

Ultrathin Boron Growth onto Nanodiamond Surfaces via Electrophilic Boron Precursors

 , ,
, ,
Abstract
:
1. Introduction
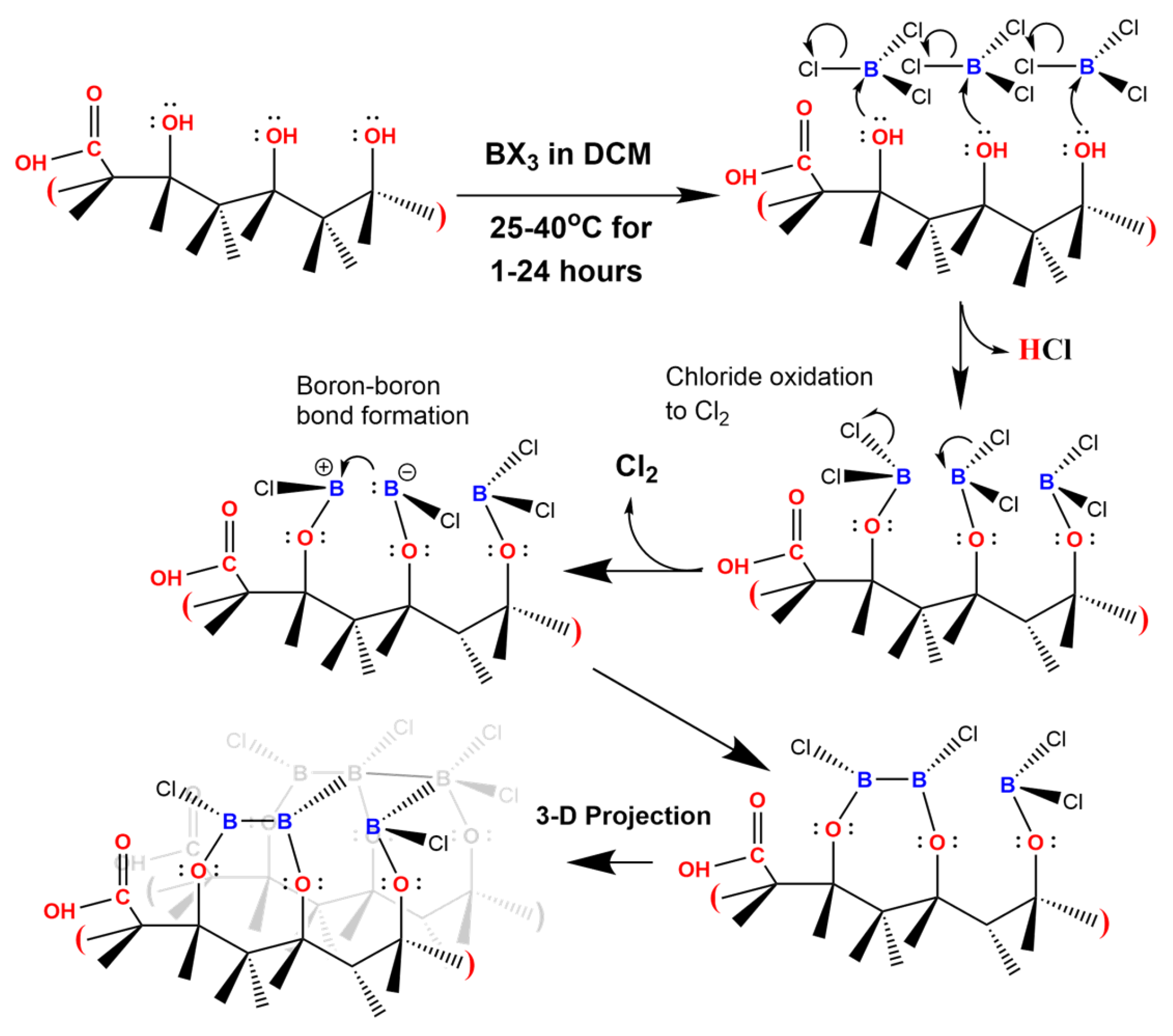
2. Materials and Methods
Open Air and Inert Atmosphere DRIFTS
3. Results and Discussion
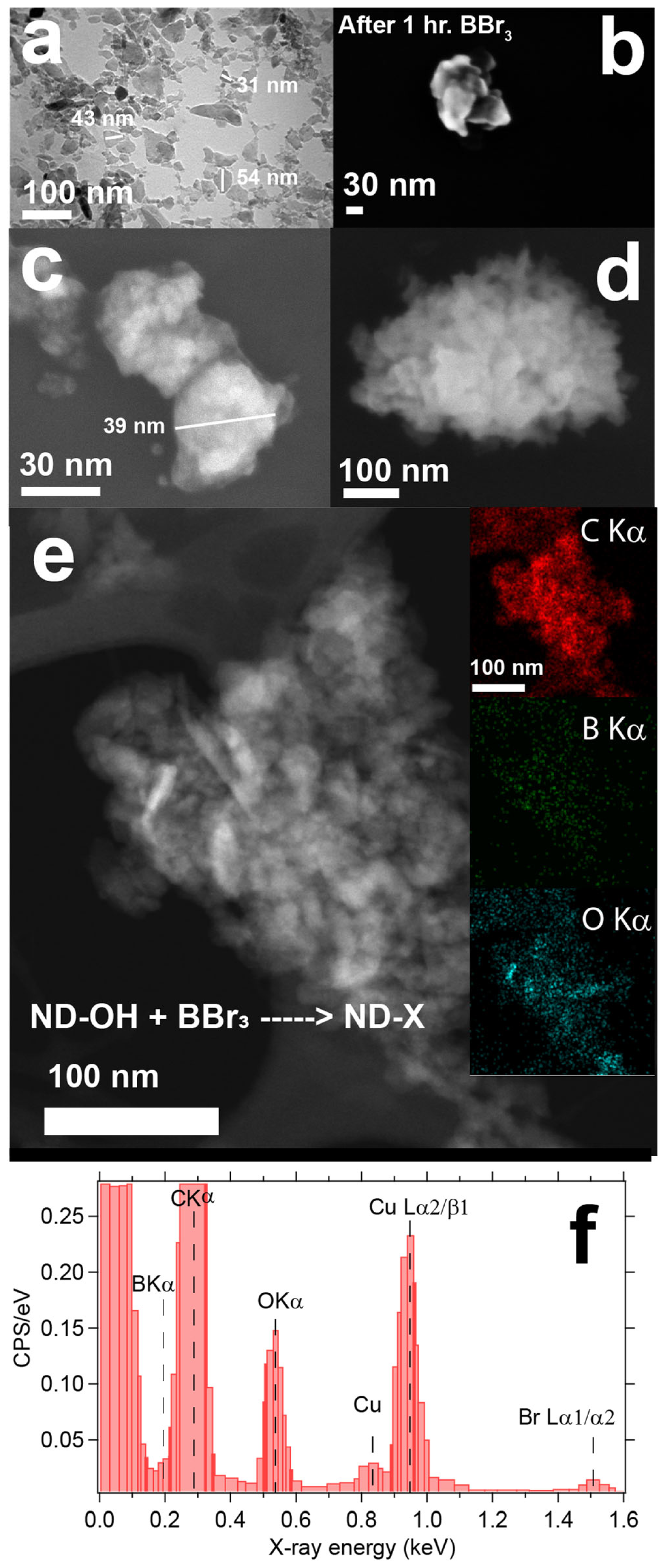
3.1. Morphology Changes after Boron Chemistry on ND Cores and Limitations of EDS Mapping to Confirm Boron
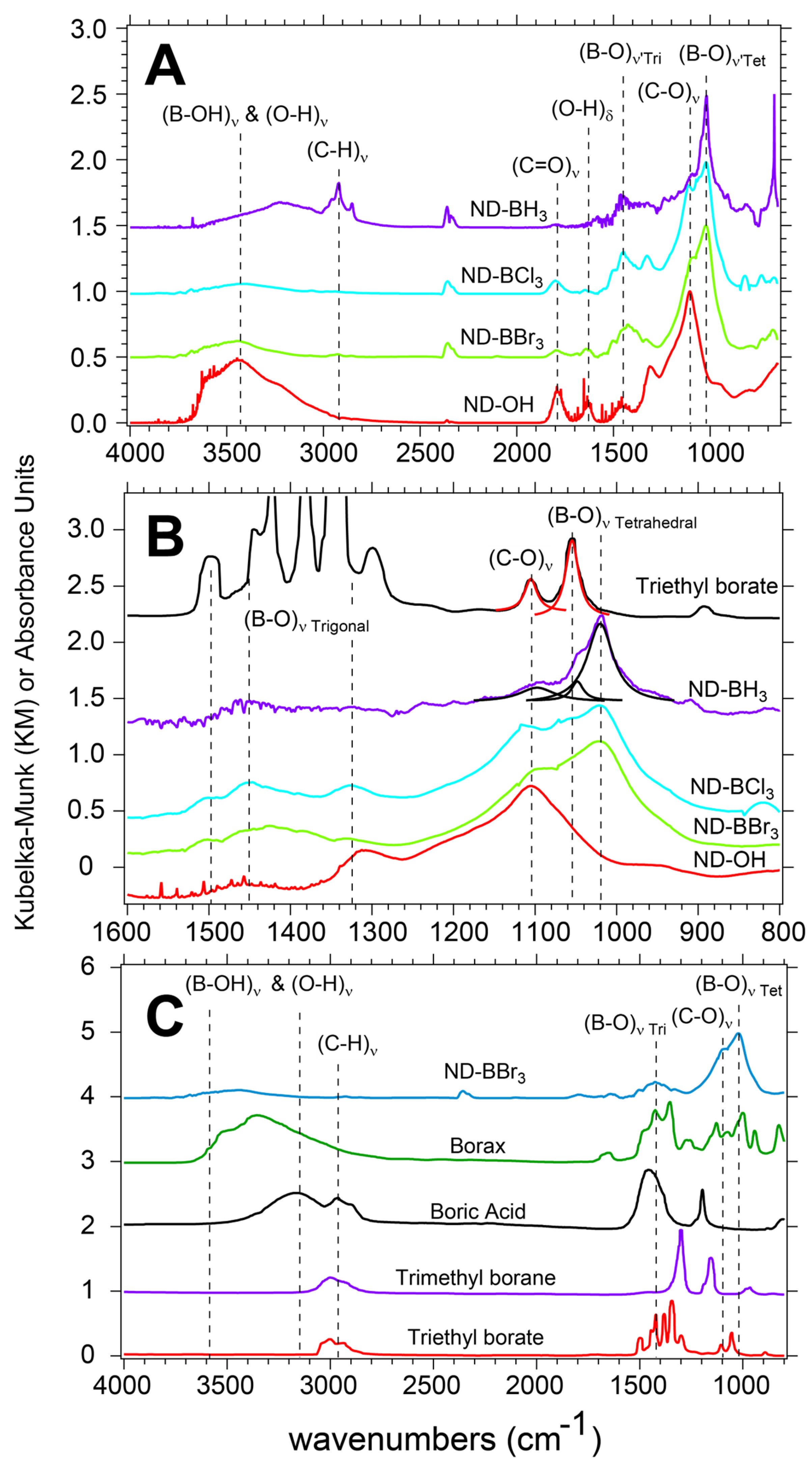
3.2. Starting ND-OH Vibrational Structure Consistent with Alcohols and a Surface Free of Adsorbed Water
Trigonal and Tetrahedral Boron Centers on ND Surfaces Post Reaction
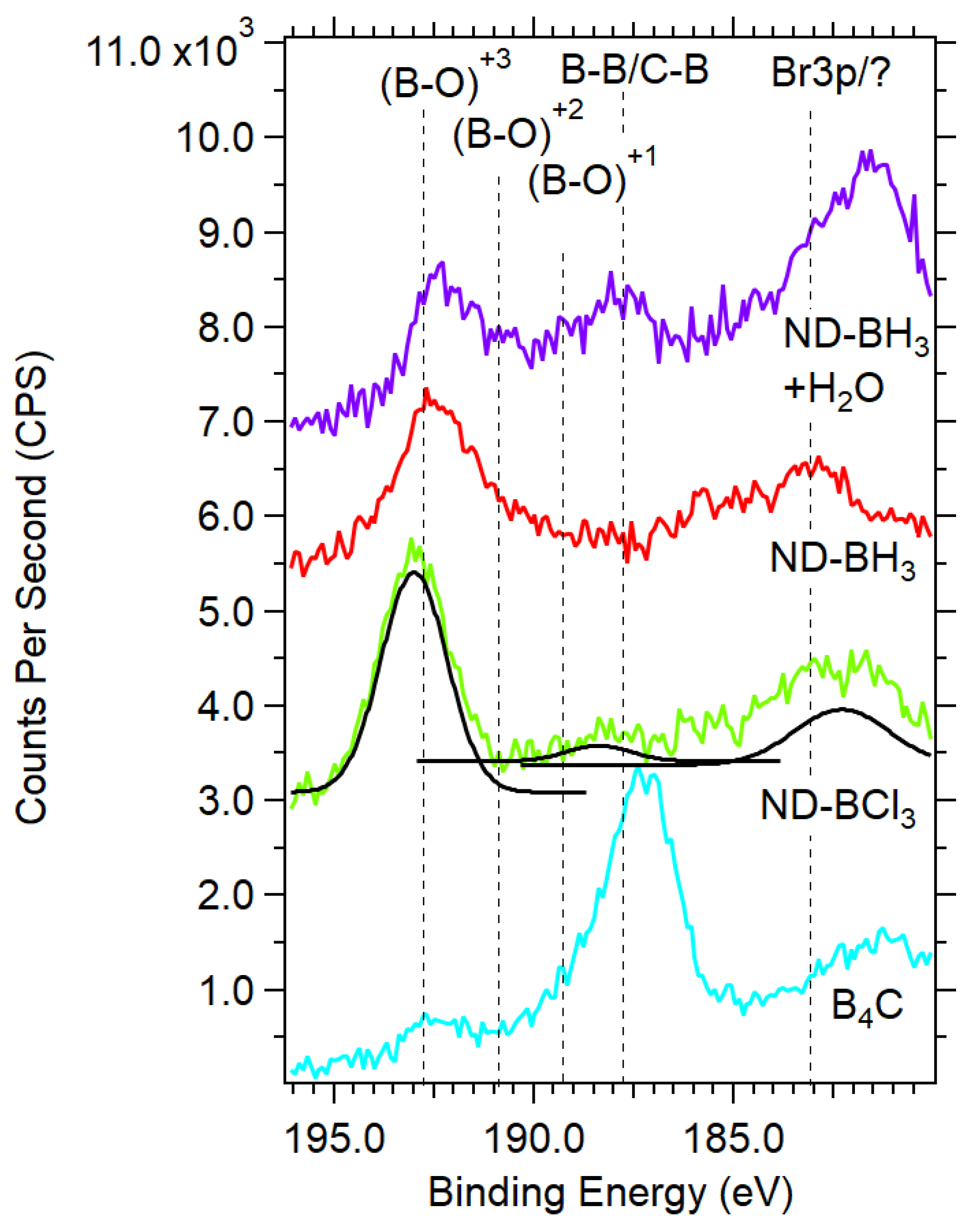
3.3. XPS Reveals Boron Templated ND Samples Contain B-O, B-B, and B-C Bonding Environments
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mannix, A.J.; Zhou, X.F.; Kiraly, B.; Wood, J.D.; Alducin, D.; Myers, B.D.; Liu, X.L.; Fisher, B.L.; Santiago, U.; Guest, J.R.; et al. Synthesis of borophenes: Anisotropic, two-dimensional boron polymorphs. Science 2015, 350, 1513–1516. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Zhang, J.; Zhong, Q.; Li, W.; Li, S.; Li, H.; Cheng, P.; Meng, S.; Chen, L.; Wu, K. Experimental realization of two-dimensional boron sheets. Nat. Chem. 2016, 8, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, P.; Lee, J.M.; Kumar, P.; Vinu, A. Borophene: New Sensation in Flatland. Adv. Mater. 2020, 32, 2000531. [Google Scholar] [CrossRef] [PubMed]
- Piazza, Z.A.; Hu, H.S.; Li, W.L.; Zhao, Y.F.; Li, J.; Wang, L.S. Planar hexagonal B36 as a potential basis for extended single-atom layer boron sheets. Nat. Commun. 2014, 5, 3113. [Google Scholar] [CrossRef] [PubMed]
- Boustani, I. Systematic ab initio investigation of bare boron clusters: Determination of the geometry and electronic structures of B-n (n = 2−14). Phys. Rev. B 1997, 55, 16426–16438. [Google Scholar] [CrossRef]
