

Delivery of Therapeutic miRNA via Plasma-Polymerised Nanoparticles Rescues Diabetes-Impaired Endothelial Function




Abstract
:1. Introduction
2. Materials and Methods
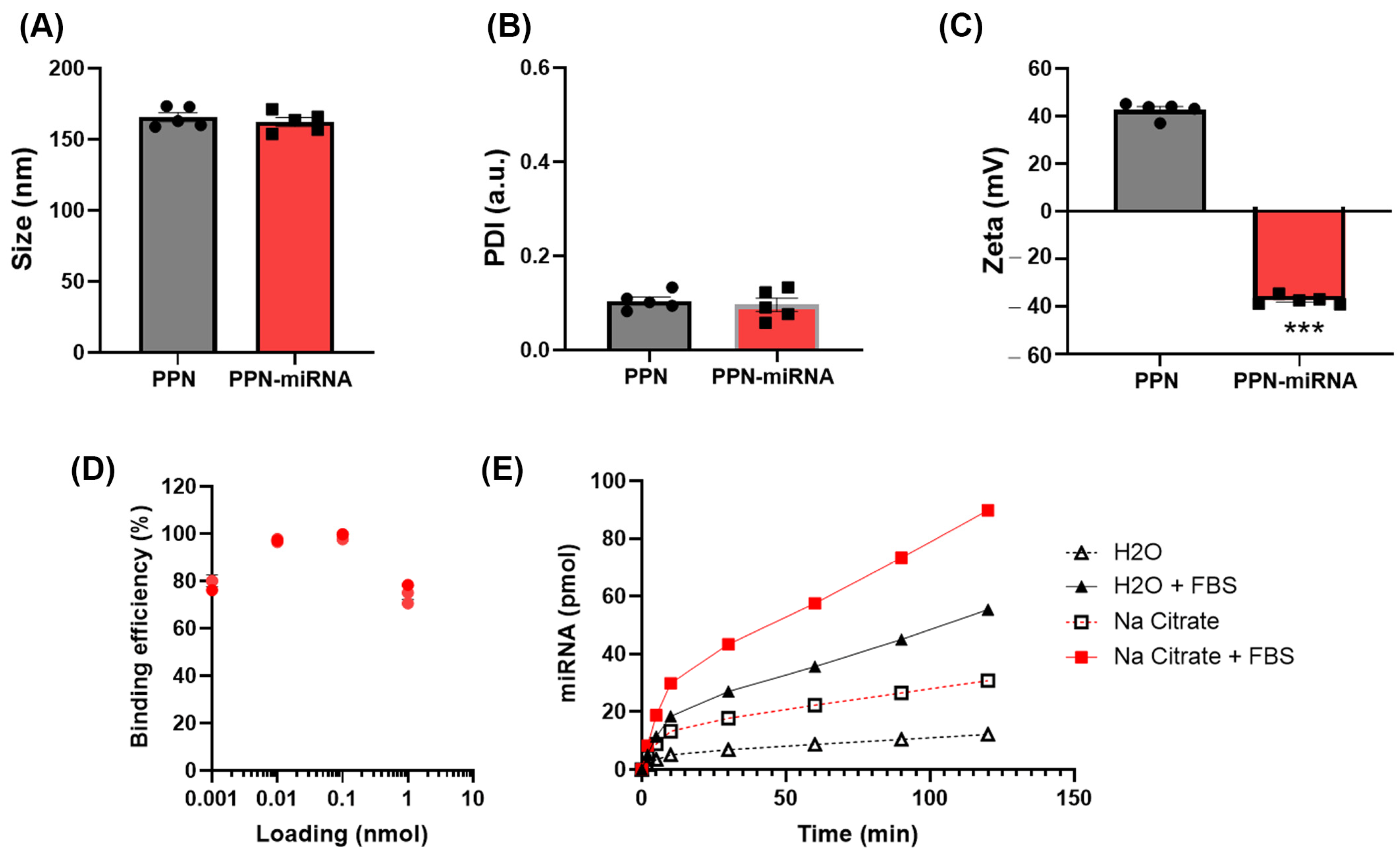
2.1. PPN Synthesis, PPN-miRNA Conjugation and Characterisation
2.2. In Vitro PPN-miRNA Binding Efficiency and miRNA Release
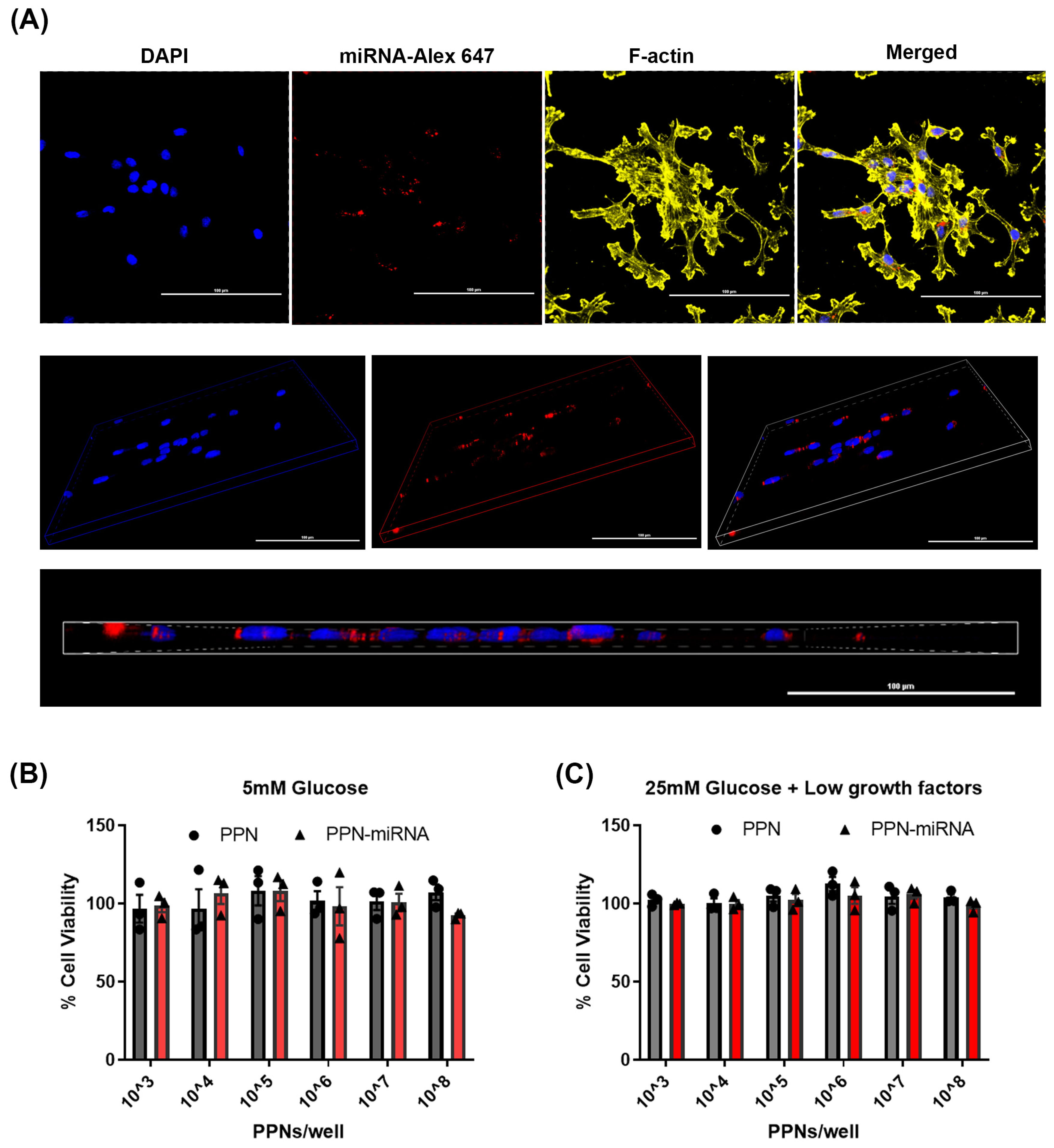
2.3. Cellular Uptake of PPN-miRNA and Its Effect on Viability
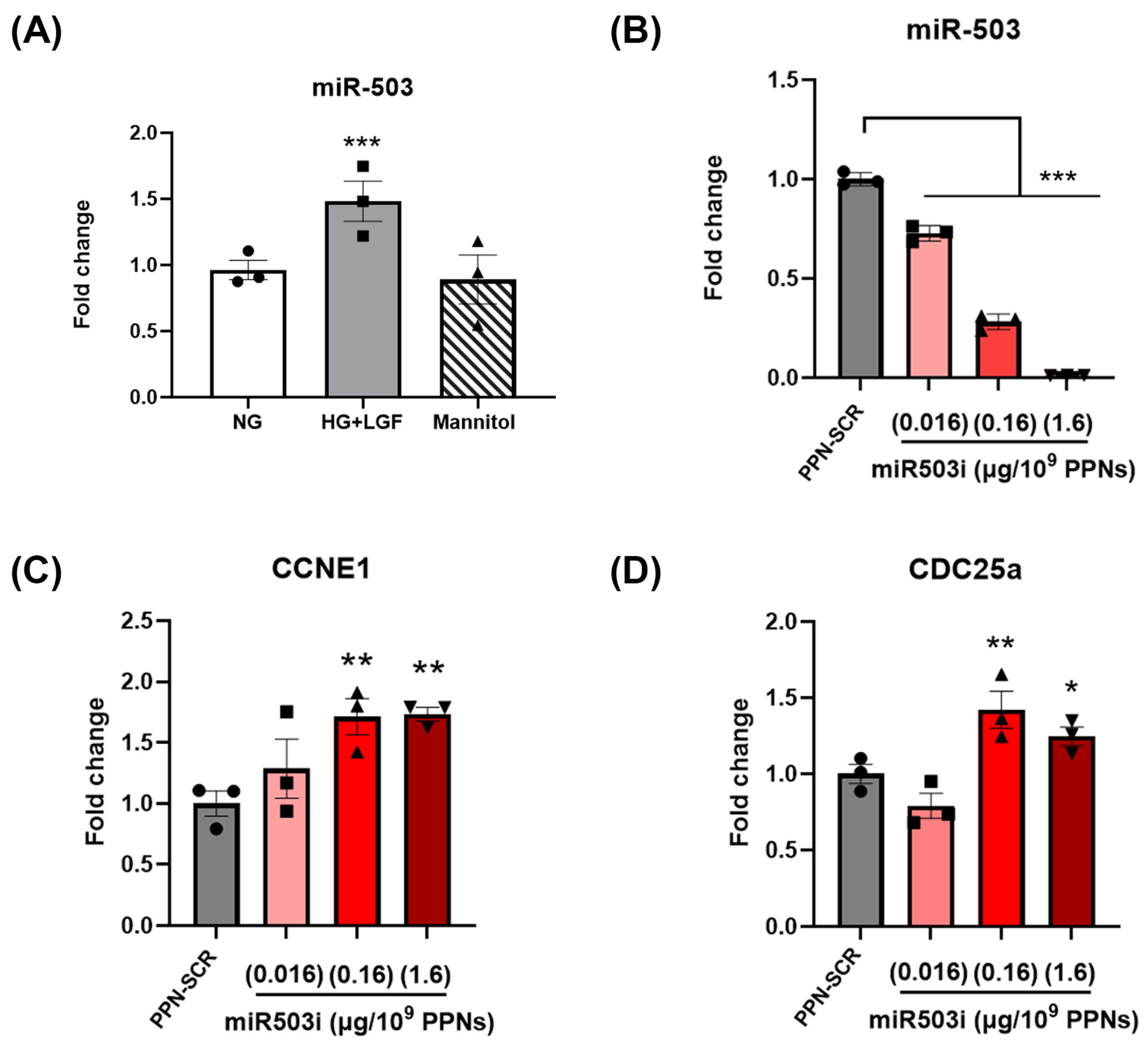
2.4. miR-503 and mRNA Quantification
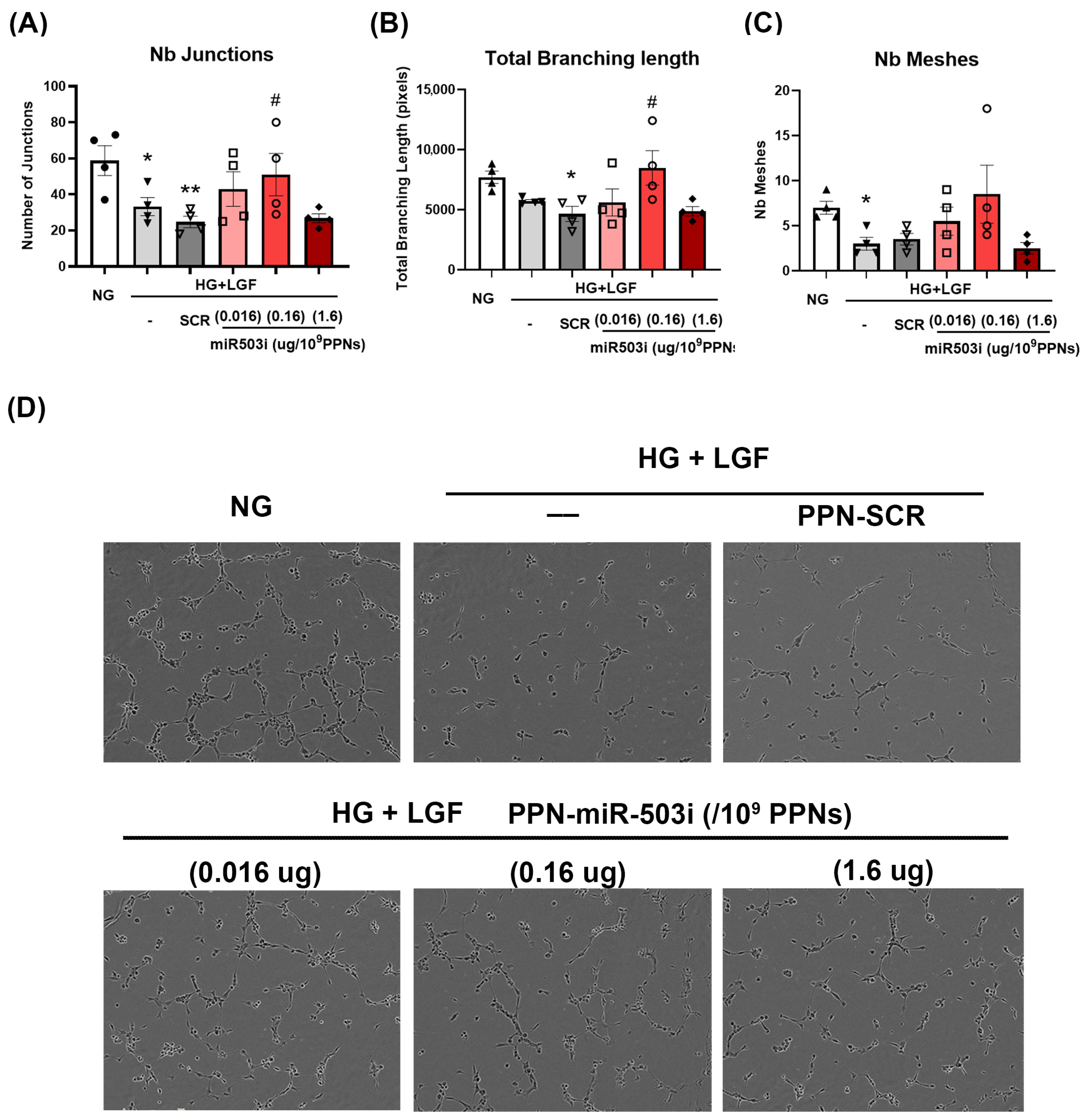
2.5. In Vitro Angiogenic Function: Tubulogenesis and Migration
2.6. Hindlimb Ischemia (HLI)
2.7. Immunohistochemistry
2.8. Statistical Analysis
3. Results
3.1. Characterisation of PPN-miRNA Complex
3.2. Endothelial Uptake and Cytotoxicity of PPN-miRNAs
3.3. PPN Delivery of miR-503 Inhibitor Modulates Endothelial Expression of miR-503 and Its Downstream Targets
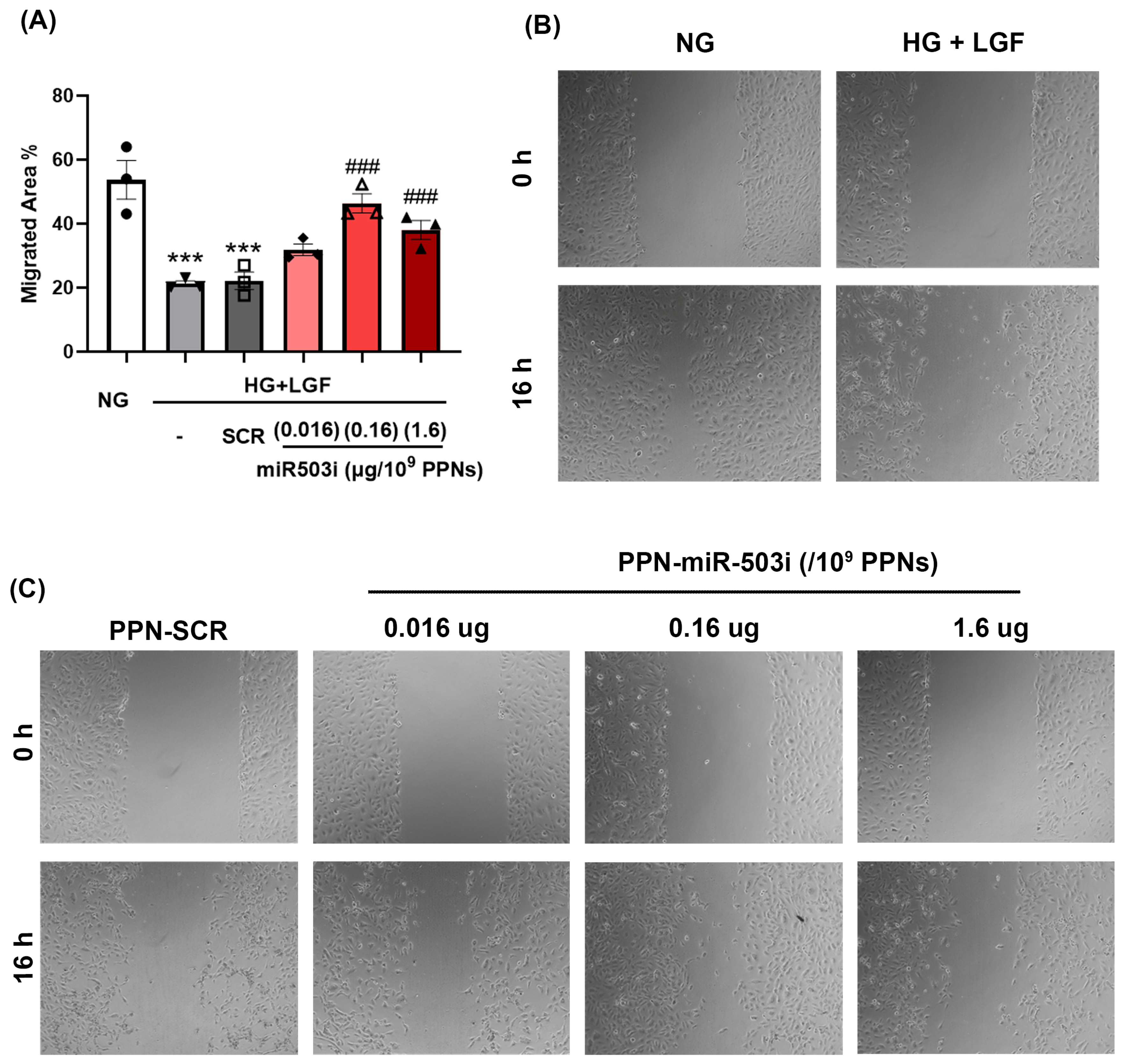
3.4. PPN Delivery of miR-503 Inhibitor Enhances Endothelial Angiogenic Function In Vitro
3.5. PPN Delivery of miR-503 Inhibitor Enhances Blood-Perfusion Recovery Post-Ischemia in Diabetes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Fridrichova, I.; Zmetakova, I. MicroRNAs Contribute to Breast Cancer Invasiveness. Cells 2019, 8, 1361. [Google Scholar] [CrossRef]
