

Exploring Spin-Phonon Coupling in Magnetic 2D Metal-Organic Frameworks


Abstract
:1. Introduction
2. Methods
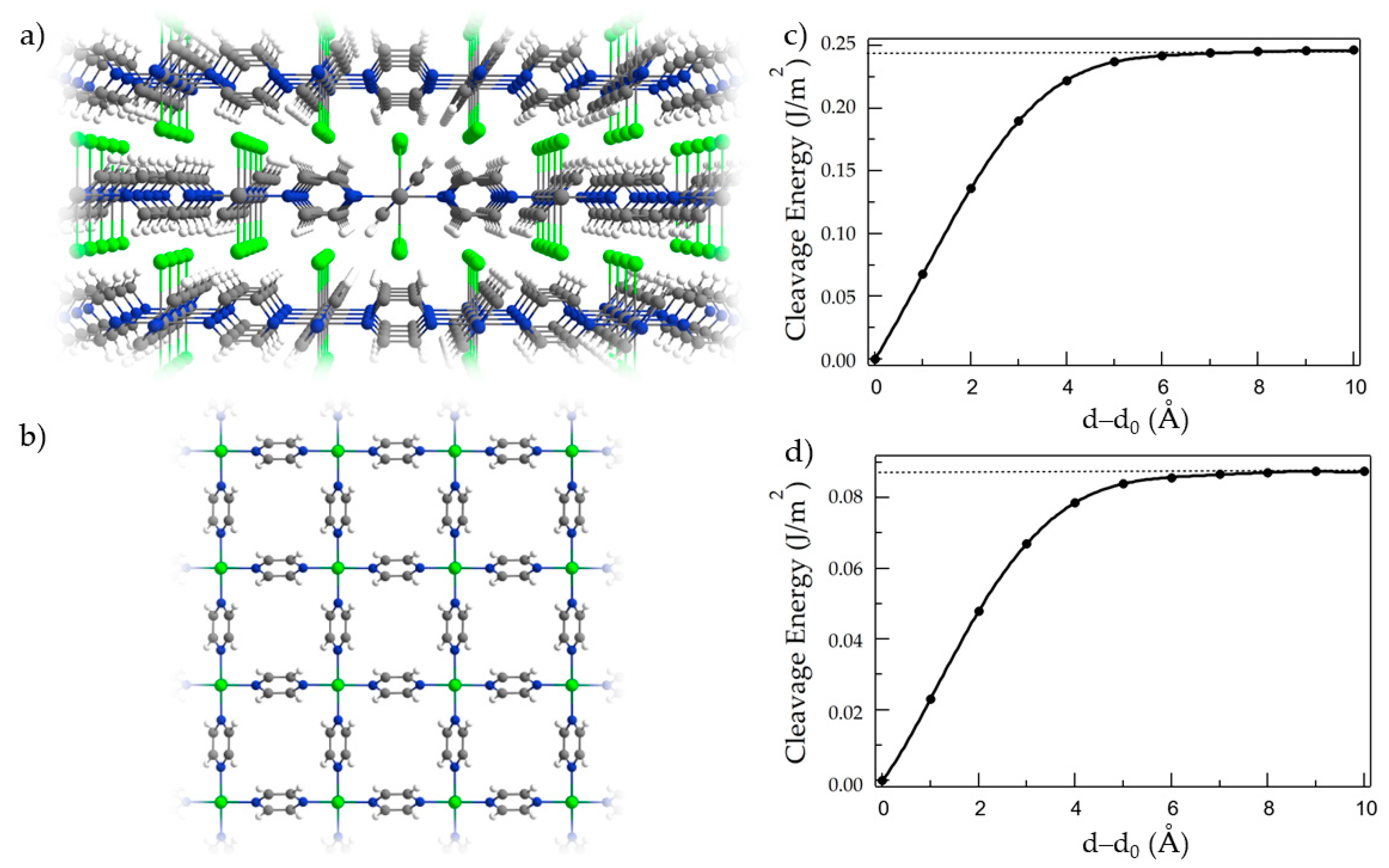
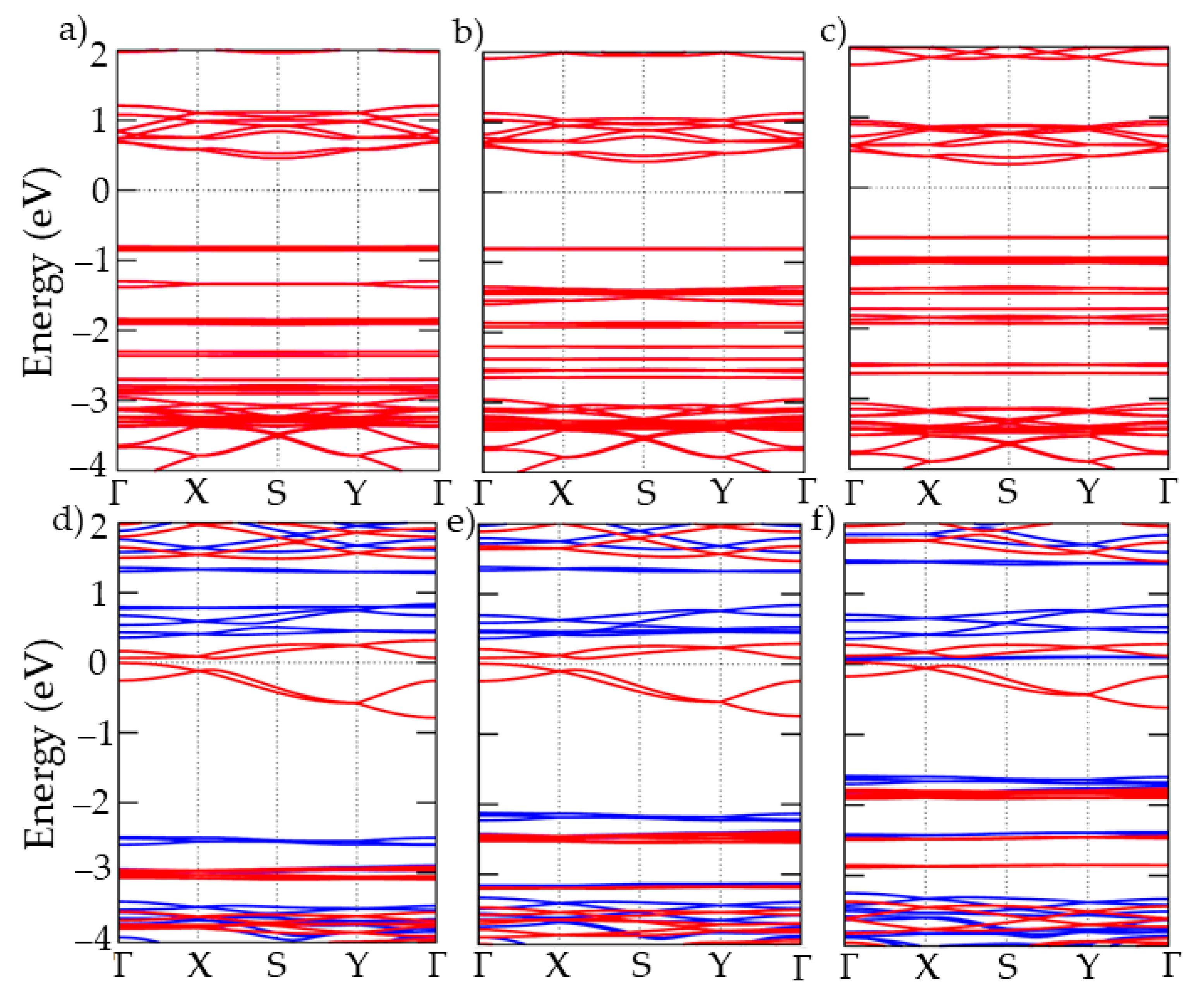
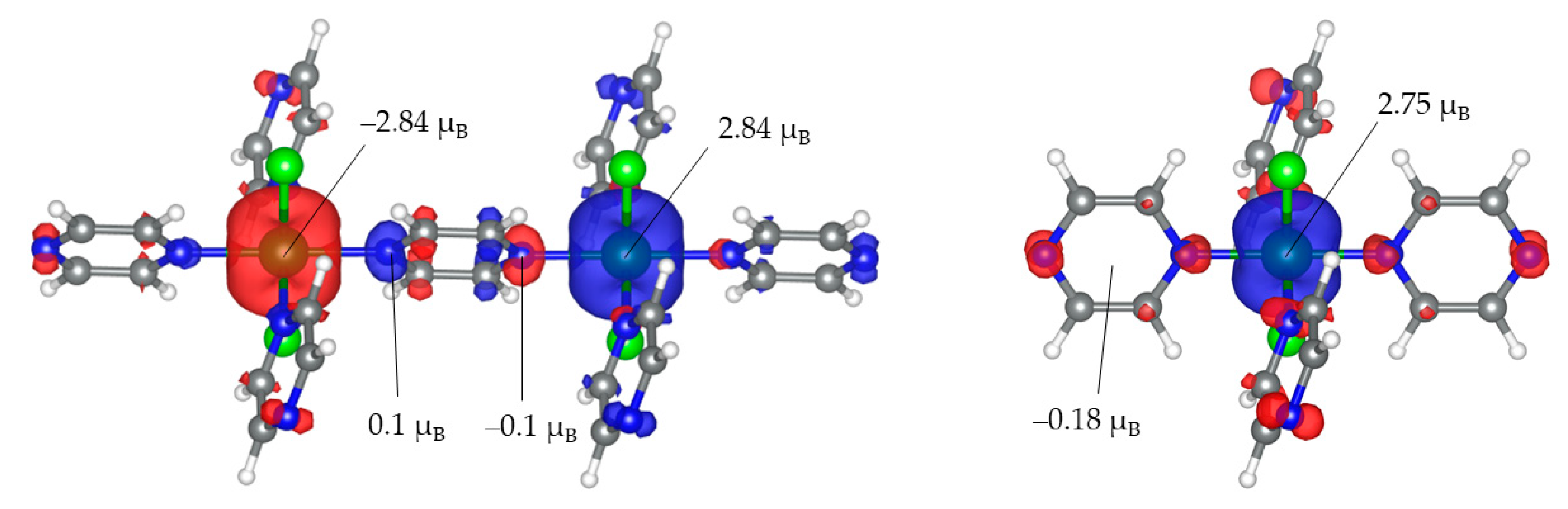
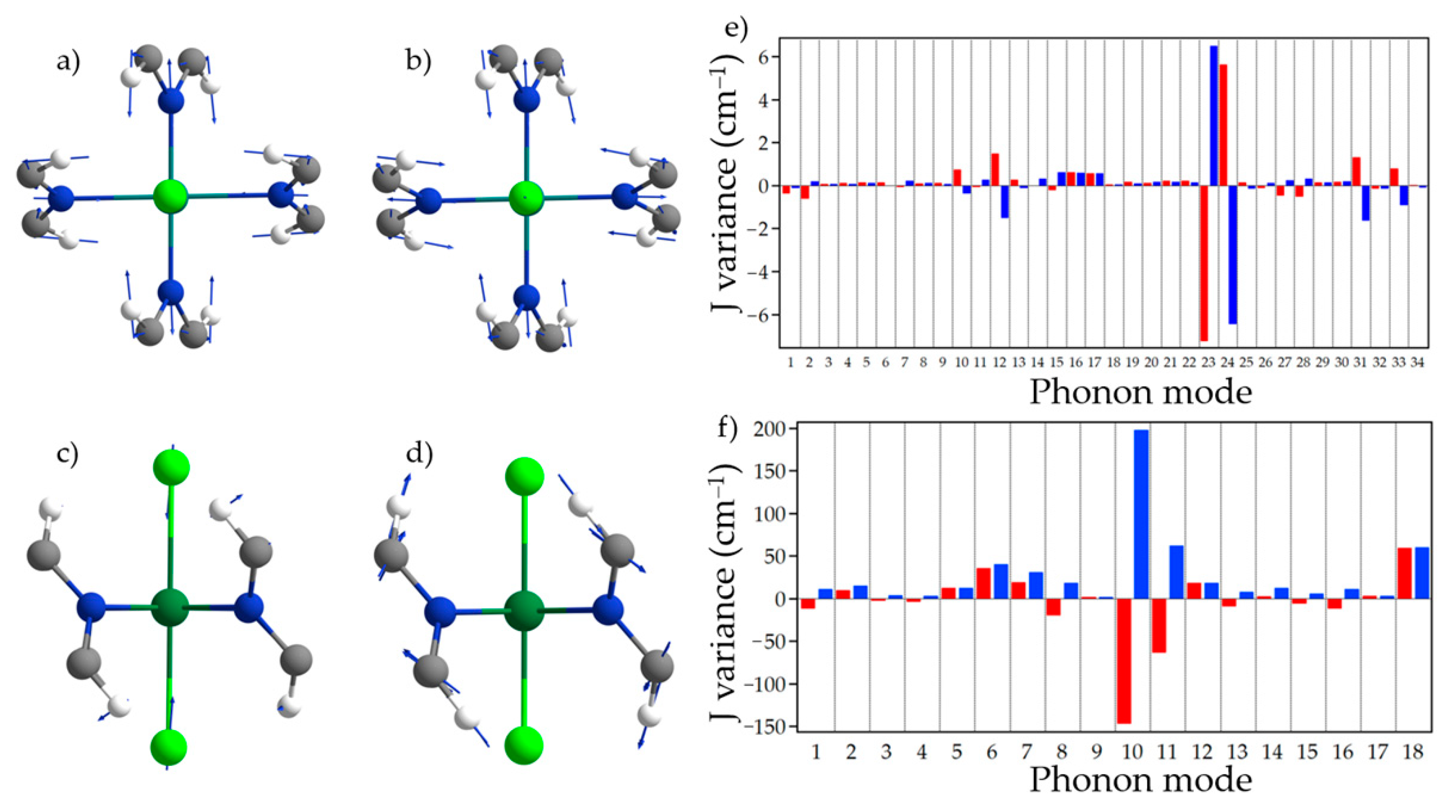
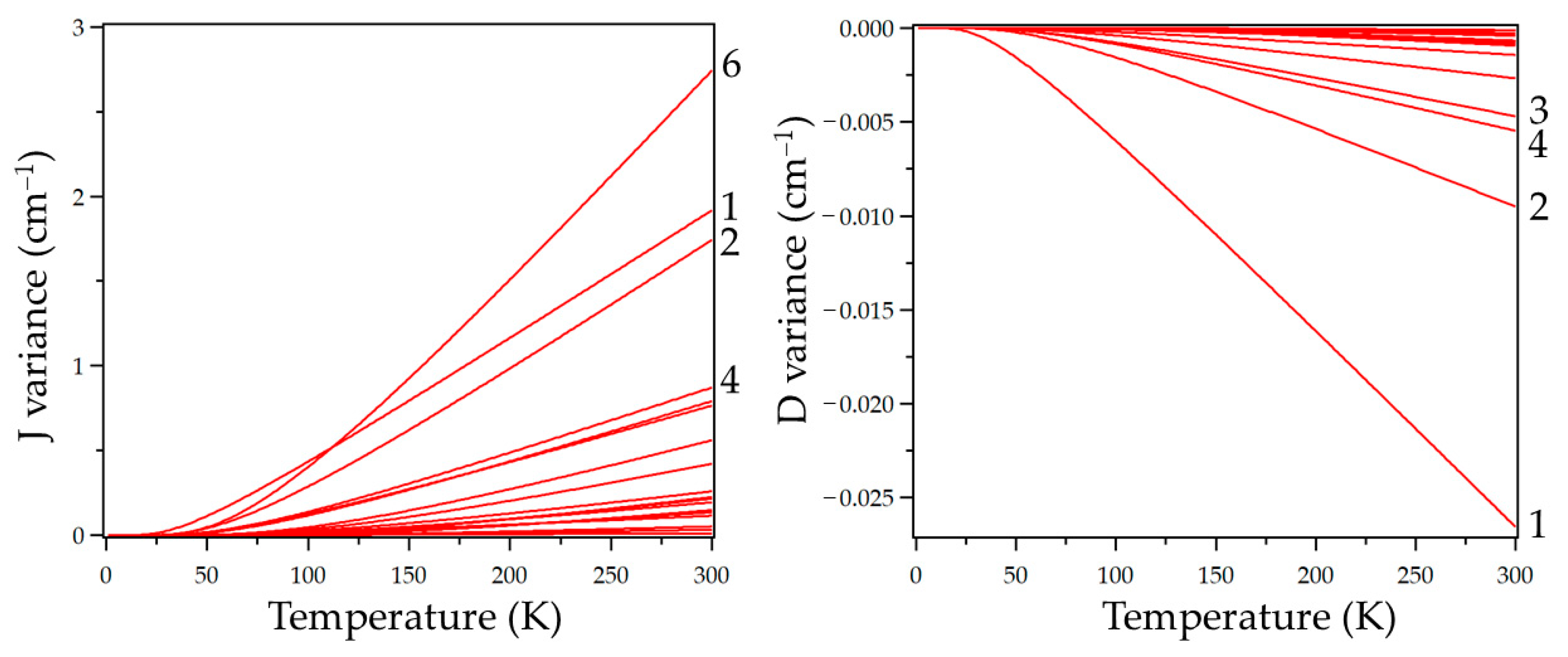
3. Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Furukawa, H.; Cordova, K.E.; O’Keeffe, M.; Yaghi, O.M. The Chemistry and Applications of Metal-Organic Frameworks. Science 2013, 341, 1230444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Xu, H.; Yu, H.; Hu, X.; Xia, J.; Zhu, Y.; Wang, F.; Wu, H.-A.; Jiang, L.; Wang, H. Ultrafast Rectifying Counter-Directional Transport of Proton and Metal Ions in Metal-Organic Framework–Based Nanochannels. Sci. Adv. 2022, 8, eabl5070. [Google Scholar] [CrossRef]
- Huang, Z.; Xiong, C.; Ying, L.; Wang, W.; Wang, S.; Ding, J.; Lu, J. Facile Synthesis of a MOF-Derived Magnetic CoAl-LDH@chitosan Composite for Pb (II) and Cr (VI) Adsorption. Chem. Eng. J. 2022, 449, 137722. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, X.; Yuan, S.; Zhou, J.; Wang, B. Challenges and Recent Advances in MOF–Polymer Composite Membranes for Gas Separation. Inorg. Chem. Front. 2016, 3, 896–909. [Google Scholar] [CrossRef]
- Cui, W.-G.; Zhang, G.-Y.; Hu, T.-L.; Bu, X.-H. Metal-Organic Framework-Based Heterogeneous Catalysts for the Conversion of C1 Chemistry: CO, CO2 and CH4. Coord. Chem. Rev. 2019, 387, 79–120. [Google Scholar] [CrossRef]
- Wu, M.-X.; Yang, Y.-W. Metal-Organic Framework (MOF)-Based Drug/Cargo Delivery and Cancer Therapy. Adv. Mater. 2017, 29, 1606134. [Google Scholar] [CrossRef]
