
The Effect of Surface Oxygen Coverage on the Oxygen Evolution Reaction over a CoFeNiCr High-Entropy Alloy

Abstract
:
1. Introduction
2. Materials and Methods
3. Results
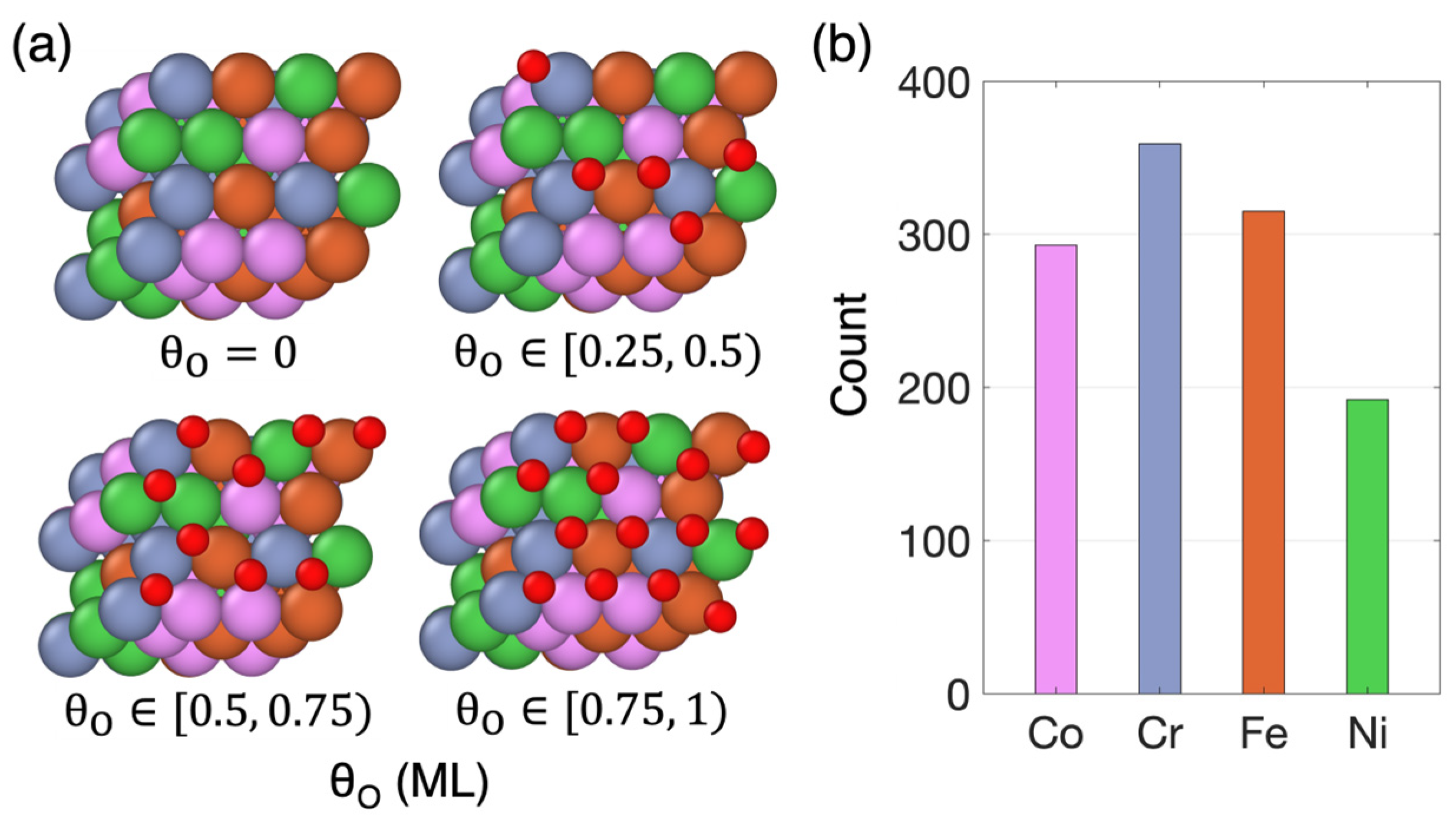
3.1. Non-Uniform Metal Oxidation
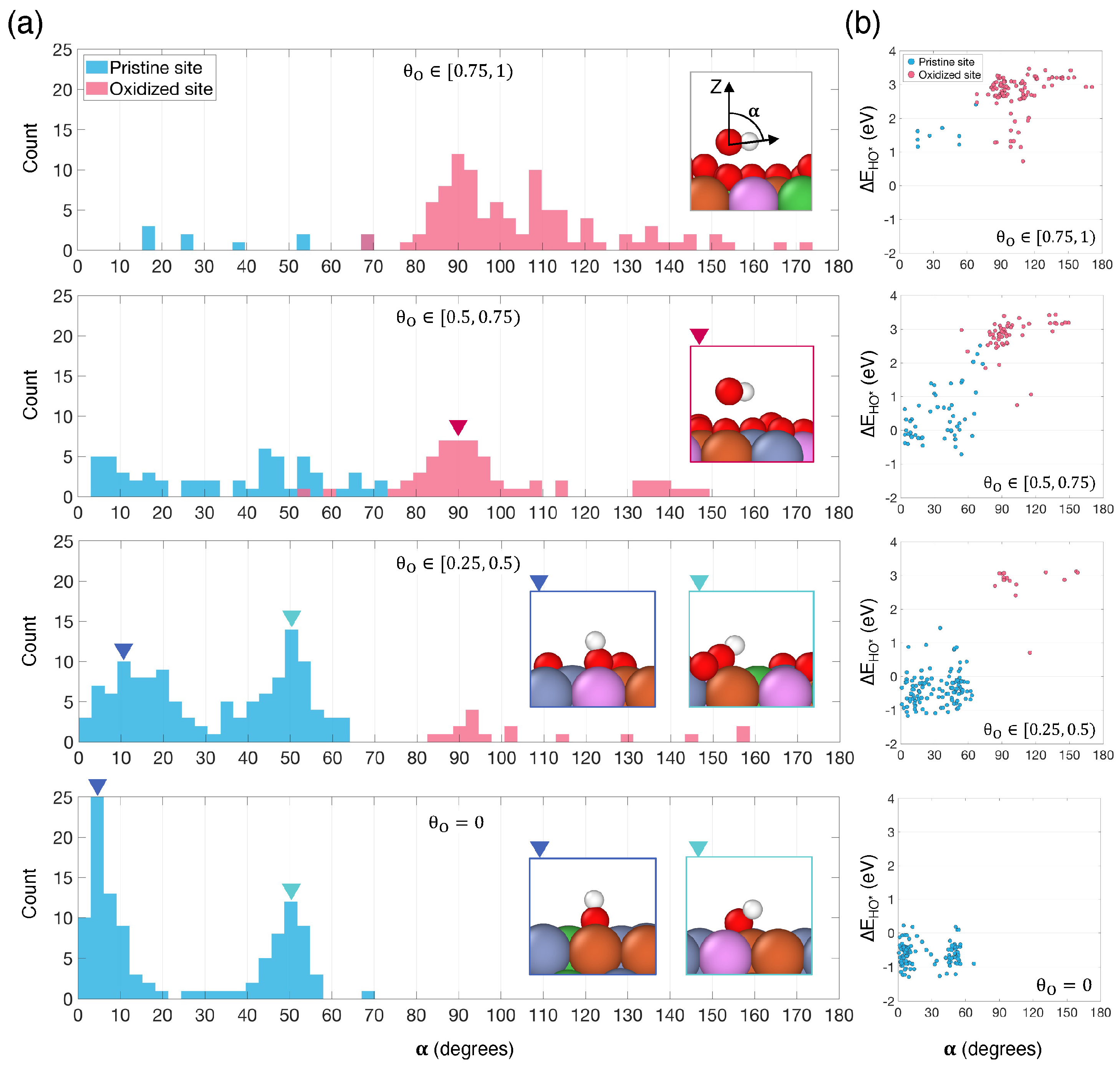
3.2. Surface Oxygen Coverage Impact on OER Intermediate Adsorption
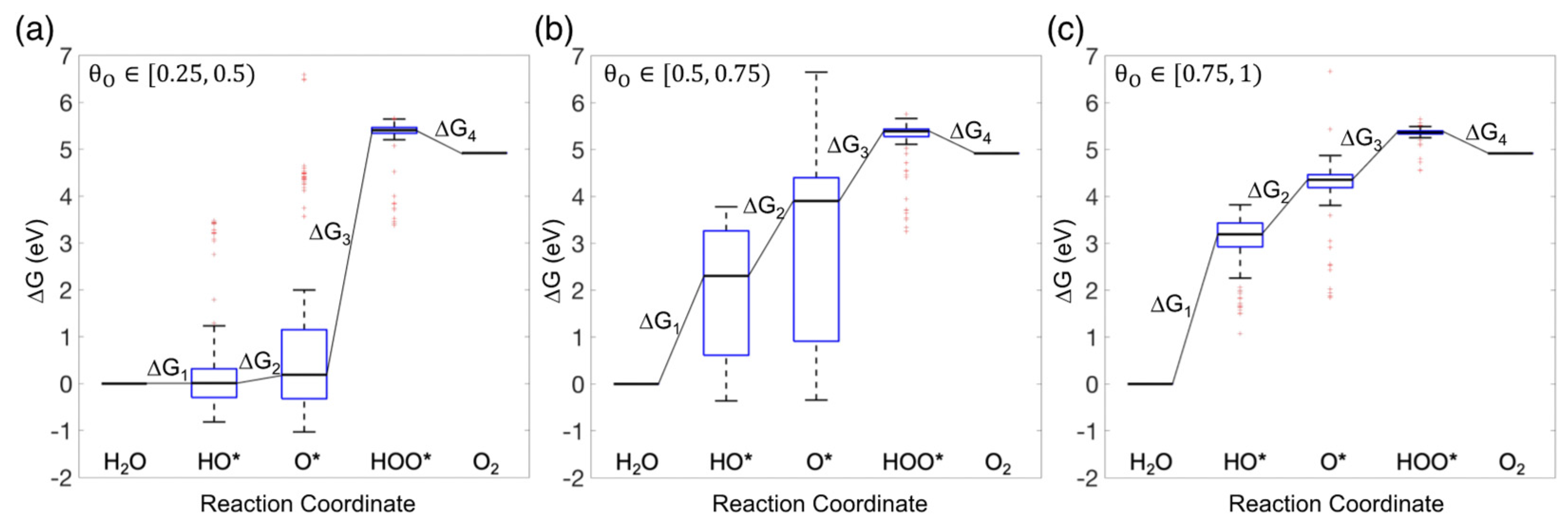
3.3. Free Energy Diagram for the OER
3.4. Theoretical Overpotential for OER
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chu, S.; Majumdar, A. Opportunities and Challenges for a Sustainable Energy Future. Nature 2012, 488, 294–303. [Google Scholar] [CrossRef]
- Bockris, J.O.; Veziroglu, T.N. Estimates of the Price of Hydrogen as a Medium for Wind and Solar Sources. Int. J. Hydrogen Energy 2007, 32, 1605–1610. [Google Scholar] [CrossRef]
- Hassan, N.S.; Jalil, A.A.; Rajendran, S.; Khusnun, N.F.; Bahari, M.B.; Johari, A.; Kamaruddin, M.J.; Ismail, M. Recent Review and Evaluation of Green Hydrogen Production via Water Electrolysis for a Sustainable and Clean Energy Society. Int. J. Hydrogen Energy 2024, 52, 420–441. [Google Scholar] [CrossRef]
- Trotochaud, L.; Boettcher, S.W. Precise Oxygen Evolution Catalysts: Status and Opportunities. Scr. Mater. 2014, 74, 25–32. [Google Scholar] [CrossRef]
- Suen, N.-T.; Hung, S.-F.; Quan, Q.; Zhang, N.; Xu, Y.-J.; Ming Chen, H. Electrocatalysis for the Oxygen Evolution Reaction: Recent Development and Future Perspectives. Chem. Soc. Rev. 2017, 46, 337–365. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Qu, Z.-W.; Zhu, H.; Kroes, G.-J.; Nørskov, J.K. Electrolysis of Water on Oxide Surfaces. J. Electroanal. Chem. 2007, 607, 83–89. [Google Scholar] [CrossRef]
- Lee, Y.; Suntivich, J.; May, K.J.; Perry, E.E.; Shao-Horn, Y. Synthesis and Activities of Rutile IrO2 and RuO2 Nanoparticles for Oxygen Evolution in Acid and Alkaline Solutions. J. Phys. Chem. Lett. 2012, 3, 399–404. [Google Scholar] [CrossRef]
