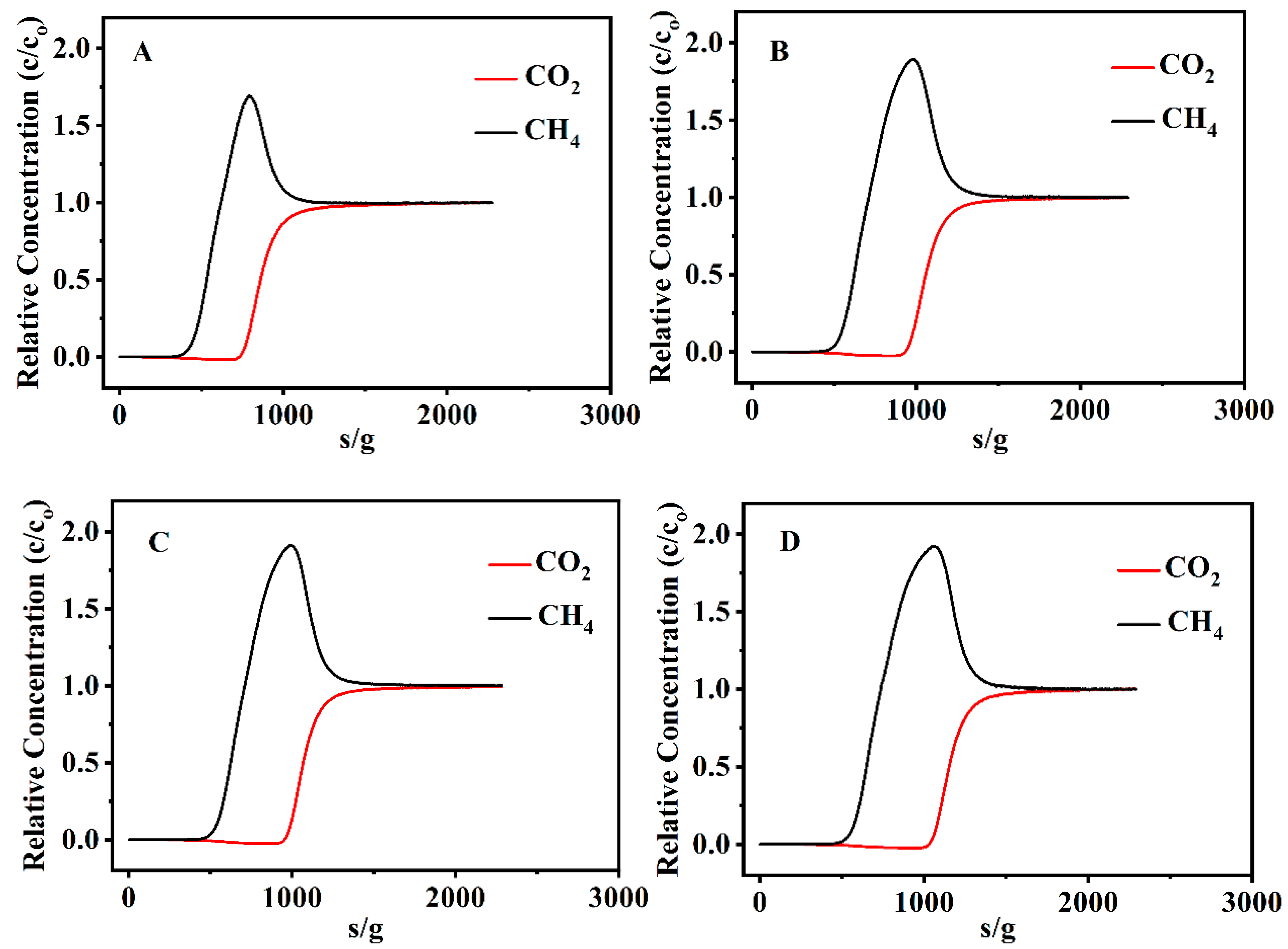
3.3. CO2 and CH4 Adsorption Performances
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alhajeri, N.S.; Dannoun, M.; Alrashed, A.; Aly, A.Z. Environmental and economic impacts of increased utilization of natural gas in the electric power generation sector: Evaluating the benefits and trade-offs of fuel switching. J. Nat. Gas. Sci. Eng. 2019, 71, 102969. [Google Scholar] [CrossRef]
- Gómez, H.O.; Calleja, M.C.; Fernández, L.A.; Kiedrzyńska, A.; Lewtak, R. Application of the CFD simulation to the evaluation of natural gas replacement by syngas in burners of the ceramic sector. Energy 2019, 185, 15–27. [Google Scholar] [CrossRef]
- Yousef, S.; Sarwar, Z.; Šereika, J.; Striūgas, N.; Krugly, E.; Danilovas, P.; Martuzevicius, D. A new industrial technology for mass production of graphene/peba membranes for CO2/CH4 selectivity with high dispersion, thermal and mechanical performance. Polymers 2020, 12, 831. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.U.; Lee, J.T.E.; Bashir, M.A.; Dissanayake, P.D.; Ok, Y.S.; Tong, Y.W.; Shariati, M.A.; Wu, S.; Ahring, B.K. Current status of biogas upgrading for direct biomethane use: A review. Renew. Sust. Energy Rev. 2021, 149, 111343. [Google Scholar] [CrossRef]
- Abad, V.; Avila, R.; Vicent, T.; Font, X. Promoting circular economy in the surroundings of an organic fraction of municipal solid waste anaerobic digestion treatment plant: Biogas production impact and economic factors. Bioresource Technol. 2019, 283, 10–17. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Li, J.; Lin, J.G.; Zhang, N.; Cao, W. Biogas energy generated from livestock manure in China: Current situation and future trends. J. Environ. Manag. 2021, 297, 113324. [Google Scholar] [CrossRef]
- Wang, X.; Yan, R.; Zhao, Y.; Cheng, S.; Han, Y.; Yang, S.; Cai, D.; Mang, H.P.; Li, Z. Biogas standard system in China. Renew. Energy 2020, 157, 1265–1273. [Google Scholar] [CrossRef]
- Sainath, K.; Kumari, P.; Bellare, J. Zeolitic imidazolate framework-8 nanoparticles coated composite hollow fiber membranes for CO2/CH4 separation. J. Environ. Chem. Eng. 2021, 9, 106052. [Google Scholar] [CrossRef]
- Naquash, A.; Qyyum, M.A.; Haider, J.; Lim, H.; Lee, M. Renewable LNG production: Biogas upgrading through CO2 solidification integrated with single-loop mixed refrigerant biomethane liquefaction process. Energy Convers. Manag. 2021, 243, 114363. [Google Scholar] [CrossRef]
- Cavaignac, R.S.; Ferreira, N.L.; Guardani, R. Techno-economic and environmental process evaluation of biogas upgrading via amine scrubbing. Renew. Energy 2021, 171, 868–880. [Google Scholar] [CrossRef]
