
Sheet-Like Morphology CuO/Co3O4 Nanocomposites for Enhanced Catalysis in Hydrogenation of CO2 to Methanol


Abstract
:1. Introduction
2. Experimental Procedures
2.1. Chemicals
2.2. Synthetic Procedures
2.2.1. Synthesis of CuO/Co3O4-IE
2.2.2. Synthesis of CuO/Co3O4-IM
2.2.3. Synthesis of Co3O4
2.3. Characterization
2.4. Catalytic Testing
3. Results
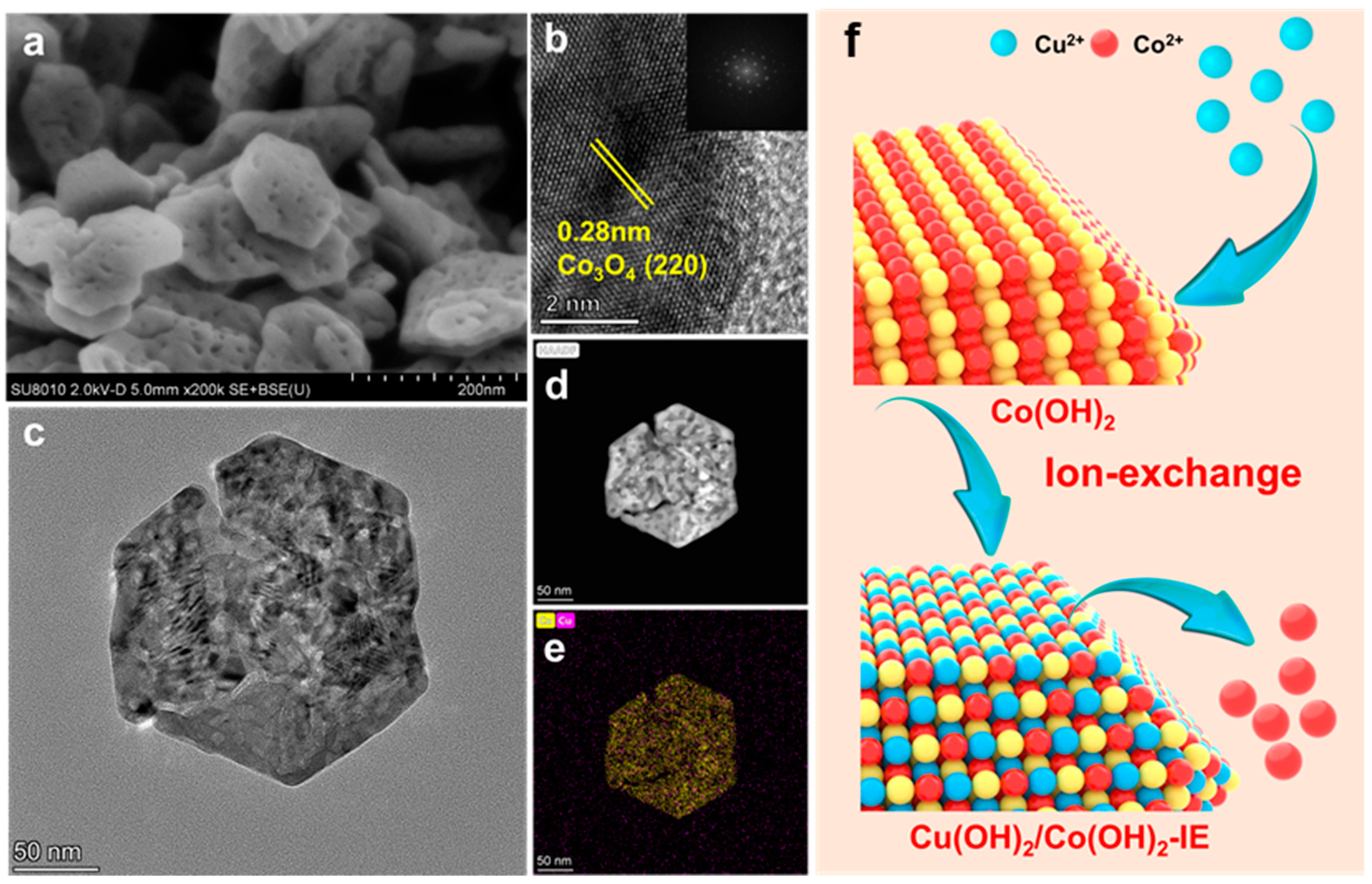
3.1. SEM and STEM
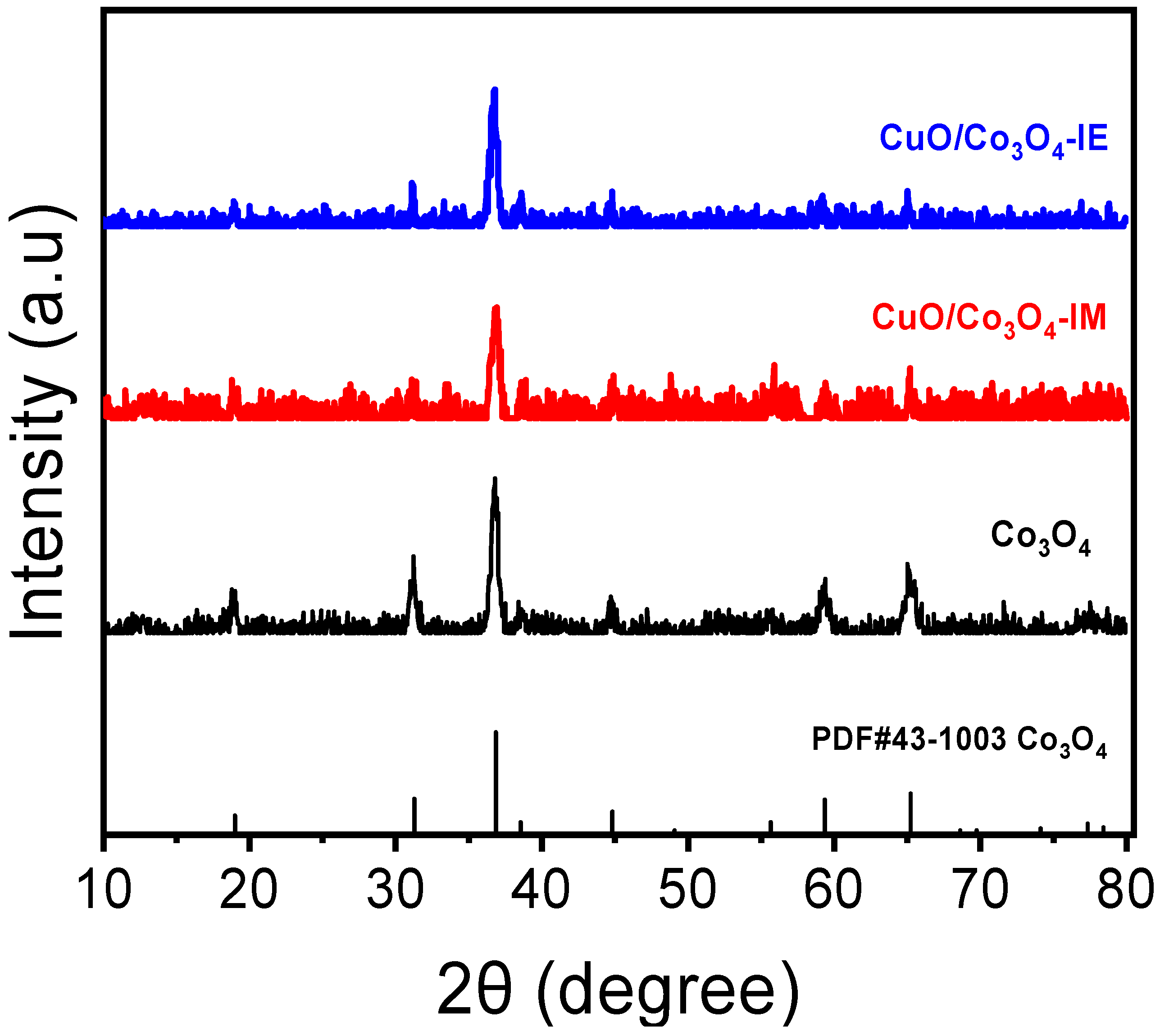
3.2. Diffraction of X-rays (XRD) and ICP
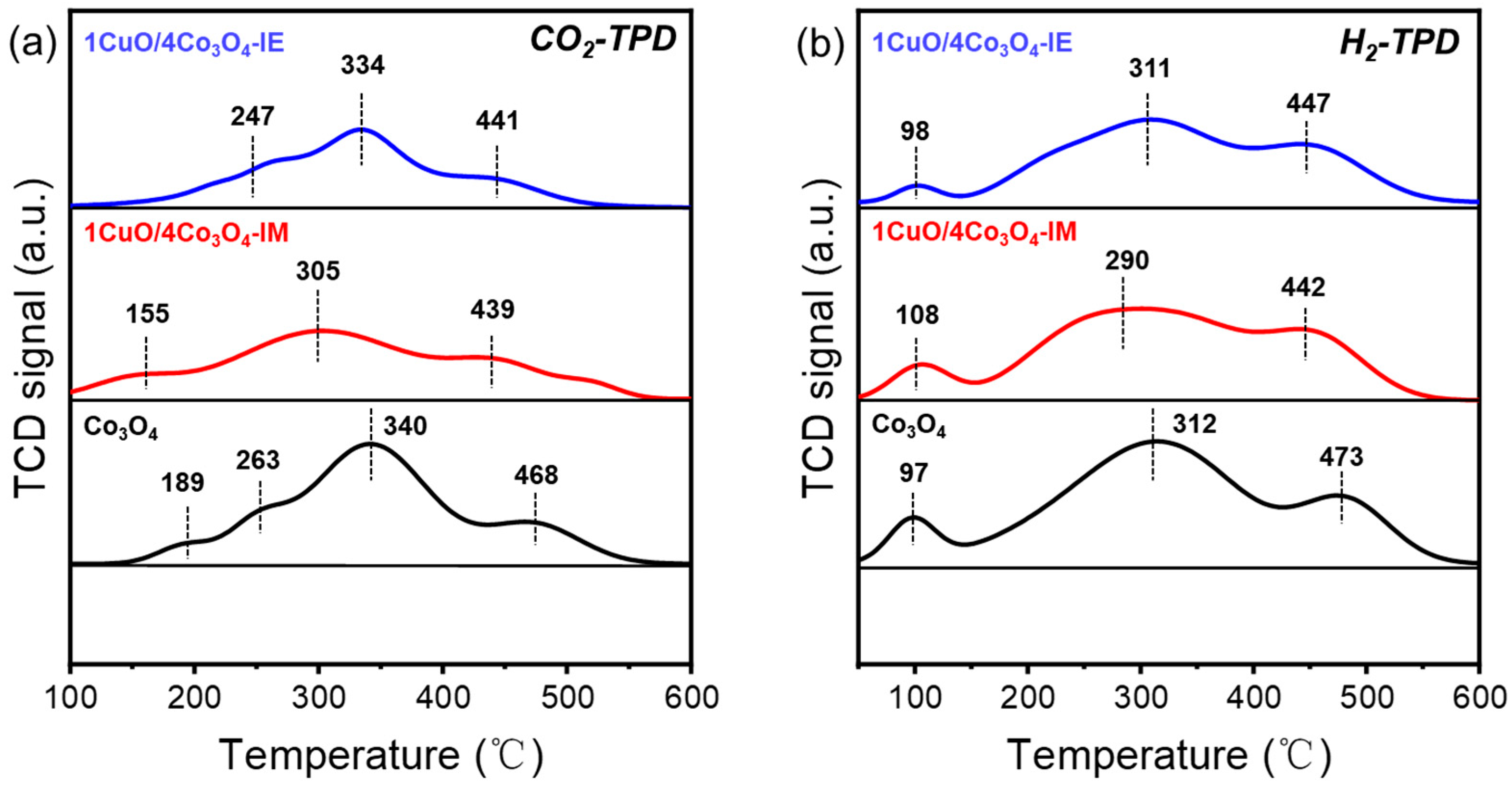
3.3. Surface Chemical Analysis and Chemical Adsorption
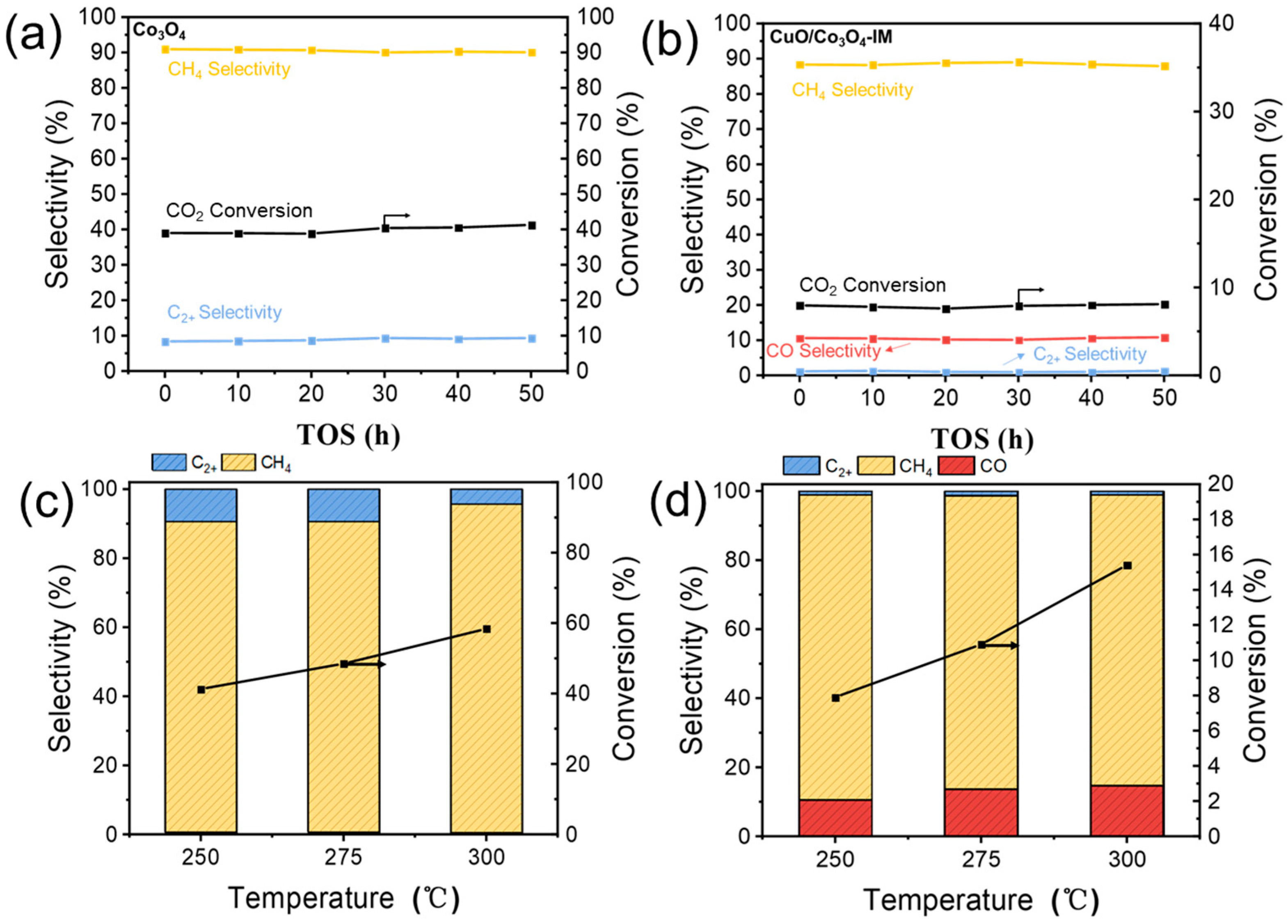
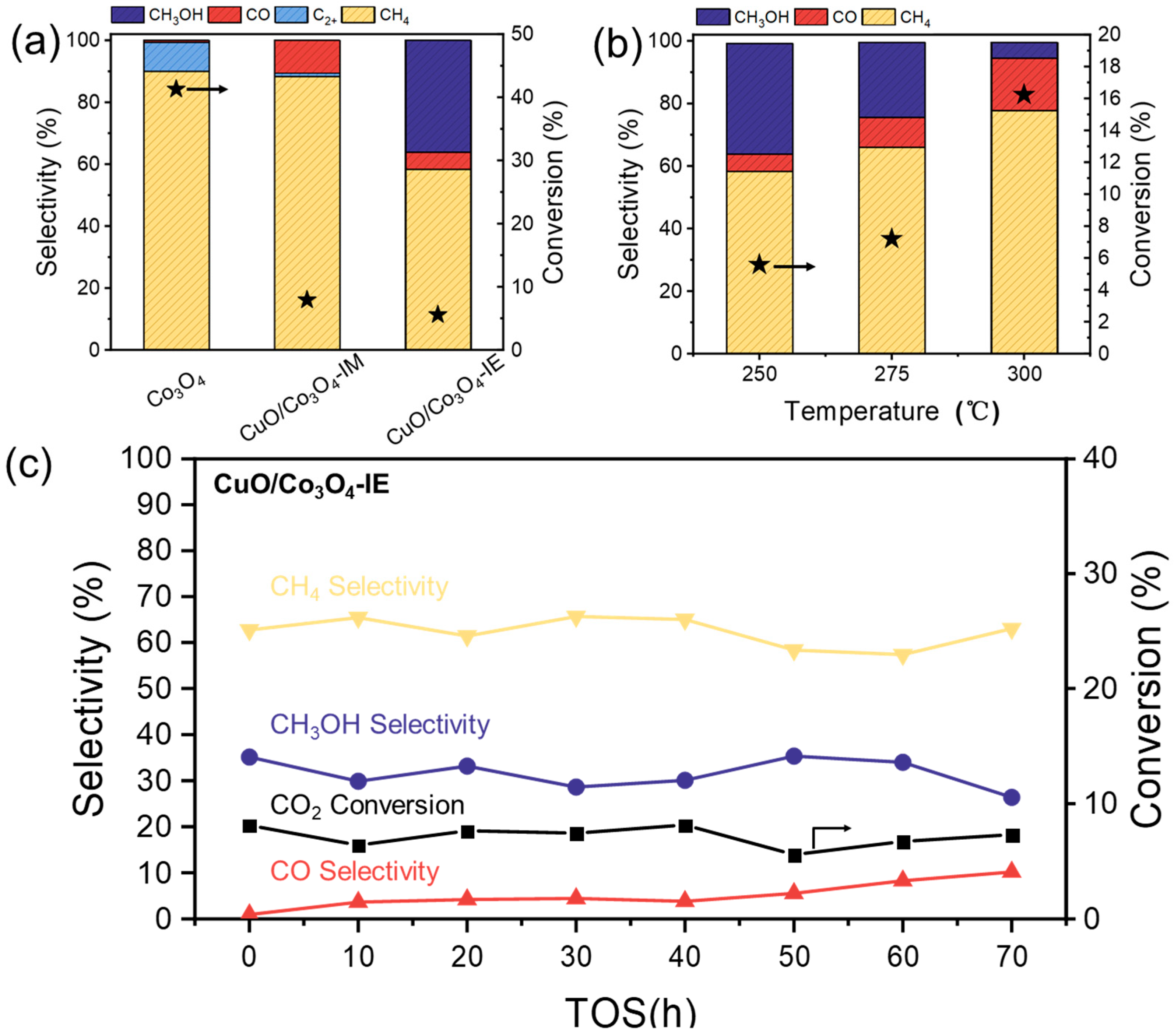
3.4. The Catalytic Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhang, Z.; Huang, X.; Chen, Z.; Zhu, J.; Endrődi, B.; Janáky, C.; Deng, D. Membrane electrode assembly for electrocatalytic CO2 reduction: Principle and application. Angew. Chem. Int. Ed. 2023, 135, e202302789. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent Advances in Catalytic Hydrogenation of Carbon Dioxide. Chem. Soc. Rev. 2011, 40, 3703. [Google Scholar] [CrossRef]
- Wei, J.; Yao, R.; Han, Y.; Ge, Q.; Sun, J. Towards the Development of the Emerging Process of CO2 Heterogenous Hydrogenation into High-Value Unsaturated Heavy Hydrocarbons. Chem. Soc. Rev. 2021, 50, 10764–10805. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.