
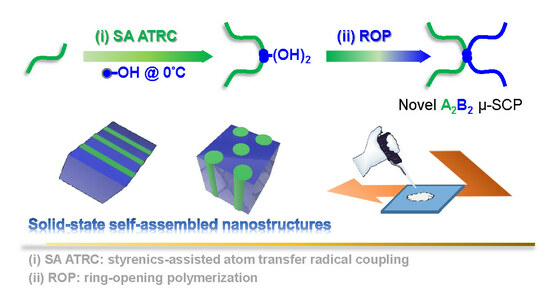
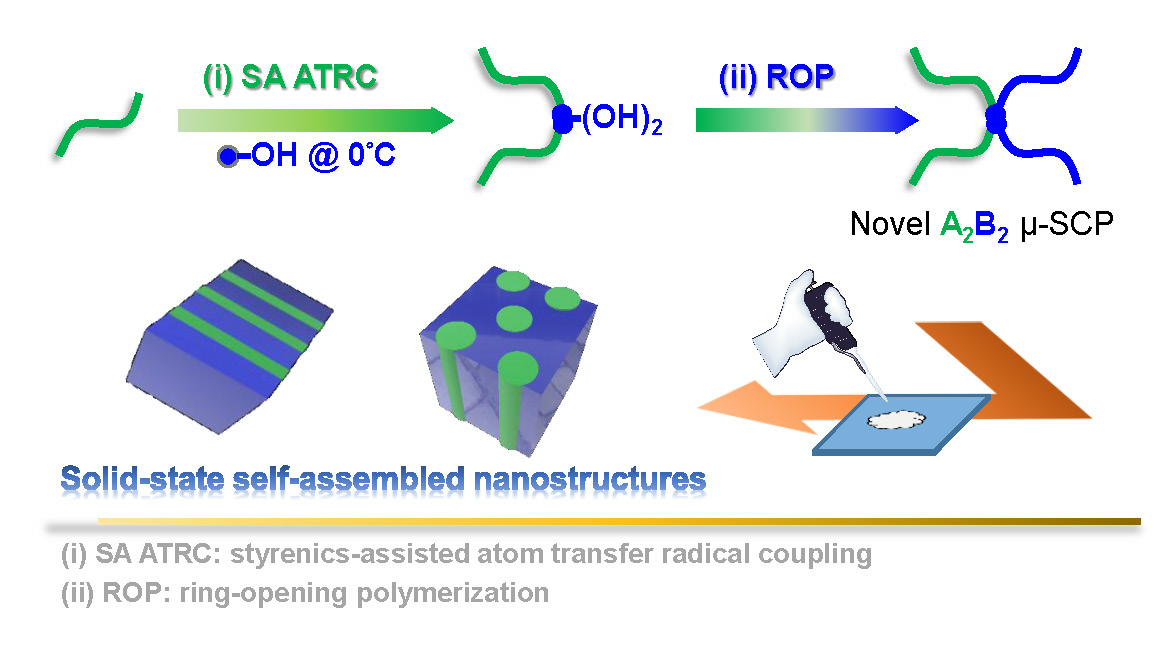
Synthesis of PDMS-μ-PCL Miktoarm Star Copolymers by Combinations (Є) of Styrenics-Assisted Atom Transfer Radical Coupling and Ring-Opening Polymerization and Study of the Self-Assembled Nanostructures

 , , and
, , and 
Abstract
:
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Characterization
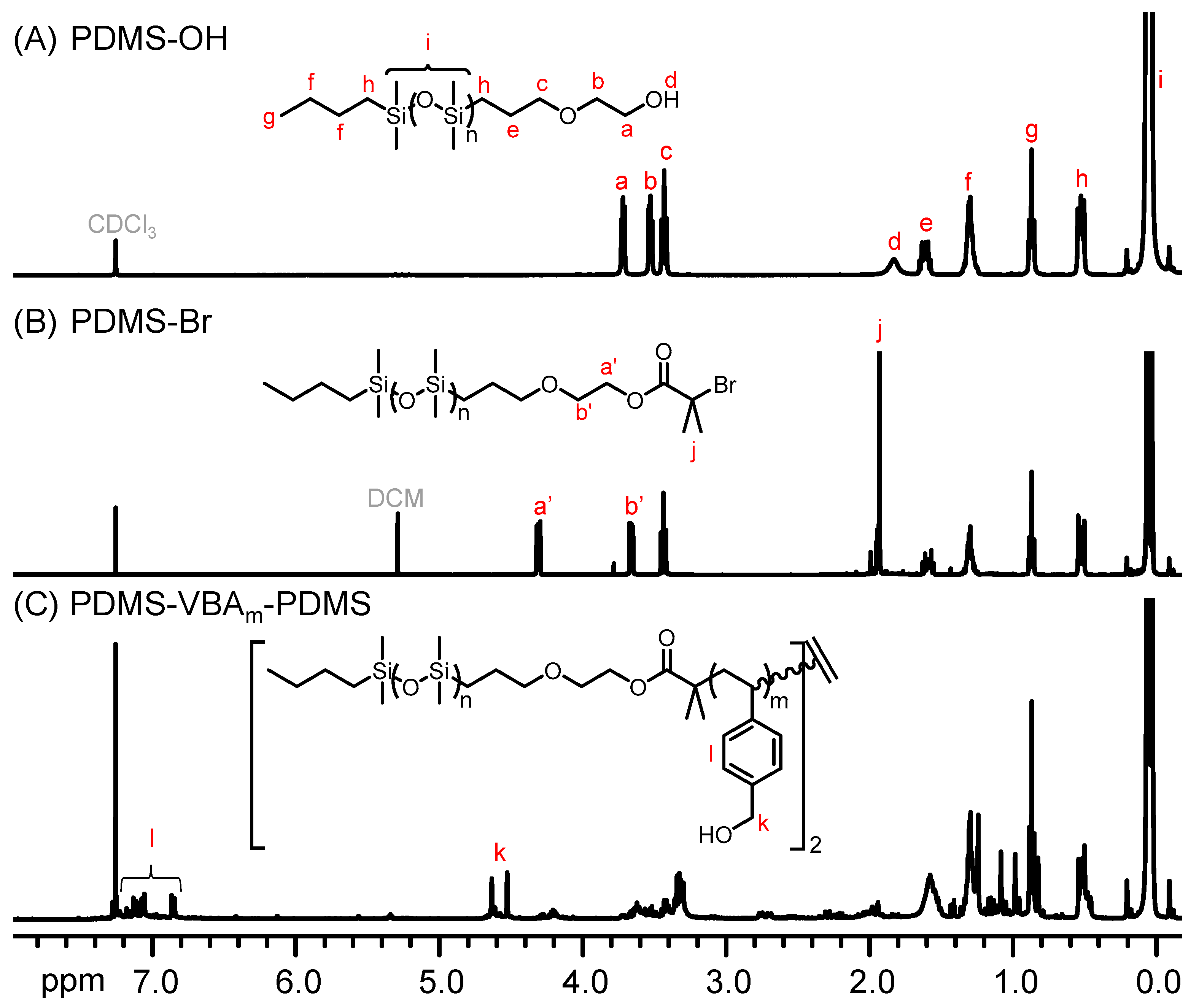
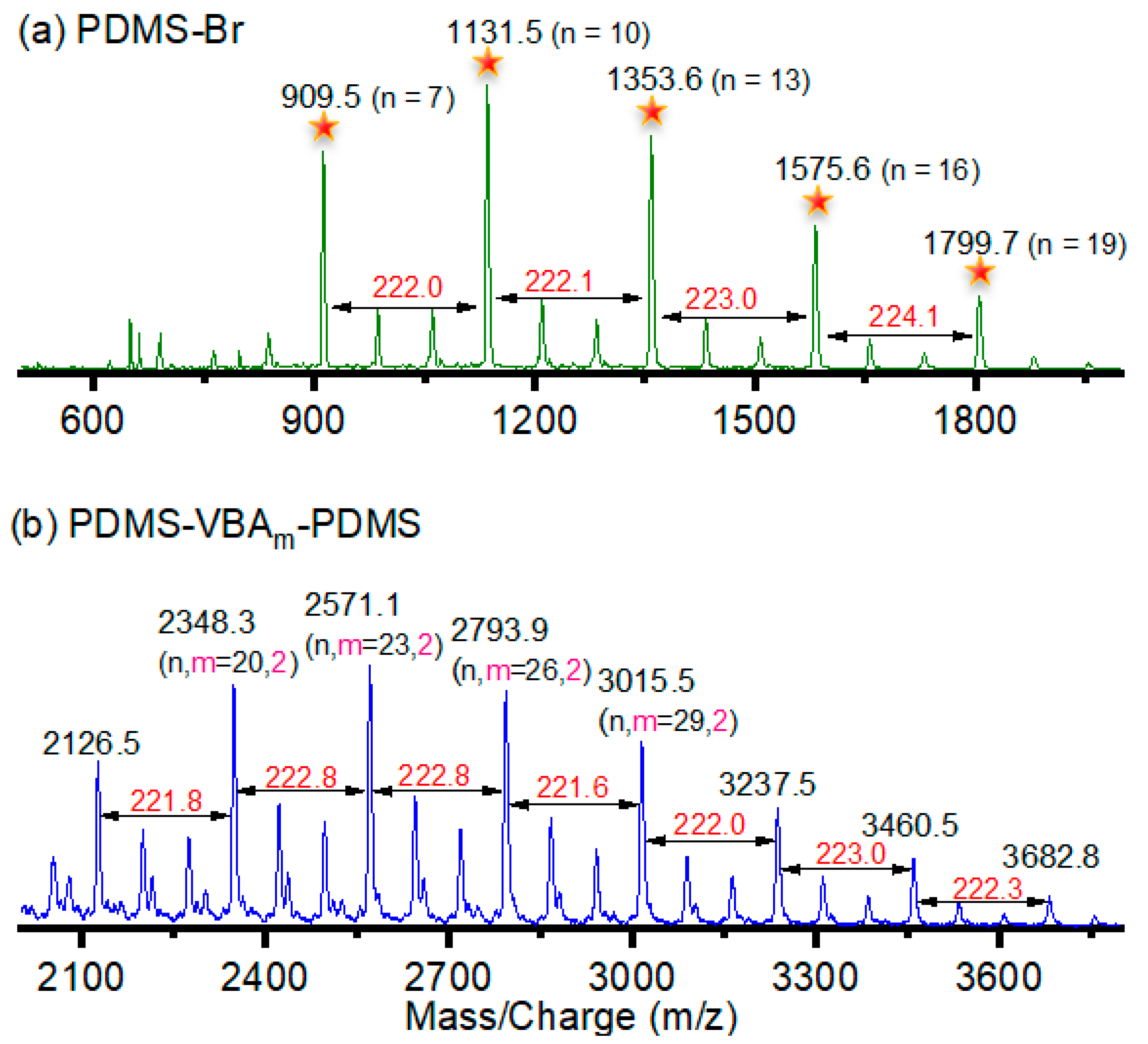
2.3. Synthesis of PDMS-Br Macroinitiator (MI)
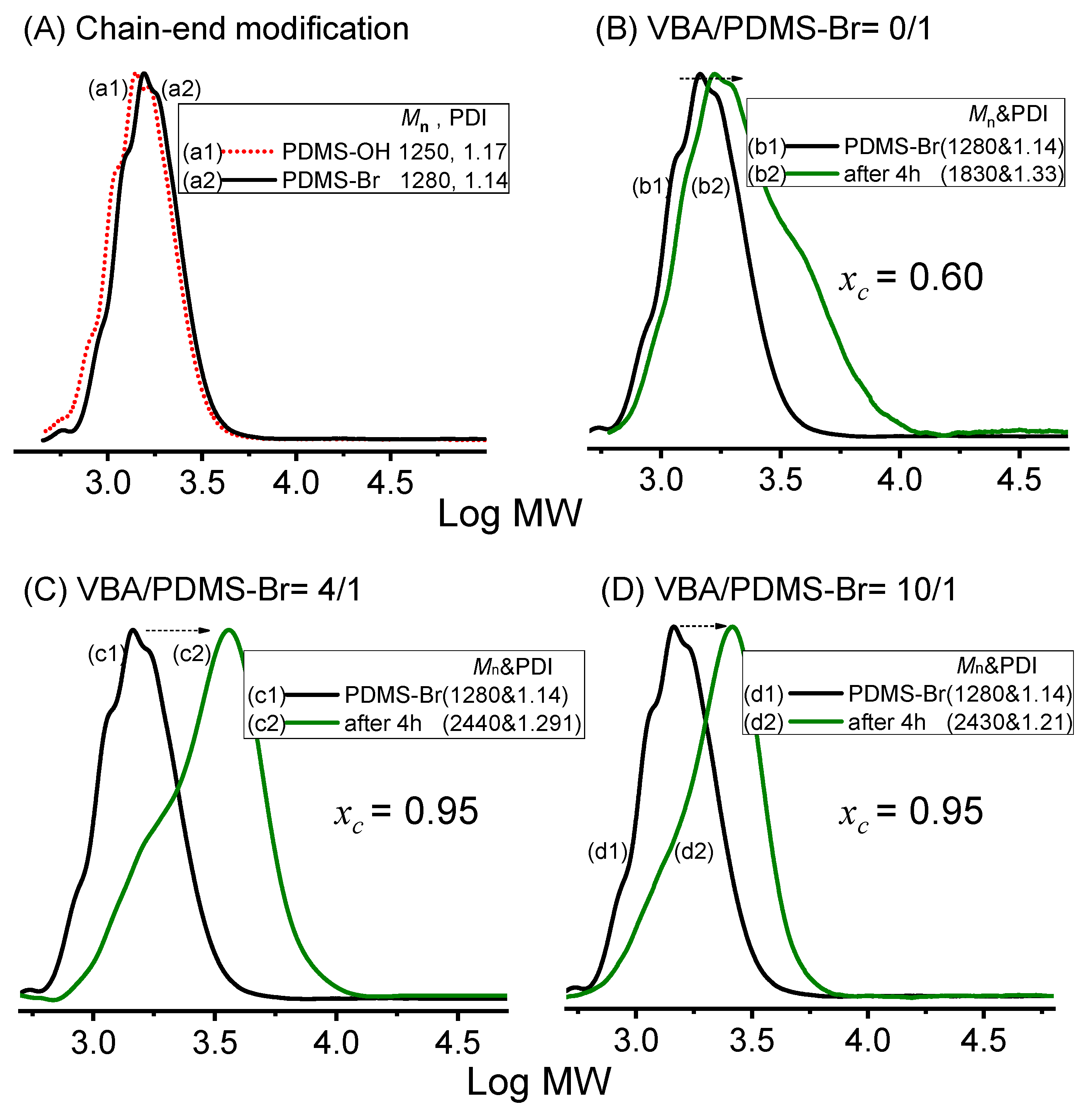
2.4. Synthesis of PDMS-VBAm-PDMS through Styrenics-Assisted Atom Transfer Radical Coupling (SA ATRC)
2.5. Chain Extension of PDMS-VBAm-PDMS with ε-CL through ROP
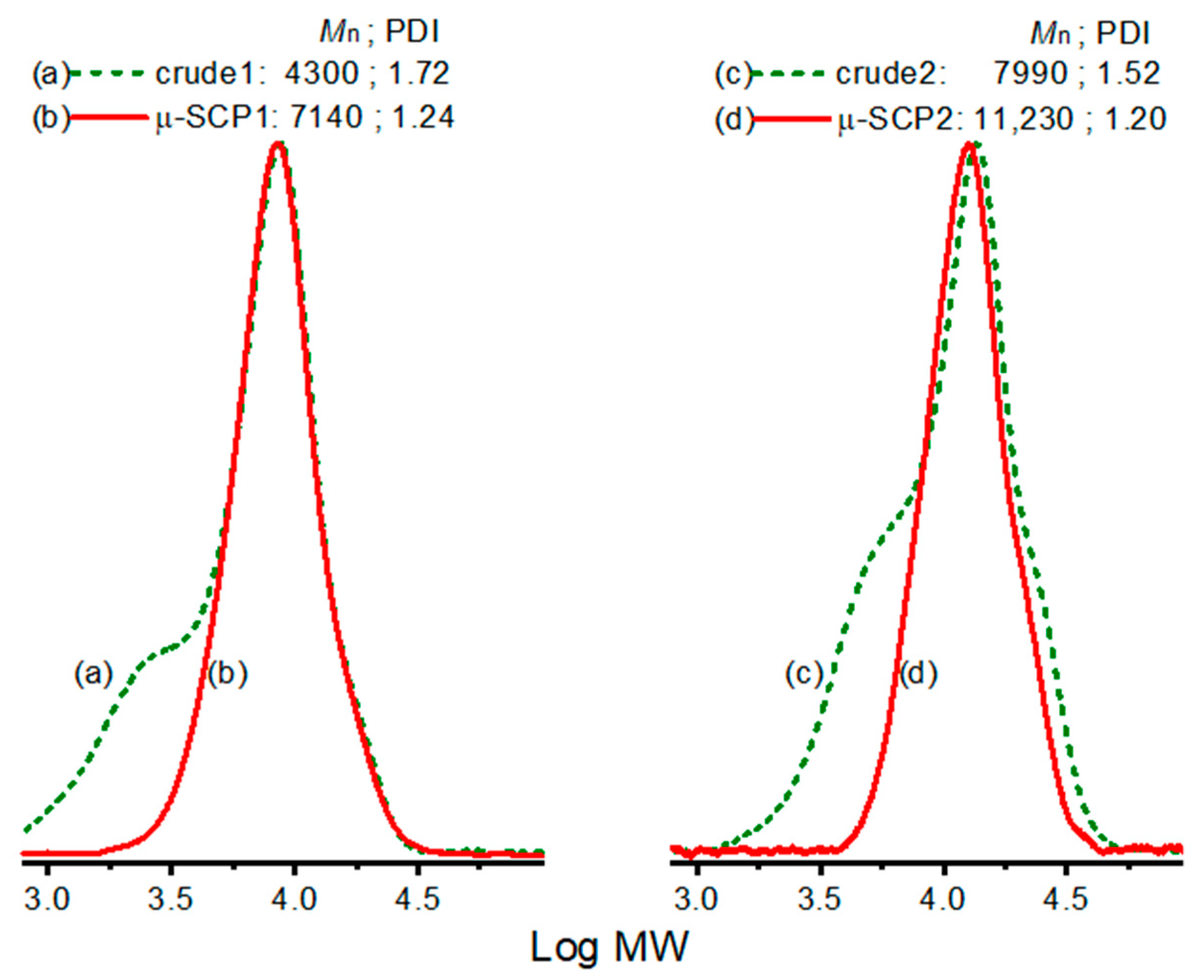
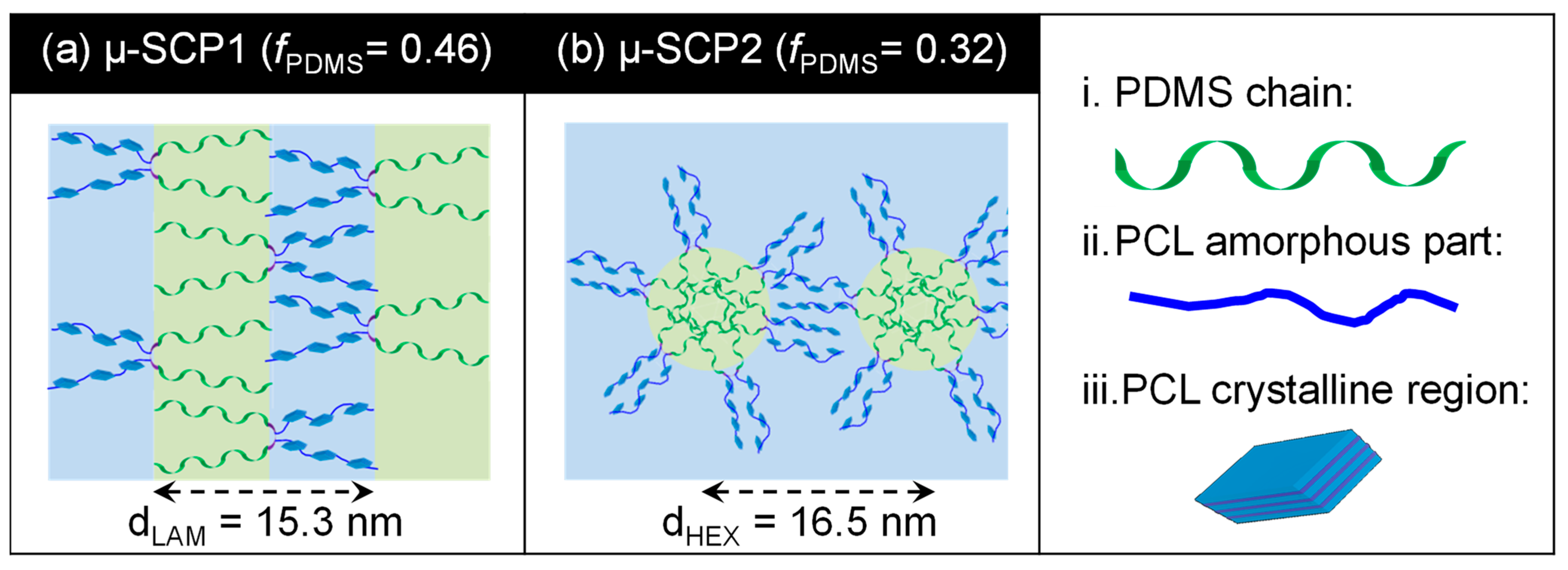
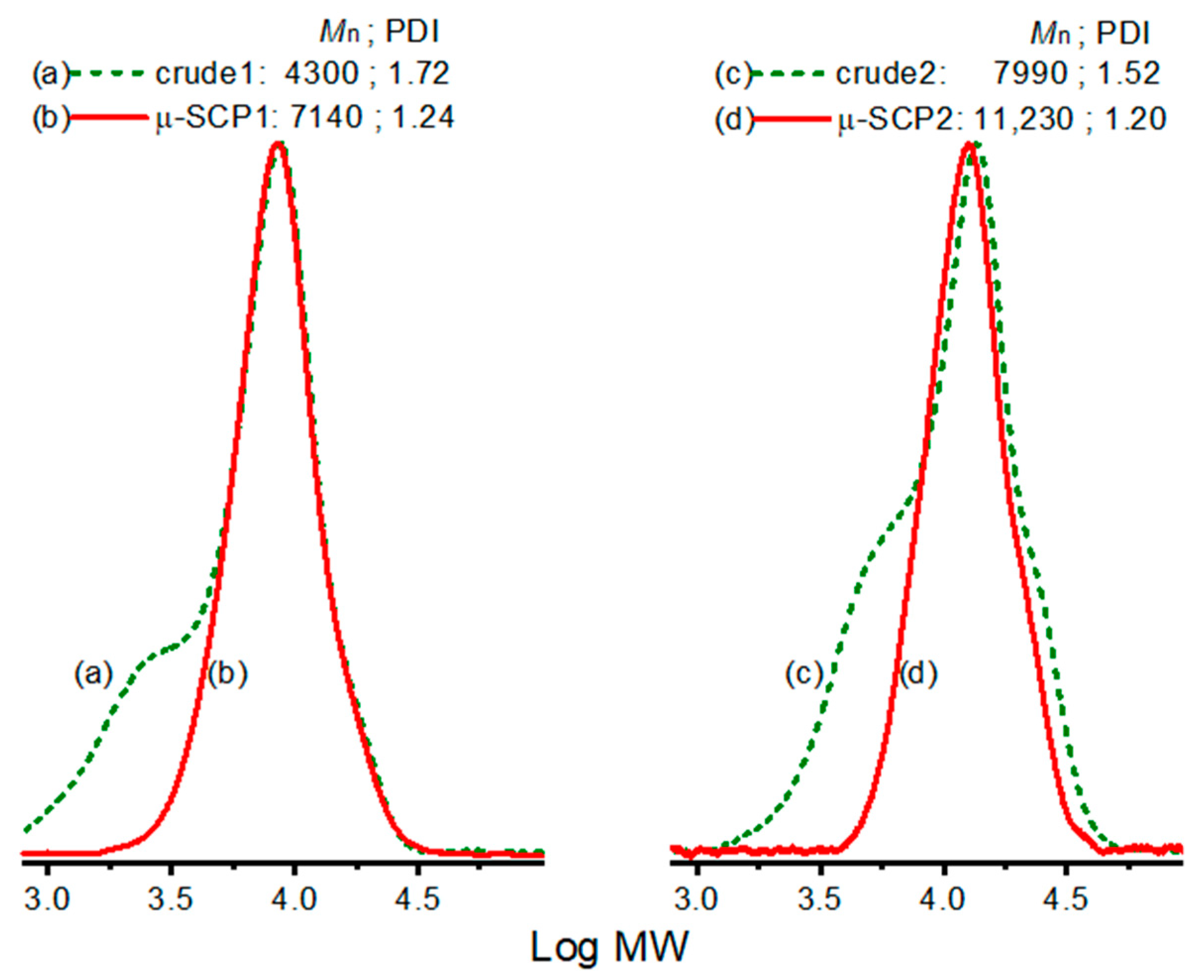
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aloorkar, N.; Kulkarni, A.; Patil, R.; Ingale, D. Star polymers: An overview. Int. J. Pharm. Sci. Nanotechnol 2012, 5, 1675–1684. [Google Scholar]
- Feng, H.; Lu, X.; Wang, W.; Kang, N.-G.; Mays, J.W. Block copolymers: Synthesis, self-assembly, and applications. Polymers 2017, 9, 494. [Google Scholar] [CrossRef] [PubMed]
- Nitta, N.; Kihara, S.I.; Haino, T. Synthesis of Supramolecular A8Bn Miktoarm Star Copolymers by Host-Guest Complexation. Angew. Chem. Int. Ed. 2023, 62, e202219001. [Google Scholar] [CrossRef]
- Gao, H.; Matyjaszewski, K. Synthesis of Low-Polydispersity Miktoarm Star Copolymers via a Simple “Arm-First” Method: Macromonomers as Arm Precursors. Macromolecules 2008, 41, 4250–4257. [Google Scholar] [CrossRef]
- Liu, M.; Blankenship, J.R.; Levi, A.E.; Fu, Q.; Hudson, Z.M.; Bates, C.M. Miktoarm Star Polymers: Synthesis and Applications. Chem. Mater. 2022, 34, 6188–6209. [Google Scholar] [CrossRef]
- Khanna, K.; Varshney, S.; Kakkar, A. Miktoarm star polymers: Advances in synthesis, self-assembly, and applications. Polym. Chem. 2010, 1, 1171–1185. [Google Scholar] [CrossRef]
- Bates, M.W.; Barbon, S.M.; Levi, A.E.; Lewis, R.M., III; Beech, H.K.; Vonk, K.M.; Zhang, C.; Fredrickson, G.H.; Hawker, C.J.; Bates, C.M. Synthesis and Self-Assembly of ABn Miktoarm Star Polymers. ACS Macro Lett. 2020, 9, 396. [Google Scholar] [CrossRef]