- Wu, L.; Wei, W.; Song, L.; Woźniak-Karczewska, M.; Chrzanowski, Ł.; Ni, B.J. Upgrading biogas produced in anaerobic digestion: Biological removal and bioconversion of CO2 in biogas. Renew. Sust. Energy Rev. 2021, 150, 111448. [Google Scholar] [CrossRef]
- Muñoz, R.; Meier, L.; Diaz, I.; Jeison, D. A review on the state-of-the-art of physical/chemical and biological technologies for biogas upgrading. Rev. Environ. Sci. Bio 2015, 14, 727–759. [Google Scholar] [CrossRef] [Green Version]
- Yousef, A.M.; El-Maghlany, W.M.; Eldrainy, Y.A.; Attia, A. New approach for biogas purification using cryogenic separation and distillation process for CO2 capture. Energy 2018, 156, 328–351. [Google Scholar] [CrossRef]
- Song, C.; Liu, Q.; Deng, S.; Li, H.; Kitamura, Y. Cryogenic-based CO2 capture technologies: State-of-the-art developments and current challenges. Renew. Sust. Energy Rev. 2019, 101, 265–278. [Google Scholar] [CrossRef]
- Zhai, H. Advanced membranes and learning scale required for cost-effective post-combustion carbon capture. iScience 2019, 13, 440–451. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Ho, W.S.W. Recent advances in polymeric membranes for CO2 capture. Chin. J. Chem. Eng. 2018, 26, 2238–2254. [Google Scholar] [CrossRef]
- Xie, K.; Fu, Q.; Qiao, G.G.; Webley, P.A. Recent progress on fabrication methods of polymeric thin film gas separation membranes for CO2 capture. J. Membrane Sci. 2019, 572, 38–60. [Google Scholar] [CrossRef]
- Cozma, P.; Wukovits, W.; Mămăligă, I.; Friedl, A.; Gavrilescu, M. Modeling and simulation of high pressure water scrubbing technology applied for biogas upgrading. Clean Technol. Environ. 2015, 17, 373–391. [Google Scholar] [CrossRef]
- Nie, H.; Jiang, H.; Chong, D.; Wu, Q.; Xu, C.; Zhou, H. Comparison of water scrubbing and propylene carbonate absorption for biogas upgrading process. Energy Fuel 2013, 27, 3239–3245. [Google Scholar] [CrossRef]
- Wu, X.; Yu, Y.; Qin, Z.; Zhang, Z. The advances of post-combustion CO2 capture with chemical solvents: Review and guidelines. Energy Procedia 2014, 63, 1339–1346. [Google Scholar] [CrossRef] [Green Version]
- Mulukutla, T.; Obuskovic, G.; Sirkar, K.K. Novel scrubbing system for post-combustion CO2 capture and recovery: Experimental studies. J. Membrane Sci. 2014, 471, 16–26. [Google Scholar] [CrossRef]
- Riboldi, L.; Bolland, O. Overview on pressure swing adsorption (PSA) as CO2 capture technology: State-of-the-art, limits and potentials. Energy Procedia 2017, 114, 2390–2400. [Google Scholar] [CrossRef]
- Riboldi, L.; Bolland, O. Evaluating Pressure Swing Adsorption as a CO2 separation technique in coal-fired power plants. Int. J. Greenh. Gas Control 2015, 39, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Xu, L.; Liu, L.; Deng, X.; Gao, Y.; Ding, Z. Ultrathin porous amine-based solid adsorbent incorporated zeolitic imidazolate framework-8 membrane for gas separation. RSC Adv. 2021, 11, 28863–28875. [Google Scholar] [CrossRef]