- Daoud, A.Z.; Mulholland, E.J.; Cole, G.; McCarthy, H.O. MicroRNAs in Pancreatic Cancer: Biomarkers, prognostic, and therapeutic modulators. BMC Cancer 2019, 19, 1130. [Google Scholar] [CrossRef]
- Eulalio, A.; Mano, M.; Ferro, M.D.; Zentilin, L.; Sinagra, G.; Zacchigna, S.; Giacca, M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 2012, 492, 376–381. [Google Scholar] [CrossRef]
- Wahlquist, C.; Jeong, D.; Rojas-Muñoz, A.; Kho, C.; Lee, A.; Mitsuyama, S.; van Mil, A.; Park, W.J.; Sluijter, J.P.G.; Doevendans, P.A.F.; et al. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature 2014, 508, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.; Montserrat, N.; Zacchigna, S.; Nivet, E.; Hishida, T.; Krause, M.N.; Kurian, L.; Ocampo, A.; Vazquez-Ferrer, E.; Rodriguez-Esteban, C.; et al. In vivo activation of a conserved microRNA program induces mammalian heart regeneration. Cell Stem Cell 2014, 15, 589–604. [Google Scholar] [CrossRef]
- Agbu, P.; Carthew, R.W. MicroRNA-mediated regulation of glucose and lipid metabolism. Nat. Rev. Mol. Cell Biol. 2021, 22, 425–438. [Google Scholar] [CrossRef]
- He, X.; Kuang, G.; Wu, Y.; Ou, C. Emerging roles of exosomal miRNAs in diabetes mellitus. Clin. Transl. Med. 2021, 11, e468. [Google Scholar] [CrossRef]
- Scherm, M.G.; Daniel, C. miRNA Regulation of T Cells in Islet Autoimmunity and Type 1 Diabetes. Curr. Diab. Rep. 2020, 20, 41. [Google Scholar] [CrossRef]