-P.; Ding, J.; Gong, W.; Argyle, M.D.; Zhong, Q.; Wang, Y.; Russell, C.K.; Xu, Z.; Russell, A.G.; Li, Q.; et al. CO2 Hydrogenation to High-Value Products via Heterogeneous Catalysis. Nat. Commun. 2019, 10, 5698. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Cheng, K.; Kang, J.; Zhou, C.; Subramanian, V.; Zhang, Q.; Wang, Y. New Horizon in C1 Chemistry: Breaking the Selectivity Limitation in Transformation of Syngas and Hydrogenation of CO2 into Hydrocarbon Chemicals and Fuels. Chem. Soc. Rev. 2019, 48, 3193–3228. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Cherevotan, A.; Peter, S.C. Thermochemical CO2 Hydrogenation to Single Carbon Products: Scientific and Technological Challenges. ACS Energy Lett. 2018, 3, 1938–1966. [Google Scholar] [CrossRef]
- Shih, C.F.; Zhang, T.; Li, J.; Bai, C. Powering the Future with Liquid Sunshine. Joule 2018, 2, 1925–1949. [Google Scholar] [CrossRef]
- Jiang, X.; Nie, X.; Guo, X.; Song, C.; Chen, J.G. Recent Advances in Carbon Dioxide Hydrogenation to Methanol via Heterogeneous Catalysis. Chem. Rev. 2020, 120, 7984–8034. [Google Scholar] [CrossRef]
- Zabilskiy, M.; Sushkevich, V.L.; Newton, M.A.; Van Bokhoven, J.A. Copper–Zinc Alloy-Free Synthesis of Methanol from Carbon Dioxide over Cu/ZnO/Faujasite. ACS Catal. 2020, 10, 14240–14244. [Google Scholar] [CrossRef]
- Martin, O.; Martín, A.J.; Mondelli, C.; Mitchell, S.; Segawa, T.F.; Hauert, R.; Drouilly, C.; Curulla-Ferré, D.; Pérez-Ramírez, J. Indium Oxide as a Superior Catalyst for Methanol Synthesis by CO2 Hydrogenation. Angew. Chem. Int. Ed. 2016, 128, 6369–6373. [Google Scholar] [CrossRef]
