
Au-Nanorods Supporting Pd and Pt Nanocatalysts for the Hydrogen Evolution Reaction: Pd Is Revealed to Be a Better Catalyst than Pt


Abstract
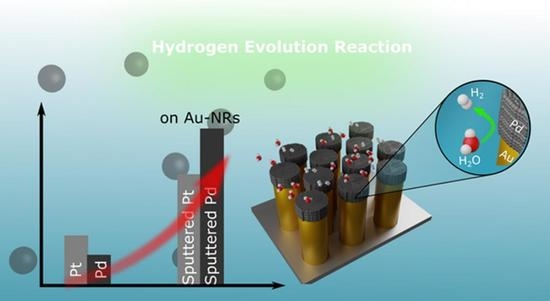
:
1. Introduction
2. Materials and Methods
2.1. Synthesis and Characterization
2.2. Computational Methods
3. Results
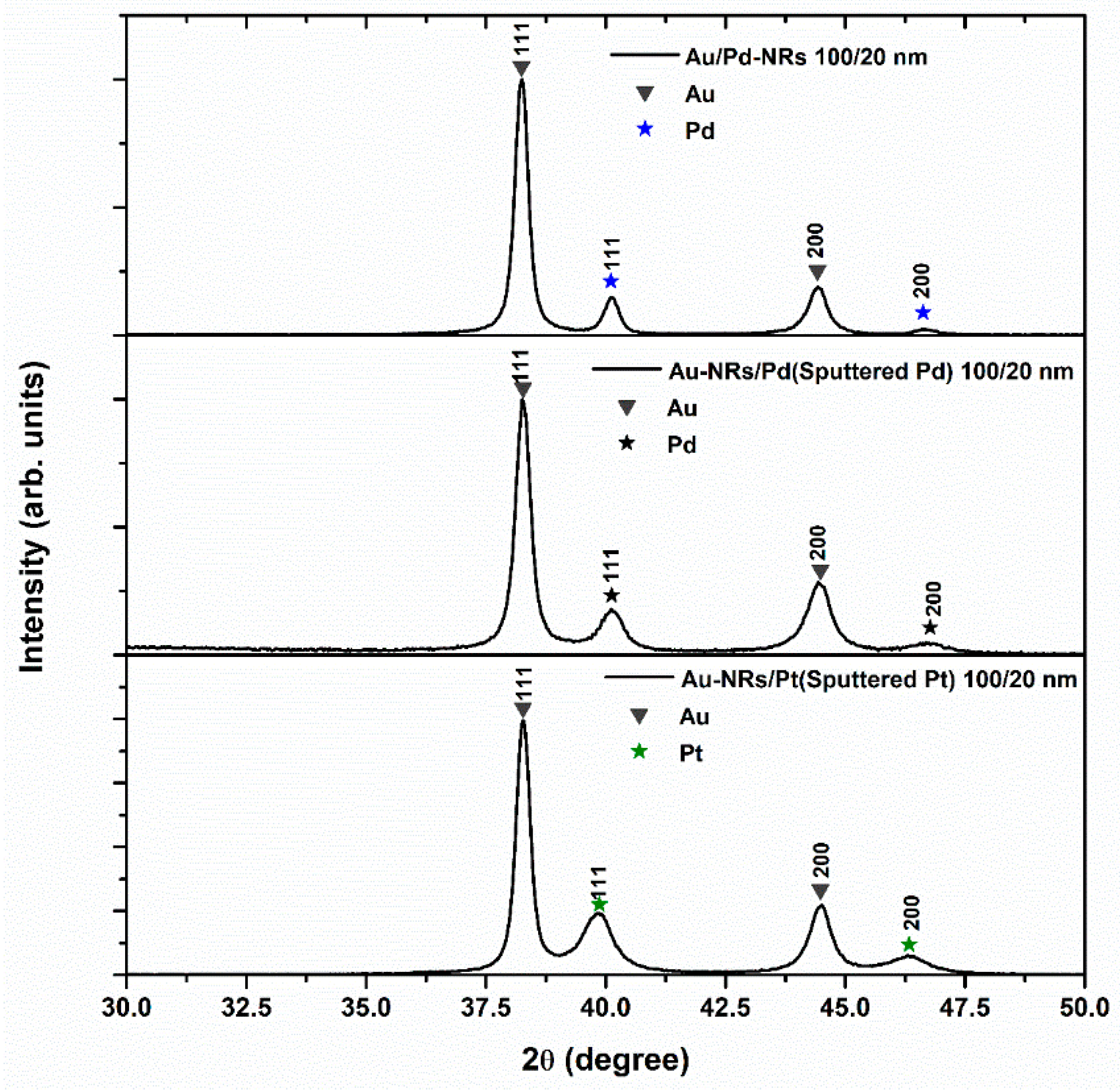
3.1. Structure and Morphology
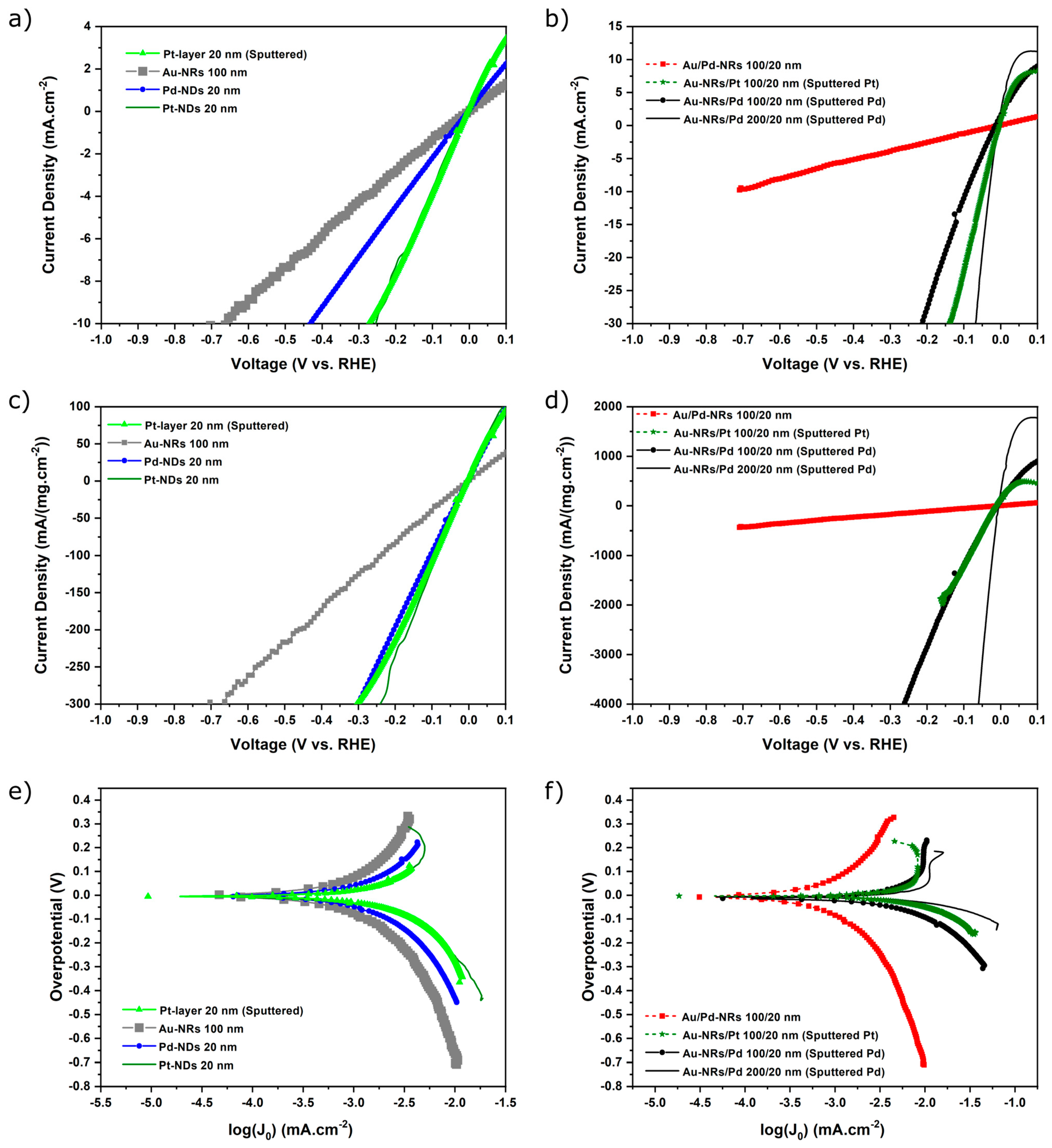
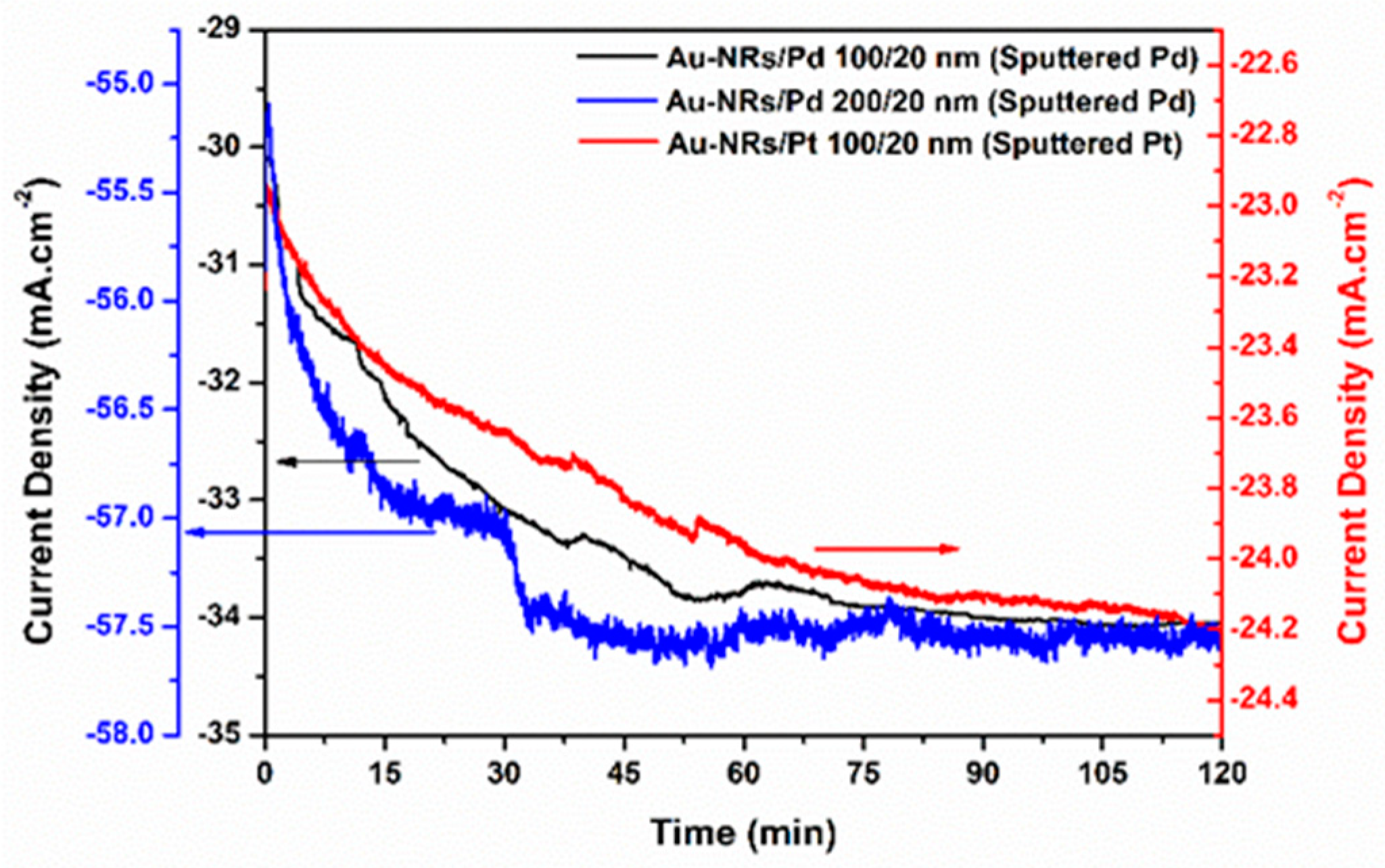
3.2. Electrochemical Investigations, HER
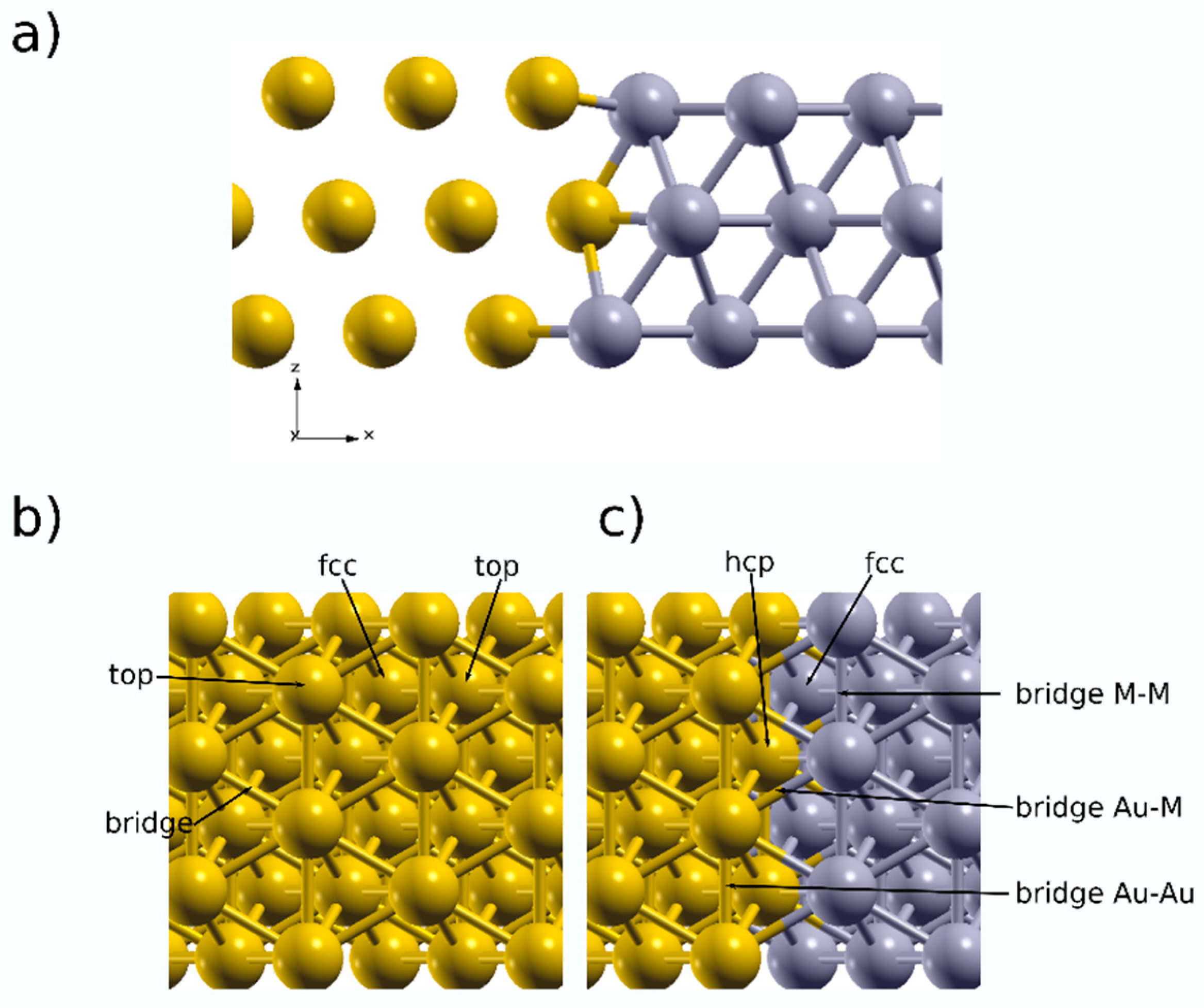
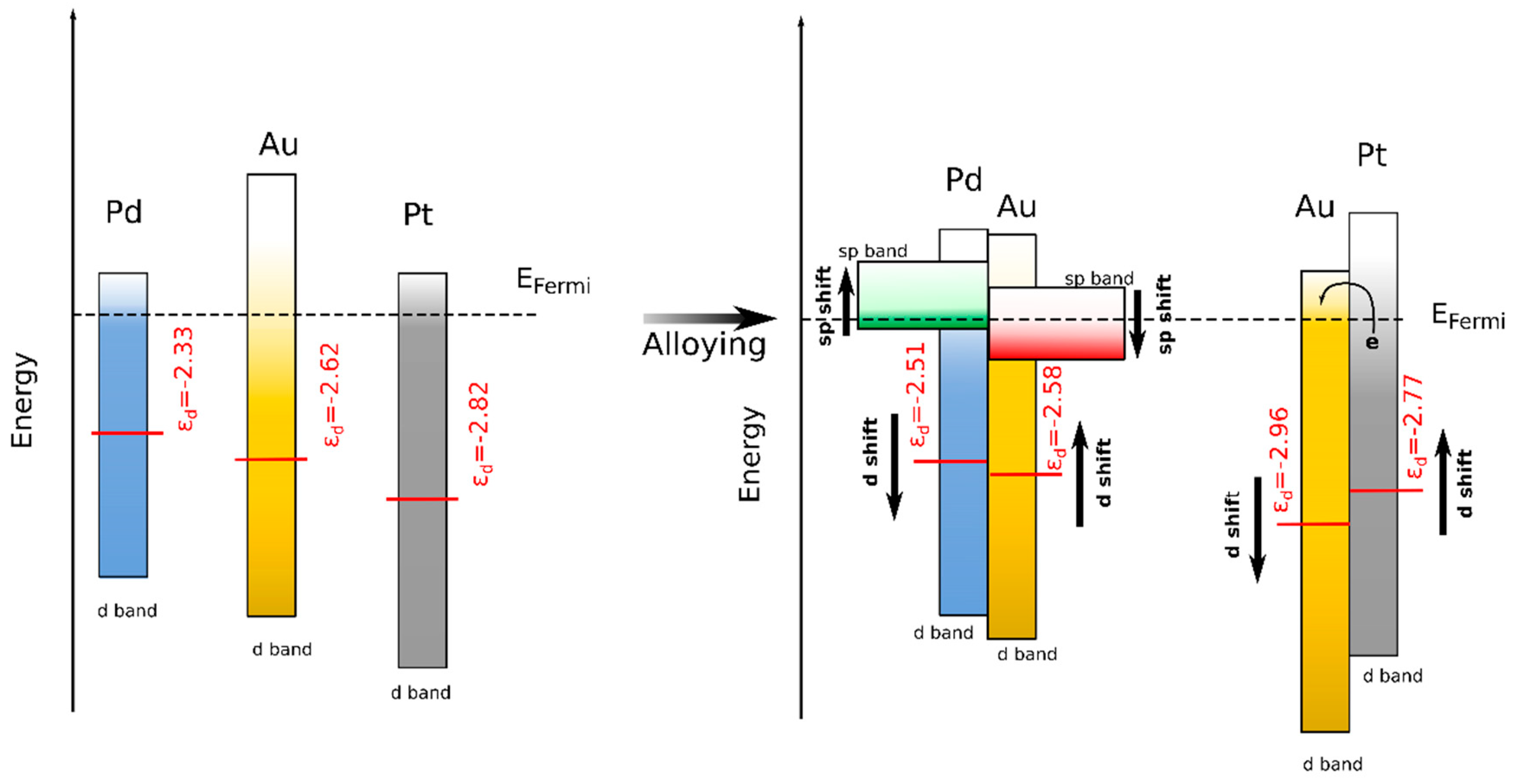
3.3. DFT Calculations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hohmeyer, O.H.; Bohm, S. Trends toward 100% renewable electricity supply in Germany and Europe: A paradigm shift in energy policies. WIREs Energy Environ. 2015, 4, 74–97. [Google Scholar] [CrossRef]
- Dincer, I.; Acar, C. Innovation in hydrogen production. Int. J. Hydrogen Energy 2017, 42, 14843–14864. [Google Scholar] [CrossRef]
- Fastmarkets. Available online: https://www.metalbulletin.com/lithium-prices-update (accessed on 1 June 2020).
- Conway, B.E.; Tilak, B.V. Interfacial processes involving electrocatalytic evolution and oxidation of H2, and the role of chemisorbed H. Electrochim. Acta 2002, 47, 3571–3594. [Google Scholar] [CrossRef]
- Bockris, J.O.; Conway, B.E. Modern Aspects of Electrochemistry; Butterworths Scientific Publications: London, UK, 1954. [Google Scholar]
- Eley, D.D.; Pines, H.; Weisz, P.B. Advance in Catalysis; Academic Press Inc.: Bronx, NY, USA, 1992. [Google Scholar]