- Gasecka, A.; Siwik, D.; Gajewska, M.; Jaguszewski, M.J.; Mazurek, T.; Filipiak, K.J.; Postuła, M.; Eyileten, C. Early Biomarkers of Neurodegenerative and Neurovascular Disorders in Diabetes. J. Clin. Med. 2020, 9, 2807. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Y.; Gao, Q.; Ping, D.; Wang, Y.; Wu, W.; Lin, X.; Fang, Y.; Zhang, J.; Shao, A. The Role of Exosomal microRNAs and Oxidative Stress in Neurodegenerative Diseases. Oxidative Med. Cell Longev. 2020, 2020, 3232869. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.Y.; Kuo, H.C. Functional roles and networks of non-coding RNAs in the pathogenesis of neurodegenerative diseases. J. Biomed. Sci. 2020, 27, 49. [Google Scholar] [CrossRef]
- Caporali, A.; Emanueli, C. MicroRNA-503 and the extended microRNA-16 family in angiogenesis. Trends Cardiovasc. Med. 2011, 21, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Caporali, A.; Meloni, M.; Völlenkle, C.; Bonci, D.; Sala-Newby, G.B.; Addis, R.; Spinetti, G.; Losa, S.; Masson, R.; Baker, A.H.; et al. Deregulation of microRNA-503 contributes to diabetes mellitus-induced impairment of endothelial function and reparative angiogenesis after limb ischemia. Circulation 2011, 123, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Pan, Q.; Xie, Z.; Chen, Y.; Wang, J.; Bihl, J.; Zhong, W.; Chen, Y.; Zhao, B.; Ma, X. Implication of MicroRNA503 in Brain Endothelial Cell Function and Ischemic Stroke. Transl. Stroke Res. 2020, 11, 1148–1164. [Google Scholar] [CrossRef]