- Bowker, M.; Lawes, N.; Gow, I.; Hayward, J.; Esquius, J.R.; Richards, N.; Smith, L.R.; Slater, T.J.A.; Davies, T.E.; Dummer, N.F.; et al. The Critical Role of βPdZn Alloy in Pd/ZnO Catalysts for the Hydrogenation of Carbon Dioxide to Methanol. ACS Catal. 2022, 12, 5371–5379. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Tang, C.; Sha, F.; Tang, S.; Wang, J.; Li, C. CO2 Hydrogenation to Methanol on ZnO-ZrO2 Solid Solution Catalysts with Ordered Mesoporous Structure. J. Catal. 2021, 396, 242–250. [Google Scholar] [CrossRef]
- Wang, L.; Guan, E.; Wang, Y.; Wang, L.; Gong, Z.; Cui, Y.; Meng, X.; Gates, B.C.; Xiao, F.-S. Silica Accelerates the Selective Hydrogenation of CO2 to Methanol on Cobalt Catalysts. Nat. Commun. 2020, 11, 1033. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-W.; Joo, O.-S.; Jung, K.-D.; Kim, H.; Han, S.-H. ZnO/Cr2O3 Catalyst for Reverse-Water-Gas-Shift Reaction of CAMERE Process. Korean J. Chem. Eng. 2000, 17, 719–722. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kuhn, J.N. CO2 Conversion by Reverse Water Gas Shift Catalysis: Comparison of Catalysts, Mechanisms and Their Consequences for CO2 Conversion to Liquid Fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Su, X.; Yang, X.; Zhao, B.; Huang, Y. Designing of Highly Selective and High-Temperature Endurable RWGS Heterogeneous Catalysts: Recent Advances and the Future Directions. J. Energy Chem. 2017, 26, 854–867. [Google Scholar] [CrossRef]
