
Human Serum Albumin Protein Corona in Prussian Blue Nanoparticles

 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of PBnps
2.3. Synthesis of HSA-Coated PBnps (PBnp@HSA)
2.4. Phosphate Buffer at pH 7.4
2.5. PBnp@HSA Stability in Phosphate Buffer at pH 7.4
2.6. PBnp Stability in Serum-like Conditions
2.7. pH-Spectrophotometric Titration of PBnp@HSA
2.8. Quantitative Determination of HSA with Folin Reagent
2.8.1. Calibration Curve
2.8.2. Determination of HSA
2.9. Determination of the PBnp Yield
2.10. Instrumentation
2.11. Statistical Analysis
3. Results and Discussion
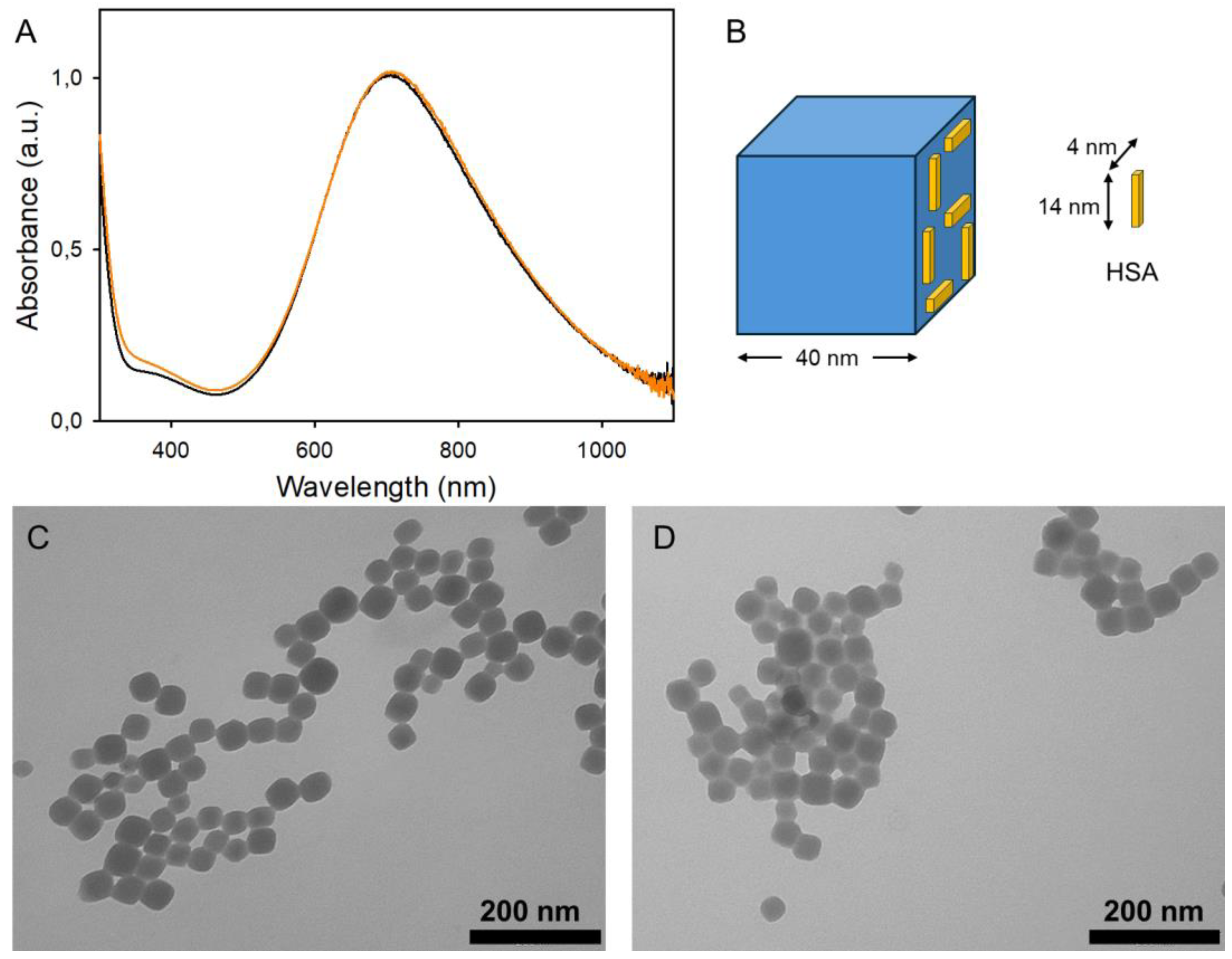
3.1. Synthesis of PBnps and Their HSA Coating
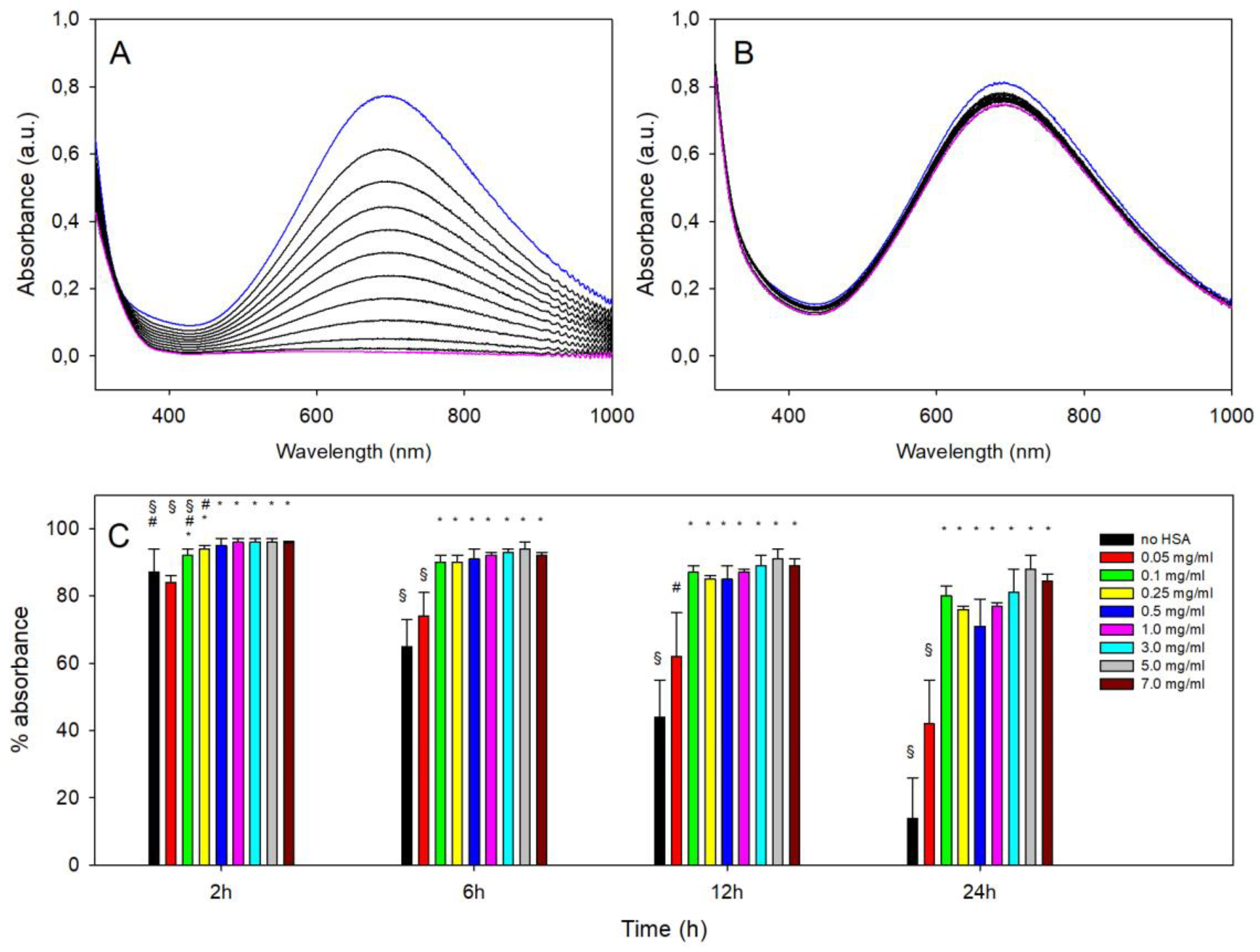
3.2. Spectral Changes with pH in PBnp@HSA
3.3. Stability vs. Decomposition of PBnp@HSA at pH 7.4 as a Function of CHSA
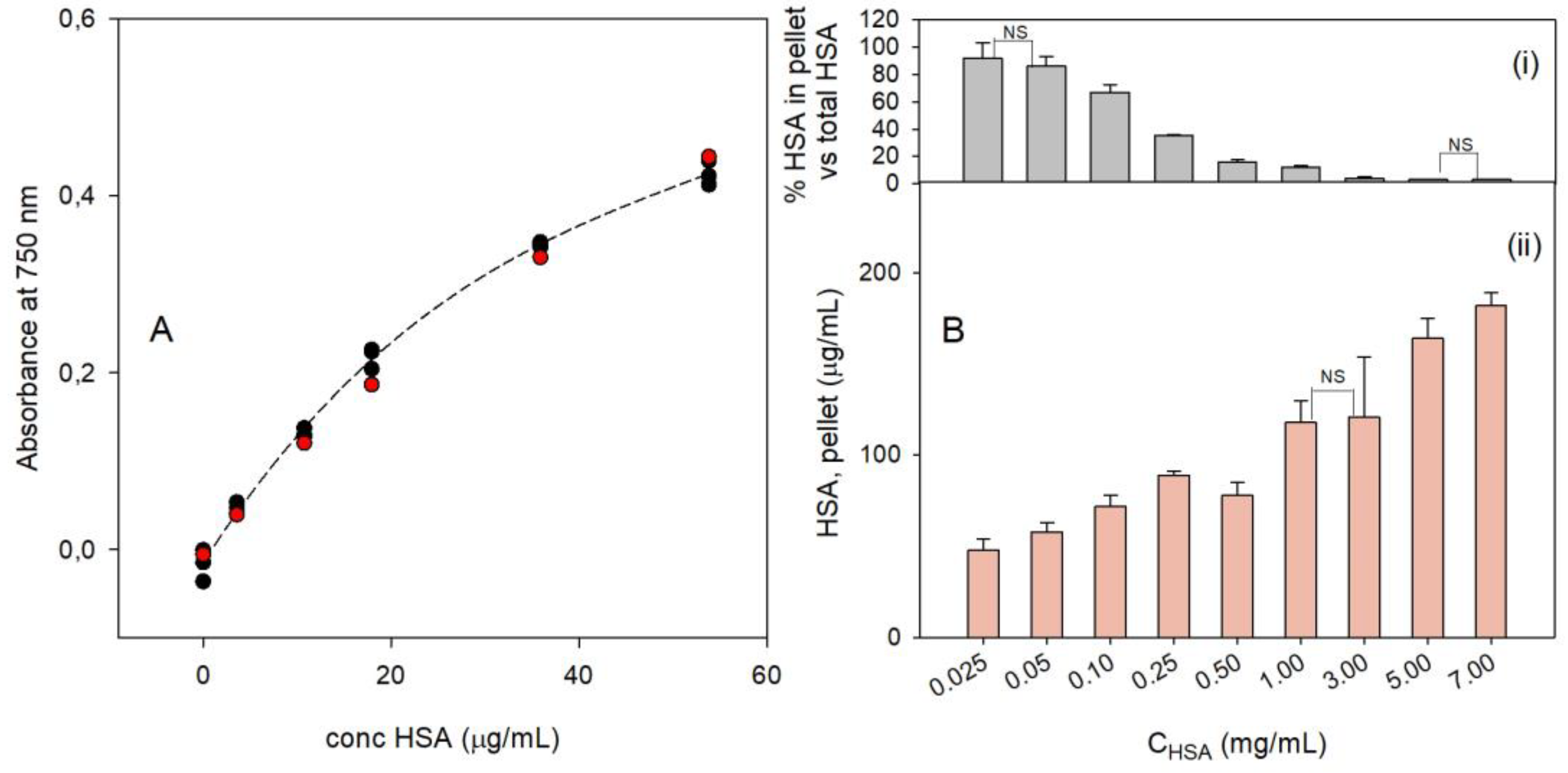
3.4. Quantitative Determination of HSA Bound to PBnps
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dacarro, G.; Taglietti, A.; Pallavicini, P. Prussian Blue Nanoparticles as a Versatile Photothermal Tool. Molecules 2018, 23, 1414. [Google Scholar] [CrossRef] [PubMed]
- Ware, M. Prussian Blue: Artists’ Pigment and Chemists’ Sponge. J. Chem. Educ. 2008, 85, 612–620. [Google Scholar] [CrossRef]
- Buser, H.J.; Schwarzenbach, D.; Petter, W.; Ludi, A. The Crystal Structure of Prussian Blue: Fe4[Fe(CN)6]3∙xH2O. Inorg. Chem. 1977, 16, 2704–2710. [Google Scholar] [CrossRef]