- Wang, C.; Liu, H. Factors influencing degradation kinetics of mRNAs and half-lives of microRNAs, circRNAs, lncRNAs in blood in vitro using quantitative PCR. Sci. Rep. 2022, 12, 7259. [Google Scholar] [CrossRef]
- Lee, S.W.L.; Paoletti, C.; Campisi, M.; Osaki, T.; Adriani, G.; Kamm, R.D.; Mattu, C.; Chiono, V. MicroRNA delivery through nanoparticles. J. Control. Release 2019, 313, 80–95. [Google Scholar] [CrossRef]
- Cullis, P.R.; Hope, M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther. 2017, 25, 1467–1475. [Google Scholar] [CrossRef]
- Silva, S.; Almeida, A.J.; Vale, N. Combination of Cell-Penetrating Peptides with Nanoparticles for Therapeutic Application: A Review. Biomolecules 2019, 9, 22. [Google Scholar] [CrossRef]
- Soares, D.C.F.; Domingues, S.C.; Viana, D.B.; Tebaldi, M.L. Polymer-hybrid nanoparticles: Current advances in biomedical applications. Biomed. Pharmacother. 2020, 131, 110695. [Google Scholar] [CrossRef]
- Zhang, N.; Xiong, G.; Liu, Z. Toxicity of metal-based nanoparticles: Challenges in the nano era. Front. Bioeng. Biotechnol. 2022, 10, 1001572. [Google Scholar] [CrossRef]