- Bhardwaj, M.; Balasubramaniam, R. Uncoupled non-linear equations method for determining kinetic parameters in case of hydrogen evolution reaction following Volmer–Heyrovsky–Tafel mechanism and Volmer–Heyrovsky mechanism. Int. J. Hydrogen Energy 2008, 33, 2178–2188. [Google Scholar] [CrossRef]
- Eftikhari, A. Electrocatalysts for hydrogen evolution reaction. Int. J. Hydrogen Energy 2017, 42, 11053–11077. [Google Scholar] [CrossRef]
- Hernandez, F.; Baltruschat, H. Hydrogen Evolution and Cu UPD at Stepped Gold Single Crystals Modified with Pd. J. Solid State Electrochem. 2007, 11, 877–885. [Google Scholar] [CrossRef]
- Steidtner, J.; Hernandez, F.; Baltruschat, H. Electrocatalytic Reactivity of Pd Monolayers and Monatomic Chains on Au. J. Phys. Chem. C 2007, 111, 12320–12327. [Google Scholar] [CrossRef]
- Kibler, L.A. Dependence of Electrocatalytic Activity on Film Thickness for the Hydrogen Evolution Reaction of Pd Overlayers on Au(1 1 1). Electrochim. Acta 2008, 53, 6824–6828. [Google Scholar] [CrossRef]
- Pluntke, Y.; Kibler, L.A.; Kolb, D.M. Unique Activity of Pd Monomers: Hydrogen Evolution at AuPd(111) Surface Alloys. Phys. Chem. Chem. Phys. 2008, 10, 3684–3688. [Google Scholar] [CrossRef]
- McKone, J.R.; Warren, E.L.; Bierman, M.J.; Boettcher, S.W.; Brunschwig, B.S.; Lewis, N.S.; Gray, H.B. Evaluation of Pt, Ni, and Ni–Mo electrocatalysts for hydrogen evolution on crystalline Si electrodes. Energy Environ. Sci. 2011, 4, 3573–3583. [Google Scholar] [CrossRef] [Green Version]