- Qin, Z.G.; Li, Y.; Gu, N. Progress in Applications of Prussian Blue Nanoparticles in Biomedicine. Adv. Healthc. Mater. 2018, 7, 1800347. [Google Scholar] [CrossRef] [PubMed]
- Busquets, M.A.; Estelrich, J. Prussian blue nanoparticles: Synthesis, surface modification, and biomedical applications. Drug Discov. Today 2020, 25, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/021626s007lbl.pdf (accessed on 30 July 2024).
- Fallingborg, J. Intraluminal pH of the human gastrointestinal tract. Dan. Med. Bull. 1999, 46, 183–196. [Google Scholar] [PubMed]
- Doveri, L.; Dacarro, G.; Diaz Fernandez, Y.A.; Razzetti, M.; Taglietti, A.; Chirico, G.; Collini, M.; Sorzabal-Bellido, I.; Esparza, M.; Ortiz-de-Solorzano, C.; et al. Prussian Blue nanoparticles: An FDA-approved substance that may quickly degrade at physiological pH. Colloid. Surface B 2023, 227, 113373. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.; Liu, W.; Feng, S.; Yue, X. Prussian blue nanoparticles operate as a new generation of photothermal ablation agents for cancer therapy. Chem. Commun. 2012, 48, 11567–11569. [Google Scholar] [CrossRef] [PubMed]
- Szigeti, K.; Hegedus, N.; Rácz, K.; Horváth, I.; Veres, D.S.; Szöllosi, D.; Futó, I.; Módos, K.; Bozó, T.; Karlinger, K.; et al. Thallium labeled citrate-coated Prussian Blue nanoparticles as potential imaging agent. Contrast Media Mol. Imaging 2018, 2018, 023604. [Google Scholar] [CrossRef] [PubMed]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Asp. Med. 2012, 33, 209–290. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Doveri, L.; Taglietti, A.; Grisoli, P.; Pallavicini, P.; Dacarro, G. Dual mode antibacterial surfaces based on Prussian blue and silver nanoparticles. Dalton Trans. 2023, 52, 452–460. [Google Scholar] [CrossRef]
- Reed, R.G.; Burrington, C.M. The Albumin Receptor Effect May Be Due to a Surface-induced Conformational Change in Albumin. J. Biol. Chem. 1989, 264, 9867–9872. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.K.; Thompson, M.R. Hydrodynamic structure of bovine serum albumin determined by transient electric birefringence. Biophys. J. 1975, 15, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Cedervall, T.; Lynch, I.; Lindman, S.; Berggård, T.; Thulin, E.; Nilsson, H.; Dawson, K.A.; Linse, S. Understanding the nanoparticle–protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc. Natl. Acad. Sci. USA 2007, 104, 2050–2055. [Google Scholar] [CrossRef] [PubMed]
- Docter, D.; Westmeier, D.; Markiewicz, M.; Stolte, S.; Knauer, S.K.; Stauber, R.H. The nanoparticle biomolecule corona: Lessons learned—Challenge accepted? Chem. Soc. Rev. 2015, 44, 6094–6121. [Google Scholar] [CrossRef] [PubMed]