- Tobarameekul, P.; Sangsuradet, S.; Chat, N.N.; Worathanakul, P. Enhancement of CO2 Adsorption Containing Zinc-ion-exchanged Zeolite NaA Synthesized from Rice Husk Ash. Appl. Sci. Eng. Prog. 2022, 15, 3640. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Lv, C.; Wu, T.; Wen, Y.; He, H.; Yu, S.; Wang, L. Adsorption Separation of CO2/CH4 from Landfill Gas by Ethanolamine-Modified Silica Gel. Water Air Soil Pollut. 2021, 232, 1–11. [Google Scholar] [CrossRef]
- El Oufir, Z.; Ramézani, H.; Mathieu, N.; Delpeux, S. Impact of adsorbent carbons and carbon surface conductivity on adsorption capacity of CO2, CH4, N2 and gas separation. Comp. Mater. Sci. 2021, 199, 110572. [Google Scholar] [CrossRef]
- Sneddon, G.; Greenaway, A.; Yiu, H.H.P. The potential applications of nanoporous materials for the adsorption, separation, and catalytic conversion of carbon dioxide. Adv. Eenergy Mater. 2014, 4, 1301873. [Google Scholar] [CrossRef] [Green Version]
- Zagho, M.M.; Hassan, M.K.; Khraisheh, M.; Al-Maadeed, M.A.A.; Nazarenko, S. A Review on Recent Advances in CO2 Separation Using Zeolite and Zeolite-like Materials as Adsorbents and Fillers in Mixed Matrix Membranes (MMMs). Chem. Eng. J. Adv. 2021, 232, 100091. [Google Scholar] [CrossRef]
- Maheshwari, S.; Jordan, E.; Kumar, S.; Bates, F.S.; Penn, R.L.; Shantz, D.F.; Tsapatsis, M. Layer structure preservation during swelling, pillaring, and exfoliation of a zeolite precursor. J. Am. Chem. Soc. 2008, 130, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Shetti, V.N.; Kim, J.; Srivastava, R.; Choi, M.; Ryoo, R. Assessment of the mesopore wall catalytic activities of MFI zeolite with mesoporous/microporous hierarchical structures. J. Catal. 2008, 254, 296–303. [Google Scholar] [CrossRef]
- Runnebaum, R.C.; Ouyang, X.; Edsinga, J.A.; Rea, T.; Arslan, I.; Hwang, S.J.; Zones, S.I.; Katz, A. Role of delamination in zeolite-catalyzed aromatic alkylation: UCB-3 versus 3-D Al-SSZ-70. ACS Catal. 2014, 4, 2364–2368. [Google Scholar] [CrossRef] [Green Version]
- Corma, A.; Fornes, V.; Pergher, S.B.; Maesen, T.L.; Buglass, J.G. Delaminated zeolite precursors as selective acidic catalysts. Nature 1998, 396, 353–356. [Google Scholar] [CrossRef]
- Choi, S.; Coronas, J.; Jordan, E.; Oh, W.; Nair, S.; Onorato, F.; Shantz, D.F.; Tsapatsis, M. Layered silicates by swelling of AMH-3 and nanocomposite membranes. Angew. Chem. Int. Ed. 2008, 47, 552–555. [Google Scholar] [CrossRef]