- Voiry, D.; Salehi, M.; Silva, R.; Fujita, T.; Chen, M.; Asefa, T.; Shenoy, V.B.; Eda, G.; Chhowalla, M. Conducting MoS2 Nanosheets as Catalysts for Hydrogen Evolution Reaction. Nano Lett. 2013, 13, 6222–6227. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, Y.; Zhu, Y.; Li, L.H.; Han, Y.; Chen, Y.; Du, A.; Jaroniec, M.; Qiao, S.Z. Hydrogen evolution by a metal-free electrocatalyst. Nat. Commun. 2014, 5, 3783. [Google Scholar] [CrossRef] [Green Version]
- Laghrissi, A.; Es-Souni, M. Layered Au-Pd-Au nanorod catalysts: Pd-layer thickness effects on catalyst performance. Int. J. Hydrogen Energy 2019, 44, 14918–14926. [Google Scholar] [CrossRef]
- Es-Souni, M.; Habouti, S. Ordered nanomaterial thin films via supported anodized alumina templates. Front. Mater. 2014, 1, 19. [Google Scholar] [CrossRef] [Green Version]
- Berger, N.; Es-Souni, M. Understanding and shaping the morphology of the barrier layer of supported porous anodized alumina on gold underlayer. Langmuir 2016, 32, 6985–6990. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter. 2009, 21, 395502. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 2006, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Kiani, A.; Hatami, S. Fabrication of platinum coated nanoporous gold film electrode: A nanostructured ultra low-platinum loading electrocatalyst for hydrogen evolution reaction. Int. J. Hydrogen Energy 2010, 35, 5202–5209. [Google Scholar] [CrossRef]