- Bezemer, G.L.; Bitter, J.H.; Kuipers, H.P.C.E.; Oosterbeek, H.; Holewijn, J.E.; Xu, X.; Kapteijn, F.; Van Dillen, A.J.; De Jong, K.P. Cobalt Particle Size Effects in the Fischer−Tropsch Reaction Studied with Carbon Nanofiber Supported Catalysts. J. Am. Chem. Soc. 2006, 128, 3956–3964. [Google Scholar] [CrossRef]
- Shimura, K.; Miyazawa, T.; Hanaoka, T.; Hirata, S. Fischer–Tropsch Synthesis over Alumina Supported Cobalt Catalyst: Effect of Promoter Addition. Appl. Catal. A Gen. 2015, 494, 1–11. [Google Scholar] [CrossRef]
- Schubert, M.; Pokhrel, S.; Thomé, A.; Zielasek, V.; Gesing, T.M.; Roessner, F.; Mädler, L.; Bäumer, M. Highly Active Co–Al2O3-Based Catalysts for CO2 Methanation with Very Low Platinum Promotion Prepared by Double Flame Spray Pyrolysis. Catal. Sci. Technol. 2016, 6, 7449–7460. [Google Scholar] [CrossRef]
- Xie, F.; Xu, S.; Deng, L.; Xie, H.; Zhou, G. CO2 Hydrogenation on Co/CeO2-δ Catalyst: Morphology Effect from CeO2 Support. Int. J. Hydrogen Energy 2020, 45, 26938–26952. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, G.; Zhu, J.; Li, W.; Wang, J.; Bian, K.; Liu, Y.; Ding, F.; Song, C.; Guo, X. Unraveling the Tunable Selectivity on Cobalt Oxide and Metallic Cobalt Sites for CO2 Hydrogenation. Chem. Eng. J. 2022, 446, 137217. [Google Scholar] [CrossRef]