- Shi, Q.; Zhu, C.; Du, D.; Lin, Y. Robust Noble Metal-Based Electrocatalysts for Oxygen Evolution Reaction. Chem. Soc. Rev. 2019, 48, 3181–3192. [Google Scholar] [CrossRef]
- Man, I.C.; Su, H.-Y.; Calle-Vallejo, F.; Hansen, H.A.; Martínez, J.I.; Inoglu, N.G.; Kitchin, J.; Jaramillo, T.F.; Nørskov, J.K.; Rossmeisl, J. Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces. ChemCatChem 2011, 3, 1159–1165. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Sato, E. Electrocatalytic Properties of Transition Metal Oxides for Oxygen Evolution Reaction. Mater. Chem. Phys. 1986, 14, 397–426. [Google Scholar] [CrossRef]
- Xin, Y.; Li, S.; Qian, Y.; Zhu, W.; Yuan, H.; Jiang, P.; Guo, R.; Wang, L. High-Entropy Alloys as a Platform for Catalysis: Progress, Challenges, and Opportunities. ACS Catal. 2020, 10, 11280–11306. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, D.; Wang, S. High-Entropy Alloys for Electrocatalysis: Design, Characterization, and Applications. Small 2022, 18, 2104339. [Google Scholar] [CrossRef]
- Ren, J.-T.; Chen, L.; Wang, H.-Y.; Yuan, Z.-Y. High-Entropy Alloys in Electrocatalysis: From Fundamentals to Applications. Chem. Soc. Rev. 2023, 52, 8319–8373. [Google Scholar] [CrossRef]
- Yeh, J.-W.; Chen, S.-K.; Lin, S.-J.; Gan, J.-Y.; Chin, T.-S.; Shun, T.-T.; Tsau, C.-H.; Chang, S.-Y. Nanostructured High-Entropy Alloys with Multiple Principal Elements: Novel Alloy Design Concepts and Outcomes. Adv. Eng. Mater. 2004, 6, 299–303. [Google Scholar] [CrossRef]
- Troparevsky, M.C.; Morris, J.R.; Kent, P.R.; Lupini, A.R.; Stocks, G.M. Criteria for Predicting the Formation of Single-Phase High-Entropy Alloys. Phys. Rev. X 2015, 5, 011041. [Google Scholar] [CrossRef]
- King, D.; Middleburgh, S.; McGregor, A.; Cortie, M. Predicting the Formation and Stability of Single Phase High-Entropy Alloys. Acta Mater. 2016, 104, 172–179. [Google Scholar] [CrossRef]
- George, E.P.; Raabe, D.; Ritchie, R.O. High-Entropy Alloys. Nat. Rev. Mater. 2019, 4, 515–534. [Google Scholar] [CrossRef]
- Löffler, T.; Ludwig, A.; Rossmeisl, J.; Schuhmann, W. What Makes High-Entropy Alloys Exceptional Electrocatalysts? Angew. Chem. Int. Ed. 2021, 60, 26894–26903. [Google Scholar] [CrossRef]