- Santos, M.; Michael, P.L.; Filipe, E.C.; Chan, A.H.; Hung, J.; Tan, R.P.; Lee, B.S.; Huynh, M.; Hawkins, C.; Waterhouse, A.; et al. Plasma Synthesis of Carbon-Based Nanocarriers for Linker-Free Immobilization of Bioactive Cargo. ACS Appl. Nano Mater. 2018, 1, 580–594. [Google Scholar] [CrossRef]
- Michael, P.L.; Lam, Y.T.; Hung, J.; Tan, R.P.; Santos, M.; Wise, S.G. Comprehensive Evaluation of the Toxicity and Biosafety of Plasma Polymerized Nanoparticles. Nanomaterials 2021, 11, 1176. [Google Scholar] [CrossRef]
- Michael, P.; Lam, Y.T.; Filipe, E.C.; Tan, R.P.; Chan, A.H.P.; Lee, B.S.L.; Feng, N.; Hung, J.; Cox, T.R.; Santos, M.; et al. Plasma polymerized nanoparticles effectively deliver dual siRNA and drug therapy in vivo. Sci. Rep. 2020, 10, 12836. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.T.; Lecce, L.; Yuen, S.C.; Wise, S.G.; Handelsman, D.J.; Karas, R.H.; Ng, M.K.C. Androgens Ameliorate Impaired Ischemia-Induced Neovascularization Due to Aging in Male Mice. Endocrinology 2019, 160, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Niiyama, H.; Huang, N.F.; Rollins, M.D.; Cooke, J.P. Murine model of hindlimb ischemia. J. Vis. Exp. 2009, 23, e1035. [Google Scholar]
- Wang, Q.; Song, Y.; Chen, J.; Li, Q.; Gao, J.; Tan, H.; Zhu, Y.; Wang, Z.; Li, M.; Yang, H.; et al. Direct in vivo reprogramming with non-viral sequential targeting nanoparticles promotes cardiac regeneration. Biomaterials 2021, 276, 121028. [Google Scholar] [CrossRef]