- Lau, K.C.; Pandey, R. Stability and electronic properties of atomistically-engineered 2D boron sheets. J. Phys. Chem. C 2007, 111, 2906–2912. [Google Scholar] [CrossRef]
- Tang, H.; Ismail-Beigi, S. Novel precursors for boron nanotubes: The competition of two-center and three-center bonding in boron sheets. Phys. Rev. Lett. 2007, 99, 115501. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.F.; Dong, X.; Oganov, A.R.; Zhu, Q.; Tian, Y.J.; Wang, H.T. Semimetallic Two-Dimensional Boron Allotrope with Massless Dirac Fermions. Phys. Rev. Lett. 2014, 112, 085502. [Google Scholar] [CrossRef]
- Penev, E.S.; Bhowmick, S.; Sadrzadeh, A.; Yakobson, B.I. Polymorphism of Two-Dimensional Boron. Nano Lett. 2012, 12, 2441–2445. [Google Scholar] [CrossRef] [PubMed]
- Rohani, P.; Kim, S.; Swihart, M.T. Boron Nanoparticles for Room-Temperature Hydrogen Generation from Water. Adv. Energy Mater. 2016, 6, 1502550. [Google Scholar] [CrossRef]
- Yang, M.; Jin, H.; Sun, Z.; Gui, R. Experimental synthesis, functionalized modifications and potential applications of monoelemental zero-dimensional boron nanomaterials. J. Mater. Chem. A 2022, 10, 5111–5146. [Google Scholar] [CrossRef]
- Li, L.; Schröder, T.; Chen, E.H.; Walsh, M.; Bayn, I.; Goldstein, J.; Gaathon, O.; Trusheim, M.E.; Lu, M.; Mower, J.; et al. Coherent spin control of a nanocavity-enhanced qubit in diamond. Nat. Commun. 2015, 6, 6173. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, W.; Hensen, B.J.; Bernien, H.; van Dam, S.B.; Blok, M.S.; Taminiau, T.H.; Tiggelman, M.J.; Schouten, R.N.; Markham, M.; Twitchen, D.J.; et al. Unconditional quantum teleportation between distant solid-state quantum bits. Science 2014, 345, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Wehner, S.; Elkouss, D.; Hanson, R. Quantum internet: A vision for the road ahead. Science 2018, 362, eaam9288. [Google Scholar] [CrossRef] [PubMed]