- Eilertsen, E.A.; Ogino, I.; Hwang, S.J.; Rea, T.; Yeh, S.; Zones, S.I.; Katz, A. Nonaqueous fluoride/chloride anion-promoted delamination of layered zeolite precursors: Synthesis and characterization of UCB-2. Chem. Mater. 2011, 23, 5404–5408. [Google Scholar] [CrossRef] [Green Version]
- Ogino, I.; Eilertsen, E.A.; Hwang, S.J.; Rea, T.; Xie, D.; Ouyang, X.; Zones, S.I.; Katz, A. Heteroatom-tolerant delamination of layered zeolite precursor materials. Chem. Mater. 2013, 25, 1502–1509. [Google Scholar] [CrossRef] [Green Version]
- Cakicioglu-Ozkan, F.; Ulku, S. The effect of HCl treatment on water vapor adsorption characteristics of clinoptilolite rich natural zeolite. Micropor. Mesopor. Mat. 2005, 77, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Sun, J.; Munir, T.; Jia, B.; Gul, A. Various morphologies of clinoptilolites synthesized in alcohol-solvent hydrothermal system and their selective adsorption of CH4 and N2. Micropor. Mesopor. Mat. 2021, 323, 111235. [Google Scholar] [CrossRef]
- Pour, A.A.; Sharifnia, S.; NeishaboriSalehi, R.; Ghodrat, M. Performance evaluation of clinoptilolite and 13X zeolites in CO2 separation from CO2/CH4 mixture. J. Nat. Gas Sci. Eng. 2015, 26, 1246–1253. [Google Scholar] [CrossRef]
- Kennedy, D.A.; Khanafer, M.; Tezel, F.H. The effect of Ag+ cations on the micropore properties of clinoptilolite and related adsorption separation of CH4 and N2 gases. Micropor. Mesopor. Mat. 2019, 281, 123–133. [Google Scholar] [CrossRef]
- Li, Z.H.; Gong, Y.J.; Pu, M.; Wu, D.; Sun, Y.H.; Wang, J.; Liu, Y.; Dong, B.Z. Determination of interface layer thickness of a pseudo two-phase system by extension of the Debye equation. J. Phys. D Appl. Phys. 2001, 34, 2085. [Google Scholar] [CrossRef]
- Li, Z.; Wu, Z.; Mo, G.; Xing, X.; Liu, P. A small-angle X-ray scattering station at Beijing synchrotron radiation facility. Instrum. Sci. Technol. 2014, 42, 128–141. [Google Scholar] [CrossRef]
- Hammersley, A.P. FIT2D: A multi-purpose data reduction, analysis and visualization program. J. Appl. Crystallogr. 2016, 49, 646–652. [Google Scholar] [CrossRef]
- Roth, W.J.; Dorset, D.L.; Kennedy, G.J. Discovery of new MWW family zeolite EMM-10: Identification of EMM-10P as the missing MWW precursor with disordered layers. Micropor. Mesopor. Mat. 2011, 142, 168–177. [Google Scholar] [CrossRef]
- Zhou, Y.; Mu, Y.; Hsieh, M.F.; Kabius, B.; Pacheco, C.; Bator, C.; Rioux, R.M.; Rimer, J.D. Enhanced surface activity of MWW zeolite nanosheets prepared via a one-step synthesis. J. Am. Chem. Soc. 2020, 142, 8211–8222. [Google Scholar] [CrossRef]
- Álvaro-Muñoz, T.; Márquez-Álvarez, C.; Sastre, E. Enhanced stability in the methanol-to-olefins process shown by SAPO-34 catalysts synthesized in biphasic medium. Catal. Today 2013, 215, 208–215. [Google Scholar] [CrossRef]