- Kundu, M.K.; Bhowmik, T.; Barman, S. Gold aerogel supported on graphitic carbon nitride: An efficient electrocatalyst for oxygen reduction reaction and hydrogen evolution reaction. J. Mater. Chem. A 2015, 3, 23120–23135. [Google Scholar] [CrossRef]
- Durst, J.; Siebel, A.; Simon, C.; Hasche, F.; Herranz, J.; Gasteiger, H.A. New insights into the electrochemical hydrogen oxidation and evolution reaction mechanism. Energy Environ. Sci. 2014, 7, 2255–2260. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.J.; Chen, L.X.; Song, P.; Wu, X.; Wang, A.J.; Yuan, J. Bimetallic AuPd nanoclusters supported on graphitic carbon nitride: One-pot synthesis and enhanced electrocatalysis for oxygen reduction and hydrogen evolution. Int. J. Hydrogen Energy 2016, 41, 8839–88469. [Google Scholar] [CrossRef]
- Conway, B.E.; Bai, L. Determination of adsorption of OPD H in the cathodic hydrogen evolution reaction at Pt in relation to electrocatalysis. J. Electroanal. Chem. Interf. Electrochem. 1986, 198, 149–175. [Google Scholar] [CrossRef]
- Shinagawa, T.; Garcia-Esparza, A.T.; Takanabe, K. Insight on Tafel slopes from a microkinetic analysis of aqueous electrocatalysis for energy conversion. Sci. Rep. 2015, 5, 13801. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.Y.; Wu, C.X.; Feng, X.J.; Tan, H.Q.; Yan, L.K.; Liu, Y.; Kang, Z.H.; Wang, E.B.; Li, Y.G. Highly efficient hydrogen evolution from seawater by a low-cost and stable CoMoP@C electrocatalyst superior to Pt/C. Energy Environ. Sci. 2017, 10, 788–798. [Google Scholar] [CrossRef]