- Dreau, A.; Tcheborateva, A.; El Mahdaoui, A.; Bonato, C.; Hanson, R. Quantum Frequency Conversion of Single Photons from a Nitrogen-Vacancy Center in Diamond to Telecommunication Wavelengths. Phys. Rev. Appl. 2018, 9, 064031. [Google Scholar] [CrossRef]
- Awschalom, D.D.; Hanson, R.; Wrachtrup, J.; Zhou, B.B. Quantum technologies with optically interfaced solid-state spins. Nat. Photonics 2018, 12, 516–527. [Google Scholar] [CrossRef]
- Maze, J.R.; Stanwix, P.L.; Hodges, J.S.; Hong, S.; Taylor, J.M.; Cappellaro, P.; Jiang, L.; Dutt, M.V.G.; Togan, E.; Zibrov, A.S.; et al. Nanoscale magnetic sensing with an individual electronic spin in diamond. Nature 2008, 455, 644–647. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M.; Cappellaro, P.; Childress, L.; Jiang, L.; Budker, D.; Hemmer, P.R.; Yacoby, A.; Walsworth, R.; Lukin, M.D. High-sensitivity diamond magnetometer with nanoscale resolution. Nat. Phys. 2008, 4, 810–816. [Google Scholar] [CrossRef]
- McGuinness, L.P.; Yan, Y.; Stacey, A.; Simpson, D.A.; Hall, L.T.; Maclaurin, D.; Prawer, S.; Mulvaney, P.; Wrachtrup, J.; Caruso, F.; et al. Quantum measurement and orientation tracking of fluorescent nanodiamonds inside living cells. Nat. Nanotechnol. 2011, 6, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Atature, M.; Englund, D.; Vamivakas, N.; Lee, S.Y.; Wrachtrup, J. Material platforms for spin-based photonic quantum technologies. Nat. Rev. Mater. 2018, 3, 38–51. [Google Scholar] [CrossRef]
- Kucsko, G.; Maurer, P.C.; Yao, N.Y.; Kubo, M.; Noh, H.J.; Lo, P.K.; Park, H.; Lukin, M.D. Nanometre-scale thermometry in a living cell. Nature 2013, 500, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Lozovoi, A.; Jayakumar, H.; Daw, D.; Vizkelethy, G.; Bielejec, E.; Doherty, M.W.; Flick, J.; Meriles, C.A. Optical activation and detection of charge transport between individual colour centres in diamond. Nat. Electron. 2021, 4, 717–724. [Google Scholar] [CrossRef]