- Lotocki, V.; Kakkar, A. Miktoarm Star Polymers: Branched Architectures in Drug Delivery. Pharmaceutics 2020, 12, 827. [Google Scholar] [CrossRef] [PubMed]
- Aghajanzadeh, M.; Zamani, M.; Rostamizadeh, K.; Sharafi, A.; Danafar, H. The role of miktoarm star copolymers in drug delivery systems. J. Macromol. Sci. Part A 2018, 55, 559–571. [Google Scholar] [CrossRef]
- Hadjichristidis, N. Synthesis of miktoarm star (μ-star) polymers. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 857. [Google Scholar] [CrossRef]
- Kalow, J.A.; Swager, T.M. Synthesis of Miktoarm Branched Conjugated Copolymers by ROMPing In and Out. ACS Macro Lett. 2015, 4, 1229–1233. [Google Scholar] [CrossRef]
- Tunca, U.; Ozyurek, Z.; Erdogan, T.; Hizal, G. Novel miktofunctional initiator for the preparation of an ABC-type miktoarm star polymer via a combination of controlled polymerization techniques. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 4228–4236. [Google Scholar] [CrossRef]
- Oliveira, A.S.R.; Mendonça, P.V.; Simões, S.; Serra, A.C.; Coelho, J.F.J. Amphiphilic well-defined degradable star block copolymers by combination of ring-opening polymerization and atom transfer radical polymerization: Synthesis and application as drug delivery carriers. J. Polym. Sci. 2021, 59, 211. [Google Scholar] [CrossRef]
- Huang, C.-F.; Aimi, J.; Lai, K.-Y. Synthesis of Novel μ-Star Copolymers with Poly(N-Octyl Benzamide) and Poly(ε-Caprolactone) Miktoarms through Chain-Growth Condensation Polymerization, Styrenics-Assisted Atom Transfer Radical Coupling, and Ring-Opening Polymerization. Macromol. Rapid Commun. 2017, 38, 1600607. [Google Scholar] [CrossRef]
- Gao, H.; Matyjaszewski, K. Arm-First Method As a Simple and General Method for Synthesis of Miktoarm Star Copolymers. J. Am. Chem. Soc. 2007, 129, 11828–11834. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.M.; McKenzie, T.G.; Fu, Q.; Wong, E.H.H.; Xu, J.; An, Z.; Shanmugam, S.; Davis, T.P.; Boyer, C.; Qiao, G.G. Star Polymers. Chem. Rev. 2016, 116, 6743. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhao, H.; Yao, L.; Zhang, L.; Cheng, Z.; Zhu, X. Photocontrolled bromine–iodine transformation reversible-deactivation radical polymerization: Facile synthesis of star copolymers and unimolecular micelles. Polym. Chem. 2021, 12, 2335–2345. [Google Scholar] [CrossRef]
