
Mechanism for Adsorption, Dissociation, and Diffusion of Hydrogen in High-Entropy Alloy AlCrTiNiV: First-Principles Calculation
 ,
,
Abstract
:1. Introduction
2. Computational Method and Model
3. Results and Discussion
3.1. Dissociation of H2 on AlCrTiNiV (100) Surface
3.2. Adsorption of Atomic H on AlCrTiNiV (100) Surface