- Monge, R.; Delord, T.; Meriles, C.A. Reversible optical data storage below the diffraction limit. Nat. Nanotechnol. 2024, 19, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Gavrilin, A.V.; Luk’yanov, I.M.; Smirnov, E.P.; Aleskovskij, V.B. Interaction of boron halides with carbon preparations. Zhurnal Obs. Khimii 1987, 57, 1350–1352. [Google Scholar]
- Shen, W.; Pan, Y.; Shen, S.; Li, H.; Zhang, Y.; Zhang, G. Electron affinity of boron-terminated diamond (001) surfaces: A density functional theory study. J. Mater. Chem. C 2019, 7, 9756–9765. [Google Scholar] [CrossRef]
- Sun, Z.; Yang, M.; Wang, X.; Wang, P.; Zhang, C.; Gao, N.; Li, H. Boron-terminated diamond (100) surfaces with promising structural and electronic properties. Phys. Chem. Chem. Phys. 2020, 22, 8060–8066. [Google Scholar] [CrossRef] [PubMed]
- Cobb, S.J.; Ayres, Z.J.; Macpherson, J.V. Boron Doped Diamond: A Designer Electrode Material for the Twenty-First Century. Annu. Rev. Anal. Chem. 2018, 11, 463–484. [Google Scholar] [CrossRef] [PubMed]
- Havlik, J.; Petrakova, V.; Kucka, J.; Raabova, H.; Panek, D.; Stepan, V.; Cilova, Z.Z.; Reineck, P.; Stursa, J.; Kucera, J.; et al. Extremely rapid isotropic irradiation of nanoparticles with ions generated in situ by a nuclear reaction. Nat. Commun. 2018, 9, 4467. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Barroso, J.; Jalife, S.; Heine, T.; Asmis, K.R.; Merino, G. Fluxional Boron Clusters: From Theory to Reality. Acc. Chem. Res. 2019, 52, 2732–2744. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.; S Hosmane, N.; Zhu, Y. Boron Chemistry for Medical Applications. Molecules 2020, 25, 828. [Google Scholar] [CrossRef] [PubMed]
- Barth, R.F.; Coderre, J.A.; Vicente, M.G.H.; Blue, T.E. Boron Neutron Capture Therapy of Cancer: Current Status and Future Prospects. Clin. Cancer Res. 2005, 11, 3987–4002. [Google Scholar] [CrossRef] [PubMed]
- Dymova, M.A.; Taskaev, S.Y.; Richter, V.A.; Kuligina, E.V. Boron neutron capture therapy: Current status and future perspectives. Cancer Commun. 2020, 40, 406–421. [Google Scholar] [CrossRef] [PubMed]