- McCrory, C.C.L.; Jung, S.; Peters, J.C.; Jaramillo, T.F. Benchmarking Heterogeneous Electrocatalysts for the Oxygen Evolution Reaction. J. Am. Chem. Soc. 2013, 135, 16977–16987. [Google Scholar] [CrossRef]
- Liu, C.; Colón, B.C.; Ziesack, M.; Silver, P.A.; Nocera, D.G. Water Splitting–Biosynthetic System with CO2 Reduction Efficiencies Exceeding Photosynthesis. Science 2016, 352, 1210–1213. [Google Scholar] [CrossRef]
- Nardi, K.L.; Yang, N.; Dickens, C.F.; Strickler, A.L.; Bent, S.F. Creating Highly Active Atomic Layer Deposited NiO Electrocatalysts for the Oxygen Evolution Reaction. Adv. Energy Mater. 2015, 5, 1500412. [Google Scholar] [CrossRef]
- Gong, M.; Li, Y.; Wang, H.; Liang, Y.; Wu, J.Z.; Zhou, J.; Wang, J.; Regier, T.; Wei, F.; Dai, H. An Advanced Ni–Fe Layered Double Hydroxide Electrocatalyst for Water Oxidation. J. Am. Chem. Soc. 2013, 135, 8452–8455. [Google Scholar] [CrossRef]
- Chen, S.; Qiao, S.-Z. Hierarchically Porous Nitrogen-Doped Graphene–NiCo2O4 Hybrid Paper as an Advanced Electrocatalytic Water-Splitting Material. ACS Nano 2013, 7, 10190–10196. [Google Scholar] [CrossRef]
- Gao, M.; Sheng, W.; Zhuang, Z.; Fang, Q.; Gu, S.; Jiang, J.; Yan, Y. Efficient Water Oxidation Using Nanostructured α-Nickel-Hydroxide as an Electrocatalyst. J. Am. Chem. Soc. 2014, 136, 7077–7084. [Google Scholar] [CrossRef]
- Chen, J.; Zheng, F.; Zhang, S.-J.; Fisher, A.; Zhou, Y.; Wang, Z.; Li, Y.; Xu, B.-B.; Li, J.-T.; Sun, S.-G. Interfacial Interaction between FeOOH and Ni–Fe LDH to Modulate the Local Electronic Structure for Enhanced OER Electrocatalysis. ACS Catal. 2018, 8, 11342–11351. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Liu, Z.; Xie, C.; Feng, S.; Liu, D.; Shao, M.; Wang, S. Layered Double Hydroxide Nanosheets with Multiple Vacancies Obtained by Dry Exfoliation as Highly Efficient Oxygen Evolution Electrocatalysts. Angew. Chem. Int. Ed. 2017, 56, 5867–5871. [Google Scholar] [CrossRef]