- Jiao, J.; Sun, J.; Ullah, R.; Bai, S.; Zhai, C. One-step synthesis of hydrophobic clinoptilolite modified by silanization for the degradation of crystal violet dye in aqueous solution. RSC Adv. 2020, 10, 22809–22818. [Google Scholar] [CrossRef]
- Shalmani, F.M.; Halladj, R.; Askari, S. An investigation of the crystallization kinetics of zeotype SAPO-34 crystals synthesized by hydrothermal and sonochemical methods. Ultrason. Sonochem. 2016, 29, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Niphadkar, P.S.; Tangale, N.P.; Joshi, P.N.; Awate, S.V. Crystallization kinetics of Sn-MFI molecular sieve formation by dry gel conversion method. Microporous Mesoporous Mater. 2013, 182, 73–80. [Google Scholar] [CrossRef]
- Svergun, D.I.; Koch, M.H.J. Small-angle scattering studies of biological macromolecules in solution. Rep. Prog. Phys. 2003, 66, 1735. [Google Scholar] [CrossRef]
- Zhai, C.; Sun, J.; Jia, B.; Gul, A.; Bai, S. A nanoprecursor method for successfully synthesizing clinoptilolite with high-crystallinity and resultant effects on CO2/CH4 selective adsorption. RSC Adv. 2021, 11, 30646–30656. [Google Scholar] [CrossRef] [PubMed]
- Narin, G.; Balköse, D.; Ülkü, S. Characterization and dehydration behavior of a natural, ammonium hydroxide, and thermally treated zeolitic tuff. Drying Technol. 2011, 29, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Corral-Capulin, N.G.; Vilchis-Nestor, A.R.; Gutiérrez-Segura, E.; Solache-Ríos, M. The influence of chemical and thermal treatments on the fluoride removal from water by three mineral structures and their characterization. J. Fluorine Chem. 2018, 213, 42–50. [Google Scholar] [CrossRef]
- Sheng, M.; Ouyang, T.; Sun, J.; Bai, S.; Wu, X. Fractal evolution of aluminosilicate sol and resulting effects on the synthesis of clinoptilolite via small angle X-ray scattering investigation. Mater. Chem. Phys. 2021, 263, 124335. [Google Scholar] [CrossRef]
- Craievich, A.F. Synchrotron SAXS studies of nanostructured materials and colloidal solutions: A review. Mater. Res. 2002, 5, 1–11. [Google Scholar] [CrossRef]
- Ramdas, S.; Klinowski, J. A simple correlation between isotropic 29Si-NMR chemical shifts and T-O-T angles in zeolite frameworks. Nature 1984, 308, 521–523. [Google Scholar] [CrossRef]
- Rivera, A.; Farıas, T.; Ruiz-Salvador, A.R.; De Ménorval, L.C. Preliminary characterization of drug support systems based on natural clinoptilolite. Microporous Mesoporous Mater. 2003, 61, 249–259. [Google Scholar] [CrossRef]
- Engelhardt, G.; Michel, D. High-Resolution Solid-State NMR of Silicates and Zeolites; Wiley: New York, NY, USA, 1987. [Google Scholar]
- Al-Yassir, N.; Akhtar, M.N.; Al-Khattaf, S. Physicochemical properties and catalytic performance of galloaluminosilicate in aromatization of lower alkanes: A comparative study with Ga/HZSM-5. J. Porous Mater. 2012, 19, 943–960. [Google Scholar] [CrossRef]