- Barth, R.F.; Vicente, M.G.; Harling, O.K.; Kiger, W.S.; Riley, K.J.; Binns, P.J.; Wagner, F.M.; Suzuki, M.; Aihara, T.; Kato, I.; et al. Current status of boron neutron capture therapy of high grade gliomas and recurrent head and neck cancer. Radiat. Oncol. 2012, 7, 146. [Google Scholar] [CrossRef] [PubMed]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd ed.; John Wiley and Sons: London, UK, 2001; p. 343. [Google Scholar]
- Osswald, S.; Yushin, G.; Mochalin, V.; Kucheyev, S.O.; Gogotsi, Y. Control of sp(2)/sp(3) carbon ratio and surface chemistry of nanodiamond powders by selective oxidation in air. J. Am. Chem. Soc. 2006, 128, 11635–11642. [Google Scholar] [CrossRef] [PubMed]
- Krueger, A.; Lang, D. Functionality is Key: Recent Progress in the Surface Modification of Nanodiamond. Adv. Funct. Mater. 2012, 22, 890–906. [Google Scholar] [CrossRef]
- Krueger, A.; Stegk, J.; Liang, Y.; Lu, L.; Jarre, G. Biotinylated nanodiamond: Simple and efficient functionalization of detonation diamond. Langmuir 2008, 24, 4200–4204. [Google Scholar] [CrossRef] [PubMed]
- Girard, H.A.; Petit, T.; Perruchas, S.; Gacoin, T.; Gesset, C.; Arnault, J.C.; Bergonzo, P. Surface properties of hydrogenated nanodiamonds: A chemical investigation. Phys. Chem. Chem. Phys. 2011, 13, 11517–11523. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Suarez, D.L. Coordination of adsorbed boron: A FTIR spectroscopic study. Environ. Sci. Technol. 1995, 29, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Morar, J.F.; Himpsel, F.J.; Hollinger, G.; Hughes, G.; Jordan, J.L. Observation of a C-1 s core exciton in diamond. Phys. Rev. Lett. 1985, 54, 1960–1963. [Google Scholar] [CrossRef] [PubMed]
- CasaXPS. CasaXPS User’s Manual; CasaXPS: Teignmouth, UK, 2001; p. 163. [Google Scholar]
- ISO 14976; Surface Chemical Analysis—Data Transfer Format. ISO: Geneve, Switzerland, 1998.