- Liu, C.; Bai, Y.; Li, W.; Yang, F.; Zhang, G.; Pang, H. In Situ Growth of Three-Dimensional MXene/Metal–Organic Framework Composites for High-Performance Supercapacitors. Angew. Chem. 2022, 134, e2021162. [Google Scholar]
- Wu, H.B.; Lou, X.W. (David) Metal-Organic Frameworks and Their Derived Materials for Electrochemical Energy Storage and Conversion: Promises and Challenges. Sci. Adv. 2017, 3, eaap9252. [Google Scholar] [CrossRef] [Green Version]
- Damron, J.T.; Ma, J.; Kurz, R.; Saalwächter, K.; Matzger, A.J.; Ramamoorthy, A. The Influence of Chemical Modification on Linker Rotational Dynamics in Metal–Organic Frameworks. Angew. Chem. 2018, 130, 8814–8817. [Google Scholar] [CrossRef]
- Tanabe, K.K.; Cohen, S.M. Postsynthetic Modification of Metal–Organic Frameworks—A Progress Report. Chem. Soc. Rev. 2011, 40, 498–519. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Natarajan, S.; Mani, P.; Pankajakshan, A. Post-Synthetic Modification of Metal–Organic Frameworks toward Applications. Adv. Funct. Mater. 2021, 31, 2006291. [Google Scholar] [CrossRef]
- Kurmoo, M. Magnetic Metal–Organic Frameworks. Chem. Soc. Rev. 2009, 38, 1353. [Google Scholar] [CrossRef]
- Thorarinsdottir, A.E.; Harris, T.D. Metal–Organic Framework Magnets. Chem. Rev. 2020, 120, 8716–8789. [Google Scholar] [CrossRef]
- Song, X.; Wang, X.; Li, Y.; Zheng, C.; Zhang, B.; Di, C.; Li, F.; Jin, C.; Mi, W.; Chen, L.; et al. 2D Semiconducting Metal–Organic Framework Thin Films for Organic Spin Valves. Angew. Chem. Int. Ed. 2020, 59, 1118–1123. [Google Scholar] [CrossRef]