- Schmidt, J.E.; Xie, D.; Davis, M.E. High-silica, heulandite-type zeolites prepared by direct synthesis and topotactic condensation. J. Mater. Chem. A 2015, 3, 12890–12897. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.E.; Xie, D.; Davis, M.E. Synthesis of the RTH-type layer: The first small-pore, two dimensional layered zeolite precursor. Chem. Sci. 2015, 6, 5955–5963. [Google Scholar] [CrossRef] [Green Version]
- Roth, W.J.; Shvets, O.V.; Shamzhy, M.; Chlubna, P.; Kubu, M.; Nachtigall, P.; Cejka, J. Postsynthesis transformation of three-dimensional framework into a lamellar zeolite with modifiable architecture. J. Am. Chem. Soc. 2011, 133, 6130–6133. [Google Scholar] [CrossRef]
- Grzybek, J.; Roth, W.J.; Gil, B.; Korzeniowska, A.; Mazur, M.; Čejka, J.; Morris, R.E. A new layered MWW zeolite obtained by direct synthesis with the bifunctional surfactant template. J. Mater. Chem. A 2019, 7, 7701–7709. [Google Scholar] [CrossRef]
- Cai, X.; Luo, Y.; Liu, B.; Cheng, H.M. Preparation of 2D material dispersions and their applications. Chem. Soc. Rev. 2018, 47, 6224–6266. [Google Scholar] [CrossRef] [PubMed]
- Baerlocher, C.; McCusker, L.B.; Meier, W.M.; Olson, D.H. Atlas of Zeolite Framework Types, 6th ed.; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Yörükoğulları, E.; Yılmaz, G.; Dikmen, S. Thermal treatment of zeolitic tuff. J. Therm. Anal. Calorim. 2010, 100, 925–928. [Google Scholar] [CrossRef]
- Peng, S.; Raza, U.; Bai, S.; Sun, J.; Wu, X. Hydrothermal Synthesis of Clinoptilolite and its Ion Exchange Performance for CH4/N2 Separation. Acta Pet. Sin. (Pet. Process. Sect.) 2019, 35, 348–358. [Google Scholar]
- Assar, E.; Meysami, A.; Zare, M. Morphology analysis and characterization of clinoptilolite/polyvinylpyrrolidone-zeolite composite nanofibers. J. Mater. Eng. Perform. 2020, 29, 4233–4240. [Google Scholar] [CrossRef]
- Wu, Q.; Zhou, Y.; Wang, T. Effects of Formic Acid on the Adsorption of Escherichia Coli K88 on Modified Clinoptilolite. Adsorpt. Sci. Technol. 2013, 31, 671–681. [Google Scholar] [CrossRef]
- Epiepang, F.E.; Li, J.; Liu, Y.; Yang, R.T. Low-pressure performance evaluation of CO2, H2O and CH4 on Li-LSX as a superior adsorbent for air prepurification. Chem. Eng. Sci. 2016, 147, 100–108. [Google Scholar] [CrossRef]
- Wei, Y.; Li, X.M.; Zhang, R.; Liu, Y.; Wang, W.X.; Ling, Y.; El-Toni, A.M.; Zhao, D.Y. Periodic mesoporous organosilica nanocubes with ultrahigh surface areas for efficient CO2 adsorption. Sci. Rep. 2016, 6, 20769. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Zhou, J.; Xian, X.; Zhang, C.; Tian, S.; Dong, Z.; Fan, M.; Cai, J. Adsorption characteristics and thermodynamic analysis of CH4 and CO2 on continental and marine shale. Transp. Porous Media 2021, 140, 763–788. [Google Scholar] [CrossRef]