- Telling, R.H.; Pickard, C.J.; Payne, M.C.; Field, J.E. Theoretical Strength and Cleavage of Diamond. Phys. Rev. Lett. 2000, 84, 5160–5163. [Google Scholar] [CrossRef] [PubMed]
- Wolcott, A.; Schiros, T.; Trusheim, M.E.; Chen, E.H.; Nordlund, D.; Diaz, R.E.; Gaathon, O.; Englund, D.; Owen, J.S. Surface Structure of Aerobically Oxidized Diamond Nanocrystals. J. Phys. Chem. C 2014, 118, 26695–26702. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.-B.; Chang, H.-C.; Wu, V.W.-K. Adsorption and hydrolytic activity of lysozyme on diamond nanocrystallites. Diam. Relat. Mater. 2007, 16, 872–876. [Google Scholar] [CrossRef]
- Melendrez, C.; Lopez-Rosas, J.A.; Stokes, C.X.; Cheung, T.C.; Lee, S.-J.; Titus, C.J.; Valenzuela, J.; Jeanpierre, G.; Muhammad, H.; Tran, P.; et al. Metastable Brominated Nanodiamond Surface Enables Room Temperature and Catalysis-Free Amine Chemistry. J. Phys. Chem. Lett. 2022, 13, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, P.J.; Lopez, K.; Arreola, A.; Len, A.; Basravi, N.; Yamaguchi, P.; Kawamura, R.; Stokes, C.X.; Melendrez, C.; Simpson, D.; et al. Quantum Diamonds at the Beach: Chemical Insights into Silica Growth on Nanoscale Diamond using Multimodal Characterization and Simulation. ACS Nanosci. Au 2023, 3, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Moddeman, W.E.; Burke, A.R.; Bowling, W.C.; Foose, D.S. Surface oxides of boron and B12O2 as determined by XPS. Surf. Interface Anal. 1989, 14, 224–232. [Google Scholar] [CrossRef]
- Jimenez, I.; Gago, R.; Albella, J.M.; Terminello, L.J. X-Ray absorption studies of bonding environments in graphitic carbon nitride. Diam. Relat. Mater. 2001, 10, 1170–1174. [Google Scholar] [CrossRef]
- Schulzendorf, M.; Hinaut, A.; Kisiel, M.; Jöhr, R.; Pawlak, R.; Restuccia, P.; Meyer, E.; Righi, M.C.; Glatzel, T. Altering the Properties of Graphene on Cu(111) by Intercalation of Potassium Bromide. ACS Nano 2019, 13, 5485–5492. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.G.; Han, D.; Park, Y.; Hyun, H.S.; Sung, H.-G.; Sohn, Y. Combustion of boron particles coated with an energetic polymer material. Korean J. Chem. Eng. 2016, 33, 3016–3020. [Google Scholar] [CrossRef]
- Jiménez, I.; Terminello, L.J.; Himpsel, F.J.; Grush, M.; Callcot, T.A. Photoemission, X-ray absorption and X-ray emission study of boron carbides. J. Electron Spectrosc. Relat. Phenom. 1999, 101–103, 611–615. [Google Scholar] [CrossRef]
- Jiménez, I.; Sutherland, D.G.J.; van Buuren, T.; Carlisle, J.A.; Terminello, L.J.; Himpsel, F.J. Photoemission and x-ray-absorption study of boron carbide and its surface thermal stability. Phys. Rev. B 1998, 57, 13167–13174. [Google Scholar] [CrossRef]
- Singleton, D.A.; Waller, S.C.; Zhang, Z.; Frantz, D.E.; Leung, S.-W. Allylboration of Alkenes with Allyldihaloboranes. J. Am. Chem. Soc. 1996, 118, 9986–9987. [Google Scholar] [CrossRef]
- Das, B.C.; Nandwana, N.K.; Das, S.; Nandwana, V.; Shareef, M.A.; Das, Y.; Saito, M.; Weiss, L.M.; Almaguel, F.; Hosmane, N.S.; et al. Boron Chemicals in Drug Discovery and Development: Synthesis and Medicinal Perspective. Molecules 2022, 27, 2615. [Google Scholar] [CrossRef] [PubMed]
- Caretti, I.; Gago, R.; Albella, J.M.; Jiménez, I. Boron carbides formed by coevaporation of B and C atoms: Vapor reactivity, BxC1−x composition, and bonding structure. Phys. Rev. B 2008, 77, 174109. [Google Scholar] [CrossRef]
- Pallier, C.; Leyssale, J.-M.; Truflandier, L.A.; Bui, A.T.; Weisbecker, P.; Gervais, C.; Fischer, H.E.; Sirotti, F.; Teyssandier, F.; Chollon, G. Structure of an Amorphous Boron Carbide Film: An Experimental and Computational Approach. Chem. Mater. 2013, 25, 2618–2629. [Google Scholar] [CrossRef]