- Ding, H.; Park, S.; Zhong, M.; Pan, X.; Pietrasik, J.; Bettinger, C.J.; Matyjaszewski, K. Facile Arm-First Synthesis of Star Block Copolymers via ARGET ATRP with ppm Amounts of Catalyst. Macromolecules 2016, 49, 6752–6760. [Google Scholar] [CrossRef]
- Kakkar, A. Miktoarm Star Polymers: From Basics of Branched Architecture to Synthesis, Self-Assembly and Applications; Royal Society of Chemistry: London, UK, 2017. [Google Scholar]
- Ito, S.; Goseki, R.; Ishizone, T.; Hirao, A. Successive synthesis of well-defined multiarmed miktoarm star polymers by iterative methodology using living anionic polymerization. Eur. Polym. J. 2013, 49, 2545–2566. [Google Scholar] [CrossRef]
- Higashihara, T.; Hayashi, M.; Hirao, A. Synthesis of well-defined star-branched polymers by stepwise iterative methodology using living anionic polymerization. Prog. Polym. Sci. 2011, 36, 323–375. [Google Scholar] [CrossRef]
- Morton, M.; Helminiak, T.E.; Gadkary, S.D.; Bueche, F. Preparation and properties of monodisperse branched polystyrene. J. Polym. Sci. 1962, 57, 471–482. [Google Scholar] [CrossRef]
- Orofino, T.; Wenger, F. Dilute Solution Properties of Branched Polymers. Polystyrene Trifunctional Star Molecules 1. J. Phys. Chem. 1963, 67, 566–575. [Google Scholar] [CrossRef]
- Mayer, R. Organized structures in amorphous styrene/cis-1,4-isoprene block copolymers: Low angle X-ray scattering and electron microscopy. Polymer 1974, 15, 137–145. [Google Scholar] [CrossRef]
- Strazielle, C.; Herz, J. Synthese et etude physicochimique de polystyrenes ramifies en “etoile” et en “peigne”. Eur. Polym. J. 1977, 13, 223. [Google Scholar] [CrossRef]
- Gervasi, J.A.; Gosnell, A.B. Synthesis and characterization of branched polystyrene. Part I. Synthesis of four- and six-branch star polystyrene. J. Polym. Sci. Part A Polym. Chem. 1966, 4, 1391. [Google Scholar] [CrossRef]
- Hsieh, H. Synthesis of radial thermoplastic elastomers. Rubber Chem. Technol. 1976, 49, 1305–1310. [Google Scholar] [CrossRef]