- Barman, A.; Gubbiotti, G.; Ladak, S.; Adeyeye, A.O.; Krawczyk, M.; Gräfe, J.; Adelmann, C.; Cotofana, S.; Naeemi, A.; Vasyuchka, V.I.; et al. The 2021 Magnonics Roadmap. J. Phys. Condens. Matter 2021, 33, 413001. [Google Scholar] [CrossRef]
- Manriquez, J.M.; Yee, G.T.; McLean, R.S.; Epstein, A.J.; Miller, J.S. A Room-Temperature Molecular/Organic-Based Magnet. Science 1991, 252, 1415–1417. [Google Scholar] [CrossRef] [PubMed]
- Perlepe, P.; Oyarzabal, I.; Mailman, A.; Yquel, M.; Platunov, M.; Dovgaliuk, I.; Rouzières, M.; Négrier, P.; Mondieig, D.; Suturina, E.A.; et al. Metal-Organic Magnets with Large Coercivity and Ordering Temperatures up to 242 °C. Science 2020, 370, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Perlepe, P.; Oyarzabal, I.; Voigt, L.; Kubus, M.; Woodruff, D.N.; Reyes-Lillo, S.E.; Aubrey, M.L.; Négrier, P.; Rouzières, M.; Wilhelm, F.; et al. From an Antiferromagnetic Insulator to a Strongly Correlated Metal in Square-Lattice MCl2(Pyrazine)2 Coordination Solids. Nat. Commun. 2022, 13, 5766. [Google Scholar] [CrossRef]
- Pedersen, K.S.; Perlepe, P.; Aubrey, M.L.; Woodruff, D.N.; Reyes-Lillo, S.E.; Reinholdt, A.; Voigt, L.; Li, Z.; Borup, K.; Rouzières, M.; et al. Formation of the Layered Conductive Magnet CrCl2(Pyrazine)2 through Redox-Active Coordination Chemistry. Nat. Chem. 2018, 10, 1056–1061. [Google Scholar] [CrossRef]