- Stacey, A.; Dontschuk, N.; Chou, J.P.; Broadway, D.A.; Schenk, A.K.; Sear, M.J.; Tetienne, J.P.; Hoffman, A.; Prawer, S.; Pakes, C.I.; et al. Evidence for Primal sp(2) Defects at the Diamond Surface: Candidates for Electron Trapping and Noise Sources. Adv. Mater. Interfaces 2019, 6, 1801449. [Google Scholar] [CrossRef]
- Ishii, I.; Hitchcook, A.P. The oscillator strengths for C1s and O1s excitation of some saturated and unsaturated organic alcohols, acids and esters. J. Electron Spectrosc. Relat. Phenom. 1988, 46, 55–84. [Google Scholar] [CrossRef]
- Chemin, A.; Levine, I.; Rusu, M.; Vaujour, R.; Knittel, P.; Reinke, P.; Hinrichs, K.; Unold, T.; Dittrich, T.; Petit, T. Surface-Mediated Charge Transfer of Photogenerated Carriers in Diamond. Small Methods 2023, 7, 2300423. [Google Scholar] [CrossRef] [PubMed]
- Sangtawesin, S.; Dwyer, B.L.; Srinivasan, S.; Allred, J.J.; Rodgers, L.V.H.; De Greve, K.; Stacey, A.; Dontschuk, N.; O’Donnell, K.M.; Hu, D.; et al. Origins of Diamond Surface Noise Probed by Correlating Single-Spin Measurements with Surface Spectroscopy. Phys. Rev. X 2019, 9, 031052. [Google Scholar] [CrossRef]
- Hitchcock, A.P.; Urquhart, S.G.; Wen, A.T.; Kilcoyne, A.L.D.; Tyliszczak, T.; Rühl, E.; Kosugi, N.; Bozek, J.D.; Spencer, J.T.; McIlroy, D.N.; et al. Inner-Shell Excitation Spectroscopy of closo-Carboranes. J. Phys. Chem. B 1997, 101, 3483–3493. [Google Scholar] [CrossRef]
- Liu, X.B.; Chen, X.; Singh, D.J.; Stern, R.A.; Wu, J.S.; Petitgirard, S.; Bina, C.R.; Jacobsen, S.D. Boron-oxygen complex yields n-type surface layer in semiconducting diamond. Proc. Natl. Acad. Sci. USA 2019, 116, 7703–7711. [Google Scholar] [CrossRef] [PubMed]
- Gerrard, W.; Lappert, M.F. Reactions of Boron Trichloride with Organic Compounds. Chem. Rev. 1958, 58, 1081–1111. [Google Scholar] [CrossRef]
- Holliday, A.K.; Massey, A.G. Boron Subhalides and Related Compounds with Boron-Boron Bonds. Chem. Rev. 1962, 62, 303–318. [Google Scholar] [CrossRef]
- Pickering, A.L.; Mitterbauer, C.; Browning, N.D.; Kauzlarich, S.M.; Power, P.P. Room temperature synthesis of surface-functionalised boron nanoparticles. Chem. Commun. 2007, 580–582. [Google Scholar] [CrossRef] [PubMed]
- Franken, L.E.; Grünewald, K.; Boekema, E.J.; Stuart, M.C.A. A Technical Introduction to Transmission Electron Microscopy for Soft-Matter: Imaging, Possibilities, Choices, and Technical Developments. Small 2020, 16, 1906198. [Google Scholar] [CrossRef] [PubMed]
- Rendler, T.; Neburkova, J.; Zemek, O.; Kotek, J.; Zappe, A.; Chu, Z.Q.; Cigler, P.; Wrachtrup, J. Optical imaging of localized chemical events using programmable diamond quantum nanosensors. Nat. Commun. 2017, 8, 14701. [Google Scholar] [CrossRef] [PubMed]
- Neburkova, J.; Vavra, J.; Cigler, P. Coating nanodiamonds with biocompatible shells for applications in biology and medicine. Curr. Opin. Solid State Mater. Sci. 2017, 21, 43–53. [Google Scholar] [CrossRef]
- Rehor, I.; Slegerova, J.; Havlik, J.; Raabova, H.; Hyvl, J.; Muchova, E.; Cigler, P. Carbon Nanomaterials for Biomedical Applications; Zhang, M., Naik, R.R., Dai, L., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; Volume 5, pp. 319–361. [Google Scholar]
- Petrakova, V.; Benson, V.; Buncek, M.; Fiserova, A.; Ledvina, M.; Stursa, J.; Cigler, P.; Nesladek, M. Imaging of transfection and intracellular release of intact, non-labeled DNA using fluorescent nanodiamonds. Nanoscale 2016, 8, 12002–12012. [Google Scholar] [CrossRef] [PubMed]
- Rehor, I.; Slegerova, J.; Kucka, J.; Proks, V.; Petrakova, V.; Adam, M.P.; Treussart, F.; Turner, S.; Bals, S.; Sacha, P.; et al. Fluorescent Nanodiamonds Embedded in Biocompatible Translucent Shells. Small 2014, 10, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Scimeca, M.; Bischetti, S.; Lamsira, H.K.; Bonfiglio, R.; Bonanno, E. Energy Dispersive X-ray (EDX) microanalysis: A powerful tool in biomedical research and diagnosis. Eur. J. Histochem. 2018, 62, 2841. [Google Scholar] [CrossRef] [PubMed]