- Liu, R.; Wang, Y.; Liu, D.; Zou, Y.; Wang, S. Water-Plasma-Enabled Exfoliation of Ultrathin Layered Double Hydroxide Nanosheets with Multivacancies for Water Oxidation. Adv. Mater. 2017, 29, 1701546. [Google Scholar] [CrossRef]
- Jin, Z.; Lv, J.; Jia, H.; Liu, W.; Li, H.; Chen, Z.; Lin, X.; Xie, G.; Liu, X.; Sun, S.; et al. Nanoporous Al-Ni-Co-Ir-Mo High-Entropy Alloy for Record-High Water Splitting Activity in Acidic Environments. Small 2019, 15, 1904180. [Google Scholar] [CrossRef]
- Qiu, H.-J.; Fang, G.; Gao, J.; Wen, Y.; Lv, J.; Li, H.; Xie, G.; Liu, X.; Sun, S. Noble Metal-Free Nanoporous High-Entropy Alloys as Highly Efficient Electrocatalysts for Oxygen Evolution Reaction. ACS Mater. Lett. 2019, 1, 526–533. [Google Scholar] [CrossRef]
- Waag, F.; Li, Y.; Rosa Ziefuß, A.; Bertin, E.; Kamp, M.; Duppel, V.; Marzun, G.; Kienle, L.; Barcikowski, S.; Gökce, B. Kinetically-Controlled Laser-Synthesis of Colloidal High-Entropy Alloy Nanoparticles. RSC Adv. 2019, 9, 18547–18558. [Google Scholar] [CrossRef]
- Huang, K.; Zhang, B.; Wu, J.; Zhang, T.; Peng, D.; Cao, X.; Zhang, Z.; Li, Z.; Huang, Y. Exploring the Impact of Atomic Lattice Deformation on Oxygen Evolution Reactions Based on a Sub-5 Nm Pure Face-Centred Cubic High-Entropy Alloy Electrocatalyst. J. Mater. Chem. A 2020, 8, 11938–11947. [Google Scholar] [CrossRef]
- Dai, W.; Lu, T.; Pan, Y. Novel and Promising Electrocatalyst for Oxygen Evolution Reaction Based on MnFeCoNi High Entropy Alloy. J. Power Sources 2019, 430, 104–111. [Google Scholar] [CrossRef]
- Guan, D.; Xu, H.; Zhang, Q.; Huang, Y.-C.; Shi, C.; Chang, Y.-C.; Xu, X.; Tang, J.; Gu, Y.; Pao, C.-W.; et al. Identifying a Universal Activity Descriptor and a Unifying Mechanism Concept on Perovskite Oxides for Green Hydrogen Production. Adv. Mater. 2023, 35, 2305074. [Google Scholar] [CrossRef]
- Guan, D.; Zhong, J.; Xu, H.; Huang, Y.-C.; Hu, Z.; Chen, B.; Zhang, Y.; Ni, M.; Xu, X.; Zhou, W.; et al. A Universal Chemical-Induced Tensile Strain Tuning Strategy to Boost Oxygen-Evolving Electrocatalysis on Perovskite Oxides. Appl. Phys. Rev. 2022, 9, 011422. [Google Scholar] [CrossRef]