- Wiig, H.; Kolmannskog, O.; Tenstad, O.; Bert, J.L. Effect of charge on interstitial distribution of albumin in rat dermis in vitro. J. Physiol. 2003, 550, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Cotton, F.A.; Wilkinson, G. Advanced Inorganic Chemistry, 5th ed.; Wiley Interscience: New York, NY, USA, 1988; Chapter 18; p. 717. [Google Scholar]
- Jancsó, A.; Paks, Z.; Mikkola, S.; Rockenbauer, A.; Gajda, T. Iron(III)- and copper(II) complexes of an asymmetric, pentadentate salen-like ligand bearing a pendant carboxylate group. J. Inorg. Biochem. 2005, 99, 1480–1489. [Google Scholar] [CrossRef] [PubMed]
- Chang-Hui, S. Diagnostic Molecular Biology, 1st ed.; Academic Press: Cambridge, MA, USA, 2019; Chapter 8; pp. 191–192. [Google Scholar]
- Lee, R.J.; Wang, S.; Low, P.S. Measurement of endosome pH following folate receptor-mediated endocytosis. Biochim. Biophys. Acta 1996, 1312, 237–242. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solution E (mL) | Bidistilled Water (mL) | Solution D (mL) | 1 N Folin Reagent (mL) | HSA Concentration (μg/mL) | |
|---|---|---|---|---|---|
| blank | 0.00 | 5.00 | 0.70 | 0.10 | 0 |
| Std 1 | 0.10 | 4.90 | 0.70 | 0.10 | 3.59 |
| Std 2 | 0.30 | 4.70 | 0.70 | 0.10 | 10.76 |
| Std 3 | 0.50 | 4.50 | 0.70 | 0.10 | 17.93 |
| Std 4 | 1.00 | 4.00 | 0.70 | 0.10 | 35.86 |
| Std 5 | 1.50 | 3.50 | 0.70 | 0.10 | 53.79 |
| Std 6 | 2.00 | 3.00 | 0.70 | 0.10 | 71.72 |
| Std 7 | 2.50 | 2.50 | 0.70 | 0.10 | 89.65 |
| Std 8 | 3.00 | 2.00 | 0.70 | 0.10 | 107.59 |
| CHSA (mg/mL) | dh (nm) | PDI 2 |
|---|---|---|