- Couturier, G.; Kirk, D.W. Electrocatalysis of the hydrogen oxidation and of the oxygen reduction reaction on Pt and some alloys in alkaline medium. Electrochim. Acta 1987, 32, 995–1005. [Google Scholar] [CrossRef]
- Markovic, N.M.; Grgur, B.N.; Ross, P.N. Temperature-dependent hydrogen electrochemistry on platinum low-index single-crystal surfaces in acidic solutions. J. Phys. Chem. 1997, 101, 5405–5413. [Google Scholar] [CrossRef]
- Yan, M.; Pan, X.; Wang, P.; Chen, F.; He, L.; Jiang, G.; Wang, J.; Liu, J.Z.; Xu, X.; Liao, X.; et al. Field-Effect Tuned Adsorption Dynamics of VSe2 Nanosheets for Enhanced Hydrogen Evolution Reaction. Nano Lett. 2017, 17, 4109–4115. [Google Scholar] [CrossRef] [PubMed]
- Ferrin, P.; Kandoi, S.; Nilekar, A.U.; Mavrikakis, M. Hydrogen adsorption, absorption and diffusion on and in transition metal surfaces: A DFT study. Surf. Sci. 2012, 606, 679–689. [Google Scholar] [CrossRef]
- Mattsson, A.E.; Armiento, R.; Schultz, P.A.; Mattsson, T.R. Nonequivalence of the generalized gradient approximations PBE and PW91. Phys. Rev. B 2006, 73, 195123. [Google Scholar] [CrossRef] [Green Version]
- Watson, G.Q.; Wells, R.P.K.; Willock, D.J.; Hutchings, G.J. A Comparison of the Adsorption and Diffusion of Hydrogen on the {111} Surfaces of Ni, Pd, and Pt from density functional theory calculations. J. Phys. Chem. B 2001, 105, 4889–4894. [Google Scholar] [CrossRef]