- He, R.; Yang, L.; Zhang, Y.; Jiang, D.; Lee, S.; Horta, S.; Liang, Z.; Lu, X.; Ostovari Moghaddam, A.; Li, J.; et al. A 3d-4d-5d High Entropy Alloy as a Bifunctional Oxygen Catalyst for Robust Aqueous Zinc–Air Batteries. Adv. Mater. 2023, 35, 2303719. [Google Scholar] [CrossRef]
- Lu, Y.; Huang, K.; Cao, X.; Zhang, L.; Wang, T.; Peng, D.; Zhang, B.; Liu, Z.; Wu, J.; Zhang, Y.; et al. Atomically Dispersed Intrinsic Hollow Sites of M-M1-M (M1 = Pt, Ir; M = Fe, Co, Ni, Cu, Pt, Ir) on FeCoNiCuPtIr Nanocrystals Enabling Rapid Water Redox. Adv. Funct. Mater. 2022, 32, 2110645. [Google Scholar] [CrossRef]
- Zhu, H.; Zhu, Z.; Hao, J.; Sun, S.; Lu, S.; Wang, C.; Ma, P.; Dong, W.; Du, M. High-Entropy Alloy Stabilized Active Ir for Highly Efficient Acidic Oxygen Evolution. Chem. Eng. J. 2022, 431, 133251. [Google Scholar] [CrossRef]
- Sharma, L.; Katiyar, N.K.; Parui, A.; Das, R.; Kumar, R.; Tiwary, C.S.; Singh, A.K.; Halder, A.; Biswas, K. Low-Cost High Entropy Alloy (HEA) for High-Efficiency Oxygen Evolution Reaction (OER). Nano Res. 2022, 15, 4799–4806. [Google Scholar] [CrossRef]
- Zhang, Z.-J.; Guo, J.-P.; Sun, S.-H.; Sun, Q.; Zhao, Y.-W.; Zhang, Y.-F.; Yu, Z.-Y.; Li, C.-S.; Sun, Y.; Zhang, M.-M.; et al. Optimized Valence State of Co and Ni in High-Entropy Alloy for High Active-Stable OER. Rare Met. 2023, 42, 3607–3613. [Google Scholar] [CrossRef]
- Yuan, G.; Wu, M.; Ruiz Pestana, L. Density Functional Theory-Machine Learning Characterization of the Adsorption Energy of Oxygen Intermediates on High-Entropy Alloys Made of Earth-Abundant Metals. J. Phys. Chem. C 2023, 127, 15809–15818. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jonsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Miracle, D.B.; Senkov, O.N. A Critical Review of High Entropy Alloys and Related Concepts. Acta Mater. 2017, 122, 448–511. [Google Scholar] [CrossRef]