- Rees, P.; Wills, J.W.; Brown, M.R.; Barnes, C.M.; Summers, H.D. The origin of heterogeneous nanoparticle uptake by cells. Nat. Commun. 2019, 10, 2341. [Google Scholar] [CrossRef]
- Yuan, S.Y.; Breslin, J.W.; Perrin, R.; Gaudreault, N.; Guo, M.; Kargozaran, H.; Wu, M.H. Microvascular permeability in diabetes and insulin resistance. Microcirculation 2007, 14, 363–373. [Google Scholar] [CrossRef]
- Wang, C.; Chen, Z.; Tang, X.; Liu, X.; Na, W.; Li, W.; Liu, T. Influences of galactose ligand on the uptake of TADF liposomes by HepG(2) cells. Photodiagnosis Photodyn. Ther. 2020, 32, 102014. [Google Scholar] [CrossRef] [PubMed]
- Torres-Pérez, S.A.; Torres-Pérez, C.E.; Pedraza-Escalona, M.; Pérez-Tapia, S.M.; Ramón-Gallegos, E. Glycosylated Nanoparticles for Cancer-Targeted Drug Delivery. Front. Oncol. 2020, 10, 605037. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Li, X.; Cui, W.; Xue, M.; Quan, Y.; Guo, X. Glucose-Modified Zein Nanoparticles Enhance Oral Delivery of Docetaxel. Pharmaceutics 2022, 14, 1361. [Google Scholar] [CrossRef]
- Masotti, A.; Miller, M.R.; Celluzzi, A.; Rose, L.; Micciulla, F.; Hadoke, P.W.; Bellucci, S.; Caporali, A. Regulation of angiogenesis through the efficient delivery of microRNAs into endothelial cells using polyamine-coated carbon nanotubes. Nanomedicine 2016, 12, 1511–1522. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Rana, T.M. Therapeutic targeting of microRNAs: Current status and future challenges. Nat. Rev. Drug Discov. 2014, 13, 622–638. [Google Scholar] [CrossRef] [PubMed]