- Liu, S.; Yang, C.; Zha, S.; Sharapa, D.; Studt, F.; Zhao, Z.; Gong, J. Moderate Surface Segregation Promotes Selective Ethanol Production in CO2 Hydrogenation Reaction over CoCu Catalysts. Angew. Chem. Int. Ed. 2022, 61, e202109027. [Google Scholar] [CrossRef] [PubMed]
- Stangeland, K.; Kalai, D.Y.; Ding, Y.; Yu, Z. Mesoporous Manganese-Cobalt Oxide Spinel Catalysts for CO2 Hydrogenation to Methanol. J. CO2 Util. 2019, 32, 146–154. [Google Scholar] [CrossRef]
- Wang, L.; Wang, L.; Zhang, J.; Liu, X.; Wang, H.; Zhang, W.; Yang, Q.; Ma, J.; Dong, X.; Yoo, S.J.; et al. Selective Hydrogenation of CO2 to Ethanol over Cobalt Catalysts. Angew. Chem. Int. Ed. 2018, 57, 6104–6108. [Google Scholar] [CrossRef]
- Khangale, P.R. Hydrogenation of CO2 to Hydrocarbons over Zirconia-Supported Cobalt Catalyst Promoted with Potassium. Catal. Lett. 2022, 152, 2745–2755. [Google Scholar] [CrossRef]
- Pustovarenko, A.; Dikhtiarenko, A.; Bavykina, A.; Gevers, L.; Russkikh, A.; Telalovic, S.; Aguilar, A.; Hazemann, J.-L.; Gascon, J. Metal Organic Framework Derived Synthesis of Cobalt Indium Catalysts for the Hydrogenation of CO2 to Methanol. ACS Catal. 2020, 10, 5064–5076. [Google Scholar] [CrossRef]
- Zhang, H.; Mao, D.; Zhang, J.; Wu, D. Tuning the CO2 Hydrogenation Path by Moderately Phosphating the Co-Al Catalyst toward Methanol Synthesis. Appl. Catal. B Environ. 2024, 340, 123257. [Google Scholar] [CrossRef]