- Herz, J.; Hert, M.; Straziel, C. Synthesis and characterization of threefold branched star-shaped polystyrenes. Makromol. Chem. 1972, 160, 213. [Google Scholar] [CrossRef]
- Uraneck, C.A.; Short, J.N. Solution-polymerized rubbers with superior breakdown properties. J. Appl. Polym. Sci. 1970, 14, 1421–1432. [Google Scholar] [CrossRef]
- Nicolas, J.; Guillaneuf, Y.; Lefay, C.; Bertin, D.; Gigmes, D.; Charleux, B. Nitroxide-mediated polymerization. Prog. Polym. Sci. 2013, 38, 63. [Google Scholar] [CrossRef]
- El Yousfi, R.; Brahmi, M.; Dalli, M.; Achalhi, N.; Azougagh, O.; Tahani, A.; Touzani, R.; El Idrissi, A. Recent Advances in Nanoparticle Development for Drug Delivery: A Comprehensive Review of Polycaprolactone-Based Multi-Arm Architectures. Polymers 2023, 15, 1835. [Google Scholar] [CrossRef]
- Sisson, A.L.; Ekinci, D.; Lendlein, A. The contemporary role of ε-caprolactone chemistry to create advanced polymer architectures. Polymer 2013, 54, 4333. [Google Scholar] [CrossRef]
- Lorenzo, A.T.; Müller, A.J.; Lin, M.-C.; Chen, H.-L.; Jeng, U.S.; Priftis, D.; Pitsikalis, M.; Hadjichristidis, N. Influence of Macromolecular Architecture on the Crystallization of (PCL)2-b-(PS)2 4-Miktoarm Star Block Copolymers in Comparison to Linear PCL-b-PS Diblock Copolymer Analogues. Macromolecules 2009, 42, 8353. [Google Scholar] [CrossRef]
- Heise, A.; Trollsås, M.; Magbitang, T.; Hedrick, J.L.; Frank, C.W.; Miller, R.D. Star Polymers with Alternating Arms from Miktofunctional μ-Initiators Using Consecutive Atom Transfer Radical Polymerization and Ring-Opening Polymerization. Macromolecules 2001, 34, 2798–2804. [Google Scholar] [CrossRef]
- Yang, L.; Zhou, H.; Shi, G.; Wang, Y.; Pan, C.-Y. Synthesis of ABCD 4-miktoarm star polymers by combination of RAFT, ROP, and “Click Chemistry”. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 6641–6653. [Google Scholar] [CrossRef]
- Sathesh, V.; Chen, J.-K.; Chang, C.-J.; Aimi, J.; Chen, Z.-C.; Hsu, Y.-C.; Huang, Y.-S.; Huang, C.-F. Synthesis of Poly(ε-caprolactone)-Based Miktoarm Star Copolymers through ROP, SA ATRC, and ATRP. Polymers 2018, 10, 858. [Google Scholar] [CrossRef]