- Dong, R.; Han, P.; Arora, H.; Ballabio, M.; Karakus, M.; Zhang, Z.; Shekhar, C.; Adler, P.; Petkov, P.S.; Erbe, A.; et al. High-Mobility Band-like Charge Transport in a Semiconducting Two-Dimensional Metal–Organic Framework. Nat. Mater. 2018, 17, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Martinez, A.; Wang, F. Optical Modulators with 2D Layered Materials. Nat. Photonics 2016, 10, 227–238. [Google Scholar] [CrossRef] [Green Version]
- López-Cabrelles, J.; Mañas-Valero, S.; Vitórica-Yrezábal, I.J.; Bereciartua, P.J.; Rodríguez-Velamazán, J.A.; Waerenborgh, J.C.; Vieira, B.J.C.; Davidovikj, D.; Steeneken, P.G.; van der Zant, H.S.J.; et al. Isoreticular Two-Dimensional Magnetic Coordination Polymers Prepared through Pre-Synthetic Ligand Functionalization. Nat. Chem. 2018, 10, 1001–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, N.; Zhou, C.H.; Tong, D.S.; Yu, W.H.; Cynthia Lin, C.X. Exfoliation of Layered Double Hydroxide Solids into Functional Nanosheets. Appl. Clay Sci. 2017, 144, 60–78. [Google Scholar] [CrossRef]
- Liu, L.; Lu, X.-Y.; Zhang, M.-L.; Ren, Y.-X.; Wang, J.-J.; Yang, X.-G. 2D MOF Nanosheets as an Artificial Light-Harvesting System with Enhanced Photoelectric Switching Performance. Inorg. Chem. Front. 2022, 9, 2676–2682. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Y.; Feng, X.; Dong, R. Conductive 2D Conjugated Metal–Organic Framework Thin Films: Synthesis and Functions for (Opto-)Electronics. Small Struct. 2022, 3, 2100210. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, J.; Gao, Y.; Hua, M.; Xia, B.; Knecht, P.; Papageorgiou, A.C.; Reichert, J.; Barth, J.V.; Xu, H.; et al. On-surface Synthesis of a Semiconducting 2D Metal–Organic Framework Cu3 (C6O6) Exhibiting Dispersive Electronic Bands. Angew. Chem. 2020, 132, 2691–2695. [Google Scholar] [CrossRef]