- Liu, B.; Wang, J.; Liu, Y.; Fang, Q.; Wu, Y.; Chen, S.; Liu, C.T. Microstructure and Mechanical Properties of Equimolar FeCoCrNi High Entropy Alloy Prepared via Powder Extrusion. Intermetallics 2016, 75, 25–30. [Google Scholar] [CrossRef]
- McKay, F.; Fang, Y.; Kizilkaya, O.; Singh, P.; Johnson, D.D.; Roy, A.; Young, D.P.; Sprunger, P.T.; Flake, J.C.; Shelton, W.A. CoCrFeNi High-Entropy Alloy as an Enhanced Hydrogen Evolution Catalyst in an Acidic Solution. J. Phys. Chem. C 2021, 125, 17008–17018. [Google Scholar] [CrossRef]
- Kühne, T.D.; Iannuzzi, M.; Del Ben, M.; Rybkin, V.V.; Seewald, P.; Stein, F.; Laino, T.; Khaliullin, R.Z.; Schütt, O.; Schiffmann, F. CP2K: An Electronic Structure and Molecular Dynamics Software Package-Quickstep: Efficient and Accurate Electronic Structure Calculations. J. Chem. Phys. 2020, 152, 194103. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, W. Comment on “Generalized Gradient Approximation Made Simple”. Phys. Rev. Lett. 1998, 80, 890. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Goedecker, S.; Teter, M.; Hutter, J. Separable Dual-Space Gaussian Pseudopotentials. Phys. Rev. B 1996, 54, 1703. [Google Scholar] [CrossRef]
- Hartwigsen, C.; Gœdecker, S.; Hutter, J. Relativistic Separable Dual-Space Gaussian Pseudopotentials from H to Rn. Phys. Rev. B 1998, 58, 3641. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. Gaussian Basis Sets for Accurate Calculations on Molecular Systems in Gas and Condensed Phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef]
- Ruiz Pestana, L.; Mardirossian, N.; Head-Gordon, M.; Head-Gordon, T. Ab Initio Molecular Dynamics Simulations of Liquid Water Using High Quality Meta-GGA Functionals. Chem. Sci. 2017, 8, 3554–3565. [Google Scholar] [CrossRef]
- Ruiz Pestana, L.; Marsalek, O.; Markland, T.E.; Head-Gordon, T. The Quest for Accurate Liquid Water Properties from First Principles. J. Phys. Chem. Lett. 2018, 9, 5009–5016. [Google Scholar] [CrossRef]
- Nilsson, S.; El Berch, J.N.; Albinsson, D.; Fritzsche, J.; Mpourmpakis, G.; Langhammer, C. The Role of Grain Boundary Sites for the Oxidation of Copper Catalysts during the CO Oxidation Reaction. ACS Nano 2023, 17, 20284–20298. [Google Scholar] [CrossRef]