- Baran, T.; Visibile, A.; Busch, M.; He, X.; Wojtyla, S.; Rondinini, S.; Minguzzi, A.; Vertova, A. Copper Oxide-Based Photocatalysts and Photocathodes: Fundamentals and Recent Advances. Molecules 2021, 26, 7271. [Google Scholar] [CrossRef]
- Xiang, Y.; Kruse, N. Cobalt–Copper Based Catalysts for Higher Terminal Alcohols Synthesis via Fischer–Tropsch Reaction. J. Energy Chem. 2016, 25, 895–906. [Google Scholar] [CrossRef]
- Xu, M.; Wang, L.; Sun, Y.; Ma, Y.; Zhu, L.; Zhu, Y.; Zhang, J.; Qiao, S.; Gao, J.; Ji, W.; et al. Synergistic Double Doping of Co and Cu Constructs Multiple Active Sites to Achieve Catalyzed Degradation of Toluene at High Humidity. Fuel 2023, 342, 127875. [Google Scholar] [CrossRef]
- Golub, K.W.; Sulmonetti, T.P.; Darunte, L.A.; Shealy, M.S.; Jones, C.W. Metal–Organic-Framework-Derived Co/Cu–Carbon Nanoparticle Catalysts for Furfural Hydrogenation. ACS Appl. Nano Mater. 2019, 2, 6040–6056. [Google Scholar] [CrossRef]
- Hao, P.; Xie, M.; Chen, S.; Li, M.; Bi, F.; Zhang, Y.; Lin, M.; Guo, X.; Ding, W.; Guo, X. Surrounded Catalysts Prepared by Ion-Exchange Inverse Loading. Sci. Adv. 2020, 6, eaay7031. [Google Scholar] [CrossRef]
- Munnik, P.; De Jongh, P.E.; De Jong, K.P. Recent Developments in the Synthesis of Supported Catalysts. Chem. Rev. 2015, 115, 6687–6718. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, W.; Zheng, X.; Chen, Y.; Wu, W.; Qiu, J.; Zhao, X.; Zhao, X.; Dai, Y.; Zeng, J. Incorporating Nitrogen Atoms into Cobalt Nanosheets as a Strategy to Boost Catalytic Activity toward CO2 Hydrogenation. Nat. Energy 2017, 2, 869–876. [Google Scholar] [CrossRef]