- Jia, B.; Xu, B.; Bing, L.; Sun, J.; Bai, S. Cationic surfactant-assisted delamination of disorderly layered clinoptilolites for selective adsorption of CO2 and CH4. J. Environ. Chem. Eng. 2022, 10, 108033. [Google Scholar] [CrossRef]
- Zhang, K.; Meng, Z.; Liu, S. Comparisons of methane adsorption/desorption, diffusion behaviors on intact coals and deformed coals: Based on experimental analysis and isosteric heat of adsorption. Energy Fuels 2021, 35, 5975–5987. [Google Scholar] [CrossRef]
- Wu, Y.; Fan, T.; Jiang, S.; Yang, X.; Ding, H.; Meng, M.; Duan, W. Methane adsorption capacities of the lower paleozoic marine shales in the Yangtze Platform, South China. Energy Fuels 2015, 29, 4160–4167. [Google Scholar] [CrossRef]
- Deng, H.; Yi, H.; Tang, X.; Yu, Q.; Ning, P.; Yang, L. Adsorption equilibrium for sulfur dioxide, nitric oxide, carbon dioxide, nitrogen on 13X and 5A zeolites. Chem. Eng. J. 2012, 188, 77–85. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | T (°C) | Induction Period | Growth Period | ||||
|---|---|---|---|---|---|---|---|
| En (kJ/mol) | lnAn | t0 (h) | kmax | Eg (kJ/mol) | lnAg | ||
| CP | 140 | 61.5 | 13.7 | 62 | 1.7 | 17.5 | 5.5 |
| 150 | 28 | 1.9 | |||||
| 170 | 18 | 2.4 | |||||
| CP-1,2-DMB-0.03 | 140 | 41.0 | 8.3 | 34 | 1.5 | 19.5 | 6.0 |
| 150 | 22 | 1.7 | |||||
| 170 | 15 | 2.2 | |||||
| CP-1,2-DMB-0.06 | 140 | 21.0 | 2.8 | 26 | 2.7 | 17.5 | 6.1 |
| 150 | 19 | 3.1 | |||||
| 170 | 17 | 3.8 | |||||
| CP-1,2-DMB-0.09 | 140 | 18.0 | 2.2 | 20 | 3.1 | 16.0 | 5.8 |
| 150 | 17 | 3.6 | |||||
| 170 | 14 | 4.3 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, J.; Jia, B.; Xu, B.; Sun, J.; Bai, S. Amphipathic Solvent-Assisted Synthetic Strategy for Random Lamellae of the Clinoptilolites with Flower-like Morphology and Thinner Nanosheet for Adsorption and Separation of CO2 and CH4. Nanomaterials 2023, 13, 1942. https://doi.org/10.3390/nano13131942
Zhou J, Jia B, Xu B, Sun J, Bai S. Amphipathic Solvent-Assisted Synthetic Strategy for Random Lamellae of the Clinoptilolites with Flower-like Morphology and Thinner Nanosheet for Adsorption and Separation of CO2 and CH4. Nanomaterials. 2023; 13(13):1942. https://doi.org/10.3390/nano13131942
Chicago/Turabian StyleZhou, Jiawei, Bingying Jia, Bang Xu, Jihong Sun, and Shiyang Bai. 2023. "Amphipathic Solvent-Assisted Synthetic Strategy for Random Lamellae of the Clinoptilolites with Flower-like Morphology and Thinner Nanosheet for Adsorption and Separation of CO2 and CH4" Nanomaterials 13, no. 13: 1942. https://doi.org/10.3390/nano13131942