- Trajkovski, M.; Hausser, J.; Soutschek, J.; Bhat, B.; Akin, A.; Zavolan, M.; Heim, M.H.; Stoffel, M. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 2011, 474, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Chamorro-Jorganes, A.; Araldi, E.; Penalva, L.O.; Sandhu, D.; Fernández-Hernando, C.; Suárez, Y. MicroRNA-16 and microRNA-424 regulate cell-autonomous angiogenic functions in endothelial cells via targeting vascular endothelial growth factor receptor-2 and fibroblast growth factor receptor-1. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2595–2606. [Google Scholar] [CrossRef]
- Polina, E.R.; Oliveira, F.M.; Sbruzzi, R.C.; Crispim, D.; Canani, L.H.; Santos, K.G. Gene polymorphism and plasma levels of miR-155 in diabetic retinopathy. Endocr. Connect. 2019, 8, 1591–1599. [Google Scholar] [CrossRef]
- Liang, L.; Wo, C.; Yuan, Y.; Cao, H.; Tan, W.; Zhou, X.; Wang, D.; Chen, R.; Shi, M.; Zhang, F.; et al. miR-124-3p improves mitochondrial function of renal tubular epithelial cells in db/db mice. FASEB J. 2023, 37, e22794. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (h) | % Alexa647+ |
|---|---|
| 2 | 54.3 ± 4.27 |
| 4 | 70.8 ± 6.29 |