- Gordin, C.; Delaite, C.; Medlej, H.; Josien-Lefebvre, D.; Hariri, K.; Rusu, M. Synthesis of ABC miktoarm star block copolymers from a new heterotrifunctional initiator by combination of ATRP and ROP. Polym. Bull. 2009, 63, 789–801. [Google Scholar] [CrossRef]
- Nuyken, O.; Pask, S.D. Ring-Opening Polymerization—An Introductory Review. Polymers 2013, 5, 361–403. [Google Scholar] [CrossRef]
- Nakamura, Y.; Arima, T.; Yamago, S. Modular Synthesis of Mid-Chain-Functionalized Polymers by Photoinduced Diene- and Styrene-Assisted Radical Coupling Reaction of Polymer-End Radicals. Macromolecules 2014, 47, 582–588. [Google Scholar] [CrossRef]
- Fan, W.J.; Nakamura, Y.; Yamago, S. Synthesis of Multivalent Organotellurium Chain-Transfer Agents by Post-modification and Their Applications in Living Radical Polymerization. Chem. Eur. J. 2016, 22, 17006–17010. [Google Scholar] [CrossRef]
- Huang, C.-F.; Huang, Y.-S.; Lai, K.-Y. Synthesis and self-assembly of Poly(N-octyl benzamide)-μ-poly(ε-caprolactone) miktoarm star copolymers displaying uniform nanofibril morphology. Polymer 2019, 178, 121582. [Google Scholar] [CrossRef]
- Lu, Y.C.; Chou, L.C.; Huang, C.F. Iron-Catalysed Atom Transfer Radical Polyaddition for the Synthesis and Modification of Novel Aliphatic Polyesters Displaying Lower Critical Solution Temperature and pH-Dependent Release Behaviors. Polym. Chem. 2019, 10, 3912. [Google Scholar] [CrossRef]
- Huang, Y.-S.; Chen, J.-K.; Chen, T.; Huang, C.-F. Synthesis of PNVP-based Copolymers with Tunable Thermosensitivity by Sequential Reversible Addition–Fragmentation Chain Transfer Copolymerization and Ring-Opening Polymerization. Polymers 2017, 9, 231. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-F.; Kuo, S.-W.; Moravčíková, D.; Liao, J.-C.; Han, Y.-M.; Lee, T.-H.; Wang, P.-H.; Lee, R.-H.; Tsiang, R.C.-C.; Mosnáček, J. Effect of Variations of CuIIX2/L, Surface Area of Cu0, Solvent, and Temperature on Atom Transfer Radical Polyaddition of 4-Vinylbenzyl 2-Bromo-2-isobutyrate Inimers. RSC Adv. 2016, 6, 51816. [Google Scholar] [CrossRef]
- Han, Y.-M.; Chen, H.-H.; Huang, C.-F. Polymerization and Degradation of Aliphatic Polyesters Synthesized by Atom Transfer Radical Polyaddition. Polym. Chem. 2015, 6, 4565–4574. [Google Scholar] [CrossRef]