- Broyden, C.G. A Class of Methods for Solving Nonlinear Simultaneous Equations. Math. Comput. 1965, 19, 577–593. [Google Scholar] [CrossRef]
- Lucas, M.S.; Belyea, D.; Bauer, C.; Bryant, N.; Michel, E.; Turgut, Z.; Leontsev, S.O.; Horwath, J.; Semiatin, S.L.; McHenry, M.E.; et al. Thermomagnetic Analysis of FeCoCrxNi Alloys: Magnetic Entropy of High-Entropy Alloys. J. Appl. Phys. 2013, 113, 17A923. [Google Scholar] [CrossRef]
- Lucas, M.S.; Mauger, L.; Muñoz, J.A.; Xiao, Y.; Sheets, A.O.; Semiatin, S.L.; Horwath, J.; Turgut, Z. Magnetic and Vibrational Properties of High-Entropy Alloys. J. Appl. Phys. 2011, 109, 07E307. [Google Scholar] [CrossRef]
- Wynblatt, P.; Chatain, D. Modeling Grain Boundary and Surface Segregation in Multicomponent High-Entropy Alloys. Phys. Rev. Mater. 2019, 3, 054004. [Google Scholar] [CrossRef]
- Ferrari, A.; Körmann, F. Surface Segregation in Cr-Mn-Fe-Co-Ni High Entropy Alloys. Appl. Surf. Sci. 2020, 533, 147471. [Google Scholar] [CrossRef]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of Crystals with the Quasi-Newton Method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Mavrikakis, M.; Rempel, J.; Greeley, J.; Hansen, L.B.; Nørskov, J.K. Atomic and Molecular Adsorption on Rh(111). J. Chem. Phys. 2002, 117, 6737–6744. [Google Scholar] [CrossRef]
- Ford, D.C.; Xu, Y.; Mavrikakis, M. Atomic and Molecular Adsorption on Pt(111). Surf. Sci. 2005, 587, 159–174. [Google Scholar] [CrossRef]
- Bai, Y.; Kirvassilis, D.; Xu, L.; Mavrikakis, M. Atomic and Molecular Adsorption on Ni(111). Surf. Sci. 2019, 679, 240–253. [Google Scholar] [CrossRef]
- Eichler, A.; Mittendorfer, F.; Hafner, J. Precursor-Mediated Adsorption of Oxygen on the (111) Surfaces of Platinum-Group Metals. Phys. Rev. B 2000, 62, 4744–4755. [Google Scholar] [CrossRef]
- Batchelor, T.A.; Pedersen, J.K.; Winther, S.H.; Castelli, I.E.; Jacobsen, K.W.; Rossmeisl, J. High-Entropy Alloys as a Discovery Platform for Electrocatalysis. Joule 2019, 3, 834–845. [Google Scholar] [CrossRef]