- Darago, L.E.; Aubrey, M.L.; Yu, C.J.; Gonzalez, M.I.; Long, J.R. Electronic Conductivity, Ferrimagnetic Ordering, and Reductive Insertion Mediated by Organic Mixed-Valence in a Ferric Semiquinoid Metal–Organic Framework. J. Am. Chem. Soc. 2015, 137, 15703–15711. [Google Scholar] [CrossRef]
- Tiana, D.; Hendon, C.H.; Walsh, A. Ligand Design for Long-Range Magnetic Order in Metal–Organic Frameworks. Chem. Commun. 2014, 50, 13990–13993. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.H.; Hou, Y.S.; Wang, Z.; Wu, R.Q. First-Principles Studies of Spin-Phonon Coupling in Monolayer Cr2Ge2Te6. Phys. Rev. B 2019, 100, 224427. [Google Scholar] [CrossRef] [Green Version]
- Moseley, D.H.; Stavretis, S.E.; Thirunavukkuarasu, K.; Ozerov, M.; Cheng, Y.; Daemen, L.L.; Ludwig, J.; Lu, Z.; Smirnov, D.; Brown, C.M.; et al. Spin–Phonon Couplings in Transition Metal Complexes with Slow Magnetic Relaxation. Nat. Commun. 2018, 9, 2572. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A Modular and Open-Source Software Project for Quantum Simulations of Materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof Exchange-Correlation Functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-Energy-Loss Spectra and the Structural Stability of Nickel Oxide: An LSDA+U Study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]