- Aimi, J.; Yasuda, T.; Huang, C.-F.; Yoshio, M.; Chen, W.-C. Fabrication of Solution-Processable OFET Memory Using a Nano-Floating Gate Based on a Phthalocyanine-Cored Star-Shaped Polymer. Mater. Adv. 2022, 3, 3128–3134. [Google Scholar] [CrossRef]
- Huang, Y.S.; Huang, C.F. Synthesis of Well-Defined PMMA-b-PDMS-b-PMMA Triblock Copolymer and Study of Its Self-Assembly Behaviors In Epoxy Resin. Eur. Polym. J. 2021, 160, 110787. [Google Scholar] [CrossRef]
- Huang, Y.-S.; Hsueh, H.-Y.; Aimi, J.; Chou, L.-C.; Lu, Y.-C.; Kuo, S.-W.; Wang, C.-C.; Chen, K.-Y.; Huang, C.-F. Effects of Various Cu(0), Fe(0), and Proanthocyanidin Reducing Agents on Fe(III)-catalysed ATRP for the Synthesis of PMMA Block Copolymers and their Self-assembly Behaviours. Polym. Chem. 2020, 11, 5147–5155. [Google Scholar] [CrossRef]
- Huang, C.-F.; Ohta, Y.; Yokoyama, A.; Yokozawa, T. Efficient Low-Temperature Atom Transfer Radical Coupling and Its Application to Synthesis of Well-Defined Symmetrical Polybenzamides. Macromolecules 2011, 44, 4140. [Google Scholar] [CrossRef]
- Lai, K.-Y.; Huang, Y.-S.; Chu, C.-Y.; Huang, C.-F. Synthesis of Poly(N-H benzamide)-b-Poly(lauryl methacrylate)-b-Poly(N-H benzamide) Symmetrical Triblock Copolymers by Combinations of CGCP, SARA ATRP, and SA ATRC. Polymer 2018, 137, 385–394. [Google Scholar] [CrossRef]
- Bunha, A.K.; Mangadlao, J.; Felipe, M.J.; Pangilinan, K.; Advincula, R. Catenated PS–PMMA Block Copolymers via Supramolecularly Templated ATRP Initiator Approach. Macromol. Rapid Commun. 2012, 33, 1214. [Google Scholar] [CrossRef]
- Arce, M.M.; Pan, C.W.; Thursby, M.M.; Wu, J.P.; Carnicom, E.M.; Tillman, E.S. Influence of Solvent on Radical Trap-Assisted Dimerization and Cyclization of Polystyrene Radicals. Macromolecules 2016, 49, 7804–7813. [Google Scholar] [CrossRef]
- Bevington, J.C.; Melville, M.H.; Taylor, R.P. The termination reaction in radical polymerizations. II. Polymerizations of styrene at 60° and of methyl methacrylate at 0 and 60°, and the copolymerization of these monomers at 60°. J. Polym. Sci. 1954, 14, 463. [Google Scholar] [CrossRef]