- Hu, M.; Linder, D.P.; Nardelli, M.G.; Striolo, A. Hydrogen adsorption on Platinum−Gold bimetallic nanoparticles: A density functional theory study. J. Phys. Chem. C 2013, 117, 15050–15060. [Google Scholar] [CrossRef] [Green Version]
- Mott, D.; Luo, J.; Smith, A.; Njoki, P.N.; Wang, L.; Zhong, C.J. Nanocrystal and surface alloy properties of bimetallic Gold-Platinum nanoparticles. Nano Express 2007, 2, 12–16. [Google Scholar] [CrossRef] [Green Version]
- Mokkath, J.H.; Schwingenschlögl, U. Catalytically Favorable Surface Patterns in Pt–Au Nanoclusters. RSC Adv. 2013, 3, 15350–15353. [Google Scholar] [CrossRef]
- Lee, Y.S.; Jeon, A.; Chung, Y.D.; Lim, K.Y.; Whang, C.N.; Oh, S.J. Charge redistribution and electronic behavior in Pd-Au alloys. J. Korean Phys. Soc. 2000, 37, 451. [Google Scholar]
- Lischka, M.; Groß, A. Hydrogen adsorption on an open metal surface: H2/Pd(210). Phys. Rev. B 2002, 65, 075420. [Google Scholar] [CrossRef] [Green Version]
- Hammer, B.; Norskov, J.K. Why gold is the noblest of all the metals. Nature 1995, 376, 238–240. [Google Scholar] [CrossRef]
- Hammer, B.; Norskov, J.K. Electronic factors determining the reactivity of metal surfaces. Surf. Sci. 1995, 343, 211–220. [Google Scholar] [CrossRef]
- Kibler, L.A.; El-Aziz, A.M.; Hoyer, R.; Kolb, D.M. Tuning Reaction Rates by Lateral Strain in a Palladium Monolayer. Angew. Chem. Int. Ed. 2005, 44, 2080–2084. [Google Scholar] [CrossRef]
- Calvo, F. Thermodynamics of Nanoalloys. Phys. Chem. Chem. Phys. 2015, 17, 27922–27939. [Google Scholar] [CrossRef]