- Yamanaka, S.; Kawakami, T.; Nagao, H.; Yamaguchi, K. Effective Exchange Integrals for Open-Shell Species by Density Functional Methods. Chem. Phys. Lett. 1994, 231, 25–33. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Glukhovtsev, M.N.; Pross, A.; McGrath, M.P.; Radom, L. Extension of Gaussian-2 (G2) Theory to Bromine- and Iodine-containing Molecules: Use of Effective Core Potentials. J. Chem. Phys. 1995, 103, 1878–1885. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Evangelisti, S.; Leininger, T.; Malrieu, J.-P. Introduction of n-Electron Valence States for Multireference Perturbation Theory. J. Chem. Phys. 2001, 114, 10252–10264. [Google Scholar] [CrossRef]
- Neese, F. The ORCA Program System. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System, Version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-Functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Björkman, T.; Gulans, A.; Krasheninnikov, A.V.; Nieminen, R.M. Van Der Waals Bonding in Layered Compounds from Advanced Density-Functional First-Principles Calculations. Phys. Rev. Lett. 2012, 108, 235502. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Wang, G.; Liu, L.; Lu, D.; Wu, H. Triaxial Magnetic Anisotropy in the Two-Dimensional Ferromagnetic Semiconductor CrSBr. Phys. Rev. B 2021, 104, 144416. [Google Scholar] [CrossRef]
- McGuire, M.A.; Dixit, H.; Cooper, V.R.; Sales, B.C. Coupling of Crystal Structure and Magnetism in the Layered, Ferromagnetic Insulator Cri3. Chem. Mater. 2015, 27, 612–620. [Google Scholar] [CrossRef]
- Setyawan, W.; Curtarolo, S. High-Throughput Electronic Band Structure Calculations: Challenges and Tools. Comput. Mater. Sci. 2010, 49, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, E.; Cano, J.; Alvarez, S.; Alemany, P. Broken Symmetry Approach to Calculation of Exchange Coupling Constants for Homobinuclear and Heterobinuclear Transition Metal Complexes. J. Comput. Chem. 1999, 20, 1391–1400. [Google Scholar] [CrossRef]