- Zammit, M.D.; Davis, T.P.; Haddleton, D.M.; Suddaby, K.G. Evaluation of the mode of termination for a thermally initiated free-radical polymerization via matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Macromolecules 1997, 30, 1915–1920. [Google Scholar] [CrossRef]
- Tang, W.; Tsarevsky, N.V.; Matyjaszewski, K. Determination of equilibrium constants for atom transfer radical polymerization. J. Am. Chem. Soc. 2006, 128, 1598–1604. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Kwak, Y.; Braunecker, W.; Tsarevsky, N.V.; Coote, M.L.; Matyjaszewski, K. Understanding atom transfer radical polymerization: Effect of ligand and initiator structures on the equilibrium constants. J. Am. Chem. Soc. 2008, 130, 10702–10713. [Google Scholar] [CrossRef]
- Tang, W.; Matyjaszewski, K. Kinetic Modeling of Normal ATRP, Normal ATRP with [CuII]0, Reverse ATRP and SR&NI ATRP. Macromol. Theory Simul. 2008, 17, 359. [Google Scholar]
- Fischer, H.; Radom, L. Factors controlling the addition of carbon-centered radicals to alkenes-an experimental and theoretical perspective. Angew. Chem. Int. Ed. 2001, 40, 1340–1371. [Google Scholar] [CrossRef]
- Yoshikawa, C.; Goto, A.; Fukuda, T. Reactions of polystyrene radicals in a monomer-free atom transfer radical polymerization system. e-Polymers 2002, 2, 172–183. [Google Scholar] [CrossRef]
- Moad, G.; Solomon, D.H.; Johns, S.R.; Willing, R.I. Fate of the initiator in the azobisisobutyronitrile-initiated polymerization of styrene. Macromolecules 1984, 17, 1094–1099. [Google Scholar] [CrossRef]
- Barth, J.; Buback, M. SP-PLP-EPR Investigations into the Chain-Length-Dependent Termination of Methyl Methacrylate Bulk Polymerization. Macromol. Rapid Commun. 2009, 30, 1805–1811. [Google Scholar] [CrossRef]
- Jang, S.; Lee, K.; Moon, H.C.; Kwak, J.; Park, J.; Jeon, G.; Lee, W.B.; Kim, J.K. Vertical Orientation of Nanodomains on Versatile Substrates through Self-Neutralization Induced by Star-Shaped Block Copolymers. Adv. Funct. Mater. 2015, 25, 5414. [Google Scholar] [CrossRef]
- Gu, X.D.; Gunkel, I.; Russell, T.P. Pattern transfer using block copolymers. Philos. Trans. R. Soc. A 2013, 371, 20120306. [Google Scholar] [CrossRef] [PubMed]