- Fierro, G.; Jacono, M.L.; Inversi, M.; Dragone, R.; Porta, P. TPR and XPS Study of Cobalt–Copper Mixed Oxide Catalysts: Evidence of a Strong Co–Cu Interaction. Top. Catal. 2000, 10, 39–48. [Google Scholar] [CrossRef]
- Zhang, H.; Mao, D.; Zhang, J.; Wu, D. Regulating the Crystal Structure of Layered Double Hydroxide-Derived Co-In Catalysts for Highly Selective CO2 Hydrogenation to Methanol. Chem. Eng. J. 2023, 452, 139144. [Google Scholar] [CrossRef]
- Dostagir, N.H.M.; Rattanawan, R.; Gao, M.; Ota, J.; Hasegawa, J.; Asakura, K.; Fukouka, A.; Shrotri, A. Co Single Atoms in ZrO2 with Inherent Oxygen Vacancies for Selective Hydrogenation of CO2 to CO. ACS Catal. 2021, 11, 9450–9461. [Google Scholar] [CrossRef]
- Boyce, A.L.; Graville, S.R.; Sermon, P.A.; Vong, M.S.W. Reduction of CuO-Containing Catalysts, CuO: I: TPR and TGA. React. Kinet. Catal. Lett. 1991, 44, 1–11. [Google Scholar] [CrossRef]
- Appel, A.M.; Bercaw, J.E.; Bocarsly, A.B.; Dobbek, H.; DuBois, D.L.; Dupuis, M.; Ferry, J.G.; Fujita, E.; Hille, R.; Kenis, P.J.A.; et al. Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chem. Rev. 2013, 113, 6621–6658. [Google Scholar] [CrossRef]
- Din, I.U.; Alotaibi, M.A.; Alharthi, A.I.; Al-Shalwi, M.N.; Alshehri, F. Green Synthesis Approach for Preparing Zeolite Based Co-Cu Bimetallic Catalysts for Low Temperature CO2 Hydrogenation to Methanol. Fuel 2022, 330, 125643. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Co (wt%) | Cu (wt%) | Co:Cu (mol/mol) | O (wt%) [a] |