- Chen, L.; Stone, M.B.; Kolesnikov, A.I.; Winn, B.; Shon, W.; Dai, P.; Chung, J.H. Massless Dirac Magnons in the Two Dimensional van Der Waals Honeycomb Magnet CrCl3. 2D Mater. 2022, 9, 015006. [Google Scholar] [CrossRef]
- Escalera-Moreno, L.; Suaud, N.; Gaita-Ariño, A.; Coronado, E. Determining Key Local Vibrations in the Relaxation of Molecular Spin Qubits and Single-Molecule Magnets. J. Phys. Chem. Lett. 2017, 8, 1695–1700. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Malissa, H.; Stolley, R.M.; Singh, J.; Groesbeck, M.; Popli, H.; Kavand, M.; Chong, S.K.; Deshpande, V.V.; Miller, J.S.; et al. Spin Wave Excitation, Detection, and Utilization in the Organic-Based Magnet, V(TCNE)x (TCNE = Tetracyanoethylene). Adv. Mater. 2020, 32, 2002663. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Kong, Y.; Wang, C.; Tan, H.; Duan, H.; Hu, W.; Li, G.; Lu, Y.; Li, N.; Wang, Y.; et al. Lattice Strain Induced by Linker Scission in Metal–Organic Framework Nanosheets for Oxygen Evolution Reaction. ACS Catal. 2020, 10, 5691–5697. [Google Scholar] [CrossRef]
- Esteras, D.L.; Rybakov, A.; Ruiz, A.M.; Baldoví, J.J. Magnon Straintronics in the 2D van Der Waals Ferromagnet CrSBr from First-Principles. Nano Lett. 2022, 22, 8771–8778. [Google Scholar] [CrossRef] [PubMed]