| 0.025 | 320(8) | 0.37(0.01) |
| 0.050 | 291(3) | 0.23(0.03) |
| 0.10 | 240(1) | 0.21(0.04) |
| 0.25 | 183(1) | 0.18(0.02) |
| 0.50 | 176(2) | 0.17(0.02) |
| 1.0 | 142(1) | 0.19(0.02) |
| 3.0 | 123(2) | 0.22(0.01) |
| 5.0 | 112(2) | 0.20(0.01) |
| 7.0 | 113(2) | 0.21(0.02) |
| CHSA (μg/mL) | p-CHSA a (μg/mL) | s-CHSA a (μg/mL) | % HSAE/C |
|---|---|---|---|
| 25 | 48(6) | 4.4(0.6) | 210 |
| 50 | 58(5) | 9(3) | 134 |
| 100 | 72(6) | 35(6) | 107 |
| 250 | 89(2) | 162(12) | 100 |
| 500 | 78(7) | 406(18) | 97 |
| 1000 | 118(12) | 899(103) | 102 |
| 3000 | 121(33) | 2566(101) | 90 |
| 5000 | 164(11) | 4574(471) | 95 |
| 7000 | 182(7) | 6627(390) | 97 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colombi, C.; Dacarro, G.; Diaz Fernandez, Y.A.; Taglietti, A.; Pallavicini, P.; Doveri, L. Human Serum Albumin Protein Corona in Prussian Blue Nanoparticles. Nanomaterials 2024, 14, 1336. https://doi.org/10.3390/nano14161336
Colombi C, Dacarro G, Diaz Fernandez YA, Taglietti A, Pallavicini P, Doveri L. Human Serum Albumin Protein Corona in Prussian Blue Nanoparticles. Nanomaterials. 2024; 14(16):1336. https://doi.org/10.3390/nano14161336
Chicago/Turabian StyleColombi, Chiara, Giacomo Dacarro, Yuri Antonio Diaz Fernandez, Angelo Taglietti, Piersandro Pallavicini, and Lavinia Doveri. 2024. "Human Serum Albumin Protein Corona in Prussian Blue Nanoparticles" Nanomaterials 14, no. 16: 1336. https://doi.org/10.3390/nano14161336