- Eom, N.; Messing, M.E.; Johansson, J.; Deppert, K. General Trends in Core–Shell Preferences for Bimetallic Nanoparticles. ACS Nano 2021, 15, 8883–8895. [Google Scholar] [CrossRef]
- Wang, Y.; Hou, M. Ordering of Bimetallic Nanoalloys Predicted from Bulk Alloy Phase Diagrams. J. Phys. Chem. C 2012, 116, 10814–10818. [Google Scholar] [CrossRef]
- Chen, R.; Yang, C.; Cai, W.; Wang, H.-Y.; Miao, J.; Zhang, L.; Chen, S.; Liu, B. Use of Platinum as the Counter Electrode to Study the Activity of Nonprecious Metal Catalysts for the Hydrogen Evolution Reaction. ACS Energy Lett. 2017, 2, 1070–1075. [Google Scholar] [CrossRef]
- Tian, M.; Cousins, C.; Beauchemin, D.; Furuya, Y.; Ohma, A.; Jerkiewicz, G. Influence of the Working and Counter Electrode Surface Area Ratios on the Dissolution of Platinum under Electrochemical Conditions. ACS Catal. 2016, 6, 5108–5116. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst Loading (mg/cm2) | Current Density at −0.1 V (mA/cm2) | Current Density at −0.1 V (mA/(mg·cm−2)) | Tafel Slope (mV/dec) | Exchange Current Density log(J0) (mA/cm2) | |
|---|---|---|---|---|---|
| Pt-layer 20 nm (Sputtered) | 0.063 | −3.88 | −107.6 | 57 | −3.61 |
| Au-NRs 100 nm | 0.044 | −1.24 | −37.3 | 139 | −3.56 |
| Pt-NDs 20 nm | 0.041 | −3.69 | −109.9 | 53 | −3.51 |
| Pd-NDs 20 nm | 0.031 | −2.19 | −95.6 | 96 | −3.63 |
| Au/Pd NRs 100/20 nm | 0.029 | −1.58 | −55.69 | 94 | −3.80 |
| Au-NRs/Pt (Sputtered) 100/20 nm | 0.022 | −22.12 | −1249.4 | 24 | −3.34 |