|---|---|---|---|---|
| 1CuO/4Co3O4-IM | 46.0 | 11.6 | 3.7 | 42.4% |
| 1CuO/4Co3O4-IE | 54.5 | 17.9 | 3.0 | 27.6% |
| Catalysts | XCO2 (%) | Selectivity (%) | |||
|---|---|---|---|---|---|
| CO | CH4 | C2-C4 | CH3OH | ||
| Co3O4 | 41.3 | 0.7 | 90.0 | 9.3 | ~ |
| 1CuO/4Co3O4-IM | 7.9 | 10.6 | 88.3 | 1.1 | ~ |
| 1CuO/4Co3O4-IE | 5.6 | 5.5 | 58.4 | 0.8 | 35.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheng, Z.; Zhou, H.; Zhang, Y.; Li, J.; Wang, L. Sheet-Like Morphology CuO/Co3O4 Nanocomposites for Enhanced Catalysis in Hydrogenation of CO2 to Methanol. Nanomaterials 2023, 13, 3153. https://doi.org/10.3390/nano13243153
Sheng Z, Zhou H, Zhang Y, Li J, Wang L. Sheet-Like Morphology CuO/Co3O4 Nanocomposites for Enhanced Catalysis in Hydrogenation of CO2 to Methanol. Nanomaterials. 2023; 13(24):3153. https://doi.org/10.3390/nano13243153
Chicago/Turabian StyleSheng, Zhenteng, Hui Zhou, Yuhua Zhang, Jinlin Li, and Li Wang. 2023. "Sheet-Like Morphology CuO/Co3O4 Nanocomposites for Enhanced Catalysis in Hydrogenation of CO2 to Methanol" Nanomaterials 13, no. 24: 3153. https://doi.org/10.3390/nano13243153
APA StyleSheng, Z., Zhou, H., Zhang, Y., Li, J., & Wang, L. (2023). Sheet-Like Morphology CuO/Co3O4 Nanocomposites for Enhanced Catalysis in Hydrogenation of CO2 to Methanol. Nanomaterials, 13(24), 3153. https://doi.org/10.3390/nano13243153