| 24 | 73.8 ± 2.46 |
| PPN-miRNA (pmol) | % Alexa647+ |
|---|---|
| 25 | 43.3 ± 3.27 |
| 50 | 52.6 ± 4.29 |
| 100 | 55.7 ± 4.73 |
| 200 | 49.4 ± 5.67 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lam, Y.T.; Lee, B.S.L.; Hung, J.; Michael, P.; Santos, M.; Tan, R.P.; Liu, R.; Wise, S.G. Delivery of Therapeutic miRNA via Plasma-Polymerised Nanoparticles Rescues Diabetes-Impaired Endothelial Function. Nanomaterials 2023, 13, 2360. https://doi.org/10.3390/nano13162360
Lam YT, Lee BSL, Hung J, Michael P, Santos M, Tan RP, Liu R, Wise SG. Delivery of Therapeutic miRNA via Plasma-Polymerised Nanoparticles Rescues Diabetes-Impaired Endothelial Function. Nanomaterials. 2023; 13(16):2360. https://doi.org/10.3390/nano13162360
Chicago/Turabian StyleLam, Yuen Ting, Bob S. L. Lee, Juichien Hung, Praveesuda Michael, Miguel Santos, Richard P. Tan, Renjing Liu, and Steven G. Wise. 2023. "Delivery of Therapeutic miRNA via Plasma-Polymerised Nanoparticles Rescues Diabetes-Impaired Endothelial Function" Nanomaterials 13, no. 16: 2360. https://doi.org/10.3390/nano13162360
APA StyleLam, Y. T., Lee, B. S. L., Hung, J., Michael, P., Santos, M., Tan, R. P., Liu, R., & Wise, S. G. (2023). Delivery of Therapeutic miRNA via Plasma-Polymerised Nanoparticles Rescues Diabetes-Impaired Endothelial Function. Nanomaterials, 13(16), 2360. https://doi.org/10.3390/nano13162360