- Lo, T.Y.; Dehghan, A.; Georgopanos, P.; Avgeropoulos, A.; Shi, A.C.; Ho, R.M. Orienting Block Copolymer Thin Films via Entropy. Macromolecules 2016, 49, 624. [Google Scholar] [CrossRef]
- Isono, T.; Otsuka, I.; Kondo, Y.; Halila, S.; Fort, S.; Rochas, C.; Satoh, T.; Borsali, R.; Kakuchi, T. Sub-10 nm Nano-Organization in AB2- and AB3-Type Miktoarm Star Copolymers Consisting of Maltoheptaose and Polycaprolactone. Macromolecules 2013, 46, 1461–1469. [Google Scholar] [CrossRef]
- Isono, T.; Otsuka, I.; Suemasa, D.; Rochas, C.; Satoh, T.; Borsali, R.; Kakuchi, T. Synthesis, Self-Assembly, and Thermal Caramelization of Maltoheptaose-Conjugated Polycaprolactones Leading to Spherical, Cylindrical, and Lamellar Morphologies. Macromolecules 2013, 46, 8932. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rate Constant | Value (M−1 s−1) | Reference |
|---|---|---|
| kact,EBiB | 13 | [55,56,57] |
| kdea,EBiB | 5.1 × 105 | [55,56,57] |
| kt,MMA | 6.0 × 108 | [61] |
| kcr | 2.5 × 103 | [58] |
| kp | 42 | [55,56,57] |
| kt,St | 4.0 × 108 | [59] |
| kact,PEBr | 0.8 | [55,56,57] |
| kdea,PEBr | 1.8 × 104 | [55,56,57] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.-S.; Ejeta, D.D.; Lin, K.-Y.; Kuo, S.-W.; Jamnongkan, T.; Huang, C.-F. Synthesis of PDMS-μ-PCL Miktoarm Star Copolymers by Combinations (Є) of Styrenics-Assisted Atom Transfer Radical Coupling and Ring-Opening Polymerization and Study of the Self-Assembled Nanostructures. Nanomaterials 2023, 13, 2355. https://doi.org/10.3390/nano13162355
Huang Y-S, Ejeta DD, Lin K-Y, Kuo S-W, Jamnongkan T, Huang C-F. Synthesis of PDMS-μ-PCL Miktoarm Star Copolymers by Combinations (Є) of Styrenics-Assisted Atom Transfer Radical Coupling and Ring-Opening Polymerization and Study of the Self-Assembled Nanostructures. Nanomaterials. 2023; 13(16):2355. https://doi.org/10.3390/nano13162355
Chicago/Turabian StyleHuang, Yi-Shen, Dula Daksa Ejeta, Kun-Yi (Andrew) Lin, Shiao-Wei Kuo, Tongsai Jamnongkan, and Chih-Feng Huang. 2023. "Synthesis of PDMS-μ-PCL Miktoarm Star Copolymers by Combinations (Є) of Styrenics-Assisted Atom Transfer Radical Coupling and Ring-Opening Polymerization and Study of the Self-Assembled Nanostructures" Nanomaterials 13, no. 16: 2355. https://doi.org/10.3390/nano13162355