- McKay, F.; Ismael, T.; Robinson, A.; Kizilkaya, O.; Sprunger, P.T.; Derosa, P.A. Surface Oxidation Mechanism of CoCrFeNi High Entropy Alloy. Surf. Sci. 2022, 723, 122124. [Google Scholar] [CrossRef]
- Yang, H.; Whitten, J.L. Energetics of Hydroxyl and Influence of Coadsorbed Oxygen on Metal Surfaces. J. Phys. Chem. B 1997, 101, 4090–4096. [Google Scholar] [CrossRef]
- Verga, L.G.; Aarons, J.; Sarwar, M.; Thompsett, D.; Russell, A.E.; Skylaris, C.-K. DFT Calculation of Oxygen Adsorption on Platinum Nanoparticles: Coverage and Size Effects. Faraday Discuss. 2018, 208, 497–522. [Google Scholar] [CrossRef]
- Montemore, M.M.; van Spronsen, M.A.; Madix, R.J.; Friend, C.M. O2 Activation by Metal Surfaces: Implications for Bonding and Reactivity on Heterogeneous Catalysts. Chem. Rev. 2018, 118, 2816–2862. [Google Scholar] [CrossRef]
- Cao, A.; Nørskov, J.K. Spin Effects in Chemisorption and Catalysis. ACS Catal. 2023, 13, 3456–3462. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(ML) | Number of Individual Sites | Number of Pristine Sites | Number of Oxidized Sites | ||||
|---|---|---|---|---|---|---|---|
| Rate-Limiting Step | Rate-Limiting Step | ||||||
| 19 | N/A | 1 | 12 | 6 | N/A | N/A | |
| 31 | 2 | 1 | 13 | 14 | 1 | N/A | |
| 42 | N/A | N/A | 1 | 38 | N/A | 3 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, G.; Ruiz Pestana, L. The Effect of Surface Oxygen Coverage on the Oxygen Evolution Reaction over a CoFeNiCr High-Entropy Alloy. Nanomaterials 2024, 14, 1058. https://doi.org/10.3390/nano14121058
Yuan G, Ruiz Pestana L. The Effect of Surface Oxygen Coverage on the Oxygen Evolution Reaction over a CoFeNiCr High-Entropy Alloy. Nanomaterials. 2024; 14(12):1058. https://doi.org/10.3390/nano14121058
Chicago/Turabian StyleYuan, Geng, and Luis Ruiz Pestana. 2024. "The Effect of Surface Oxygen Coverage on the Oxygen Evolution Reaction over a CoFeNiCr High-Entropy Alloy" Nanomaterials 14, no. 12: 1058. https://doi.org/10.3390/nano14121058