| Au-NRs/Pd (Sputtered) 100/20 nm | 0.013 | −11.36 | −1205.3 | 37 | −3.58 |
| Au-NRs/Pd (Sputtered) 200/20 nm | 0.008 | −40.21 | −9327.2 | 26 | −3.11 |
| Eads (eV)/dH-S (A) | ||||
|---|---|---|---|---|
| Top | Bridge | Fcc | Hcp | |
| Au | −1.57/1.60 | −1.75/0.99 | −1.81/0.81 | −1.62/1.08 |
| Pt | −2.76/1.57 | −2.85/1.07 | −2.87/0.94 | −2.84/0.95 |
| Pd | −2.28/1.57 | −2.78/1.05 | −2.91/0.87 | −2.87/0.88 |
| Eads (eV)/dH-S (A) | |||||||
|---|---|---|---|---|---|---|---|
| Top | Bridge | Fcc | Hcp | ||||
| Au | M | Au-Au | Au-M | M-M | Au2M | 2AuM | |
| AuPt | −2.20/1.59 | −2.88/1.57 | −2.19/1.36 | −2.23/1.49 | −2.52/1.12 | −2.10/1.60 | −2.34/1.33 |
| AuPd | −2.37/1.60 | −2.51/1.56 | −2.29/1.41 | −2.25/1.65 | −2.48/1.11 | −2.28/1.50 | −2.29/1.39 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laghrissi, A.; Es-Souni, M. Au-Nanorods Supporting Pd and Pt Nanocatalysts for the Hydrogen Evolution Reaction: Pd Is Revealed to Be a Better Catalyst than Pt. Nanomaterials 2023, 13, 2007. https://doi.org/10.3390/nano13132007
Laghrissi A, Es-Souni M. Au-Nanorods Supporting Pd and Pt Nanocatalysts for the Hydrogen Evolution Reaction: Pd Is Revealed to Be a Better Catalyst than Pt. Nanomaterials. 2023; 13(13):2007. https://doi.org/10.3390/nano13132007
Chicago/Turabian StyleLaghrissi, Ayoub, and Mohammed Es-Souni. 2023. "Au-Nanorods Supporting Pd and Pt Nanocatalysts for the Hydrogen Evolution Reaction: Pd Is Revealed to Be a Better Catalyst than Pt" Nanomaterials 13, no. 13: 2007. https://doi.org/10.3390/nano13132007