- Cimpoesu, F.; Frecus, B.; Oprea, C.I.; Panait, P.; Gîrţu, M.A. Disorder, Exchange and Magnetic Anisotropy in the Room-Temperature Molecular Magnet V[TCNE]x—A Theoretical Study. Comput. Mater. Sci. 2014, 91, 320–328. [Google Scholar] [CrossRef]
- Frecus, B.; Oprea, C.I.; Panait, P.; Ferbinteanu, M.; Cimpoesu, F.; Gîrţu, M.A. Ab Initio Study of Exchange Coupling for the Consistent Understanding of the Magnetic Ordering at Room Temperature in V[TCNE]x. Theor. Chem. Acc. 2014, 133, 1–17. [Google Scholar] [CrossRef]
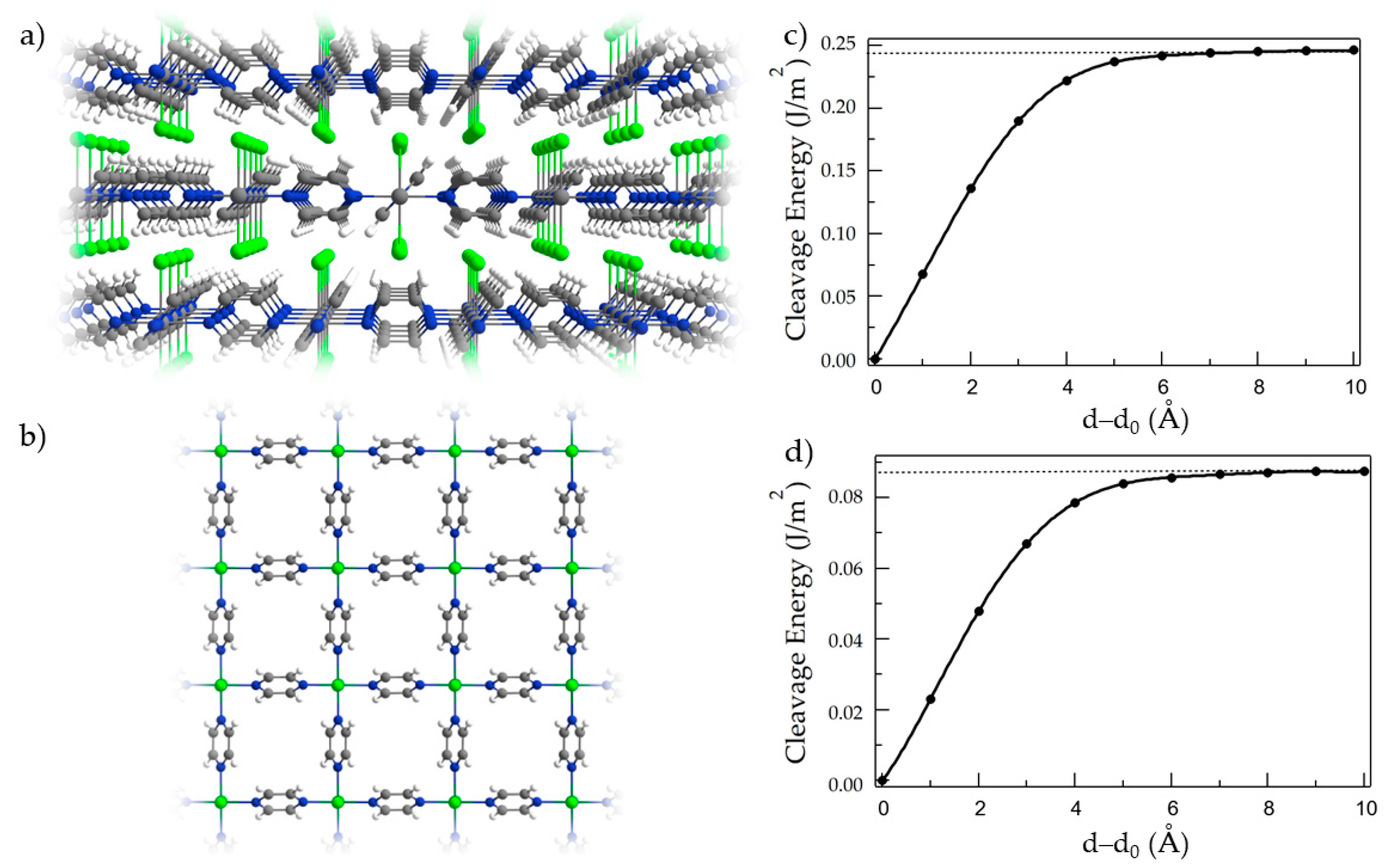
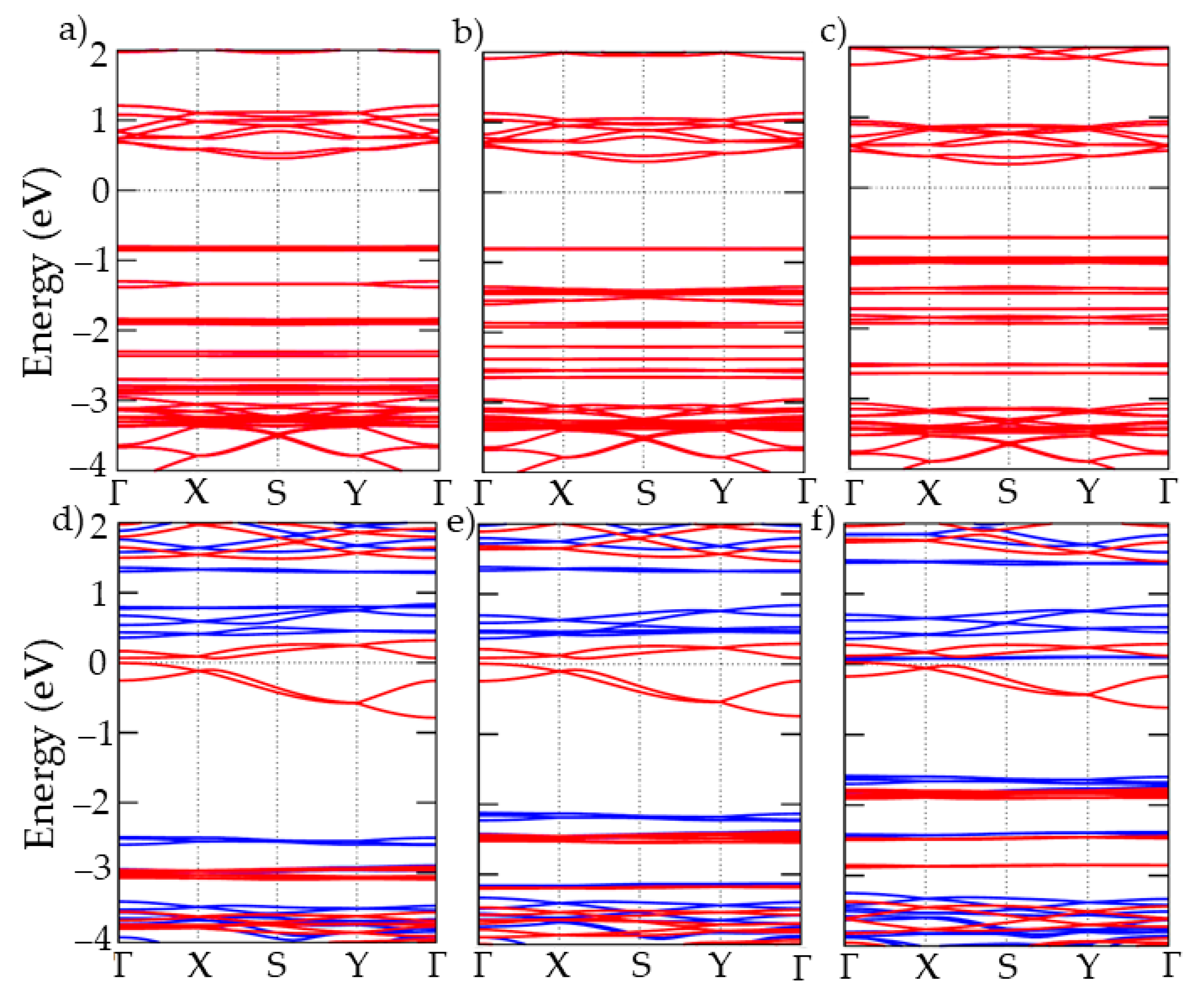

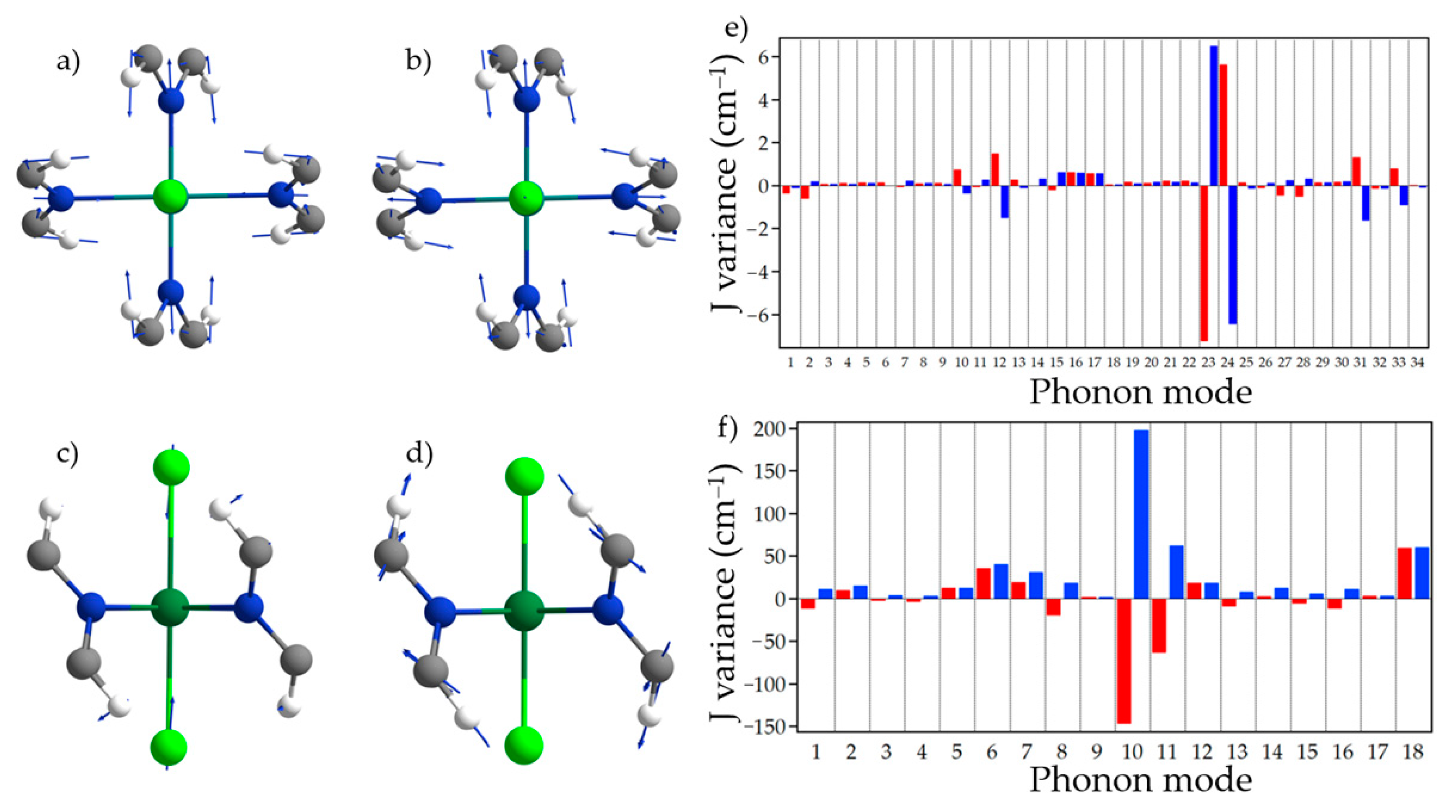
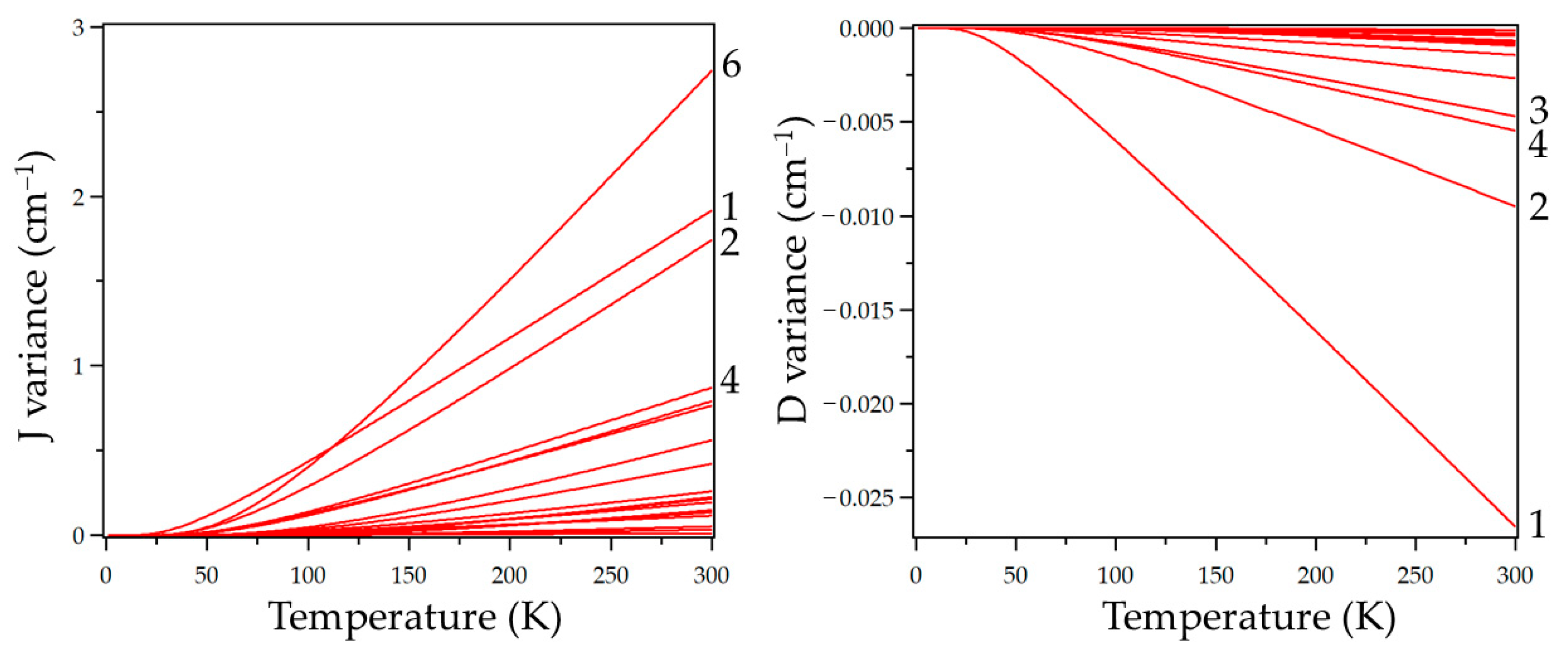

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| J (cm−1) | ||
|---|---|---|
| X | VX2(pyz)2 | CrX2(pyz)2 |
| Cl | −40.57 | −2034.13 |
| Br | −37.02 | −1480.83 |
| I | −34.43 | −549.66 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Alcalá, D.; Ruiz, A.M.; Baldoví, J.J. Exploring Spin-Phonon Coupling in Magnetic 2D Metal-Organic Frameworks. Nanomaterials 2023, 13, 1172. https://doi.org/10.3390/nano13071172
López-Alcalá D, Ruiz AM, Baldoví JJ. Exploring Spin-Phonon Coupling in Magnetic 2D Metal-Organic Frameworks. Nanomaterials. 2023; 13(7):1172. https://doi.org/10.3390/nano13071172
Chicago/Turabian StyleLópez-Alcalá, Diego, Alberto M. Ruiz, and José J. Baldoví. 2023. "Exploring Spin-Phonon Coupling in Magnetic 2D Metal-Organic Frameworks" Nanomaterials 13, no. 7: 1172. https://doi.org/10.3390/nano13071172
APA StyleLópez-Alcalá, D., Ruiz, A. M., & Baldoví, J. J. (2023). Exploring Spin-Phonon Coupling in Magnetic 2D Metal-Organic Frameworks. Nanomaterials, 13(7), 1172. https://doi.org/10.3390/nano13071172