- Egerton, R.F. Electron energy-loss spectroscopy in the TEM. Rep. Prog. Phys. 2009, 72, 016502. [Google Scholar] [CrossRef]
- Stacey, A.; Cowie, B.C.C.; Orwa, J.; Prawer, S.; Hoffman, A. Diamond C 1s core-level excitons: Surface sensitivity. Phys. Rev. B 2010, 82, 125427. [Google Scholar] [CrossRef]
- Zvi, D.R.C.U.; Weiss, A.; Jones, A.R.; Chen, L.; Golovina, I.; Yu, X.; Wang, S.; Talapin, D.V.; Flatté, M.E.; Esser-Kahn, A.P.; et al. Engineering Spin Coherence in Core-Shell Diamond Nanocrystals. arXiv 2023, arXiv:2305.03075. [Google Scholar]
- Haynes, W.M. CRC Handbook of Chemistry and Physics, 97th ed.; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Anderson, N.C.; Hendricks, M.P.; Choi, J.J.; Owen, J.S. Ligand Exchange and the Stoichiometry of Metal Chalcogenide Nanocrystals: Spectroscopic Observation of Facile Metal-Carboxylate Displacement and Binding. J. Am. Chem. Soc. 2013, 135, 18536–18548. [Google Scholar] [CrossRef] [PubMed]
- Wolcott, A.; Doyeux, V.; Nelson, C.A.; Gearba, R.; Lei, K.W.; Yager, K.G.; Dolocan, A.D.; Williams, K.; Nguyen, D.; Zhu, X.Y. Anomalously Large Polarization Effect Responsible for Excitonic Red Shifts in PbSe Quantum Dot Solids. J. Phys. Chem. Lett. 2011, 2, 795–800. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Edge | IMPF (nm) | Sensitivity Factors |
|---|---|---|---|
| Carbon (Diamond) | 1s | 3.25 | 1.0 |
| Boron (Boron Carbide) | 1s | 3.01 | 0.486 |
| Oxygen (SiO2) | 1s | 2.81 | 2.93 |
| Sample | Surface Termination | Carbon % | Boron % | Oxygen % | Halogen % |
|---|---|---|---|---|---|
| ND-OH-1 | Alcohols | 86.9 | 0.0 | 13.1 | 0.0 |
| Boron | Boron Oxide | 15.3 | 57.0 | 27.7 | 0.0 |
| Boron Carbide | Boron Oxide | 52.1 | 30.0 | 17.9 | 0.0 |
| ND-BBr3-1 | B-B, B-C, B-O | 89.7 | 3.1 | 6.5 | 0.7 |
| ND-BBr3-2 + 1 month | B-B, B-C, B-O | 69.4 | 2.5 | 27.7 | 0.4 |
| ND-BBr3-3 | B-B, B-C, B-O | 82.2 | 3.4 | 14.2 | 0.2 |
| ND-BCl3-1 | B-B, B-C, B-O | 76.6 | 2.8 | 20.4 | 0.2 |
| ND-BCl3-2 + 1 month | B-B, B-C, B-O | 90.0 | 0.6 | 9.0 | 0.4 |
| ND-BCl3-3 | B-B, B-C, B-O | 83.8 | 1.7 | 14.1 | 0.4 |
| ND-BH3-1 | B-B, B-C, B-O | 78.5 | 2.0 | 19.5 | 0.0 |
| ND-BH3-2 + water | B-B, B-C, B-O | 82.0 | 0.8 | 17.2 | 0.0 |
| ND-BH3-3 | B-B, B-C, B-O | 82.7 | 1.6 | 15.8 | 0.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Govindaraju, K.; Supreme, T.; Labunsky, D.N.; Martin, N.; Del Rosario, J.M.; Washington, A.; Uwadiale, E.O.; Adjei, S., II; Ladjadj, S.; Melendrez, C.V.; et al. Ultrathin Boron Growth onto Nanodiamond Surfaces via Electrophilic Boron Precursors. Nanomaterials 2024, 14, 1274. https://doi.org/10.3390/nano14151274
Govindaraju K, Supreme T, Labunsky DN, Martin N, Del Rosario JM, Washington A, Uwadiale EO, Adjei S II, Ladjadj S, Melendrez CV, et al. Ultrathin Boron Growth onto Nanodiamond Surfaces via Electrophilic Boron Precursors. Nanomaterials. 2024; 14(15):1274. https://doi.org/10.3390/nano14151274
Chicago/Turabian StyleGovindaraju, Krishna, Tyanna Supreme, Daniel N. Labunsky, Nicole Martin, Juan Miguel Del Rosario, Alana Washington, Ezhioghode O. Uwadiale, Solomon Adjei, II, Sandra Ladjadj, Cynthia V. Melendrez, and et al. 2024. "Ultrathin Boron Growth onto Nanodiamond Surfaces via Electrophilic Boron Precursors" Nanomaterials 14, no. 15: 1274. https://doi.org/10.3390/nano14151274
APA StyleGovindaraju, K., Supreme, T., Labunsky, D. N., Martin, N., Del Rosario, J. M., Washington, A., Uwadiale, E. O., Adjei, S., II, Ladjadj, S., Melendrez, C. V., Lee, S.-J., Altoe, M. V., Green, A., Riano, S., Sainio, S., Nordlund, D., & Wolcott, A. (2024). Ultrathin Boron Growth onto Nanodiamond Surfaces via Electrophilic Boron Precursors. Nanomaterials, 14(15), 1274. https://doi.org/10.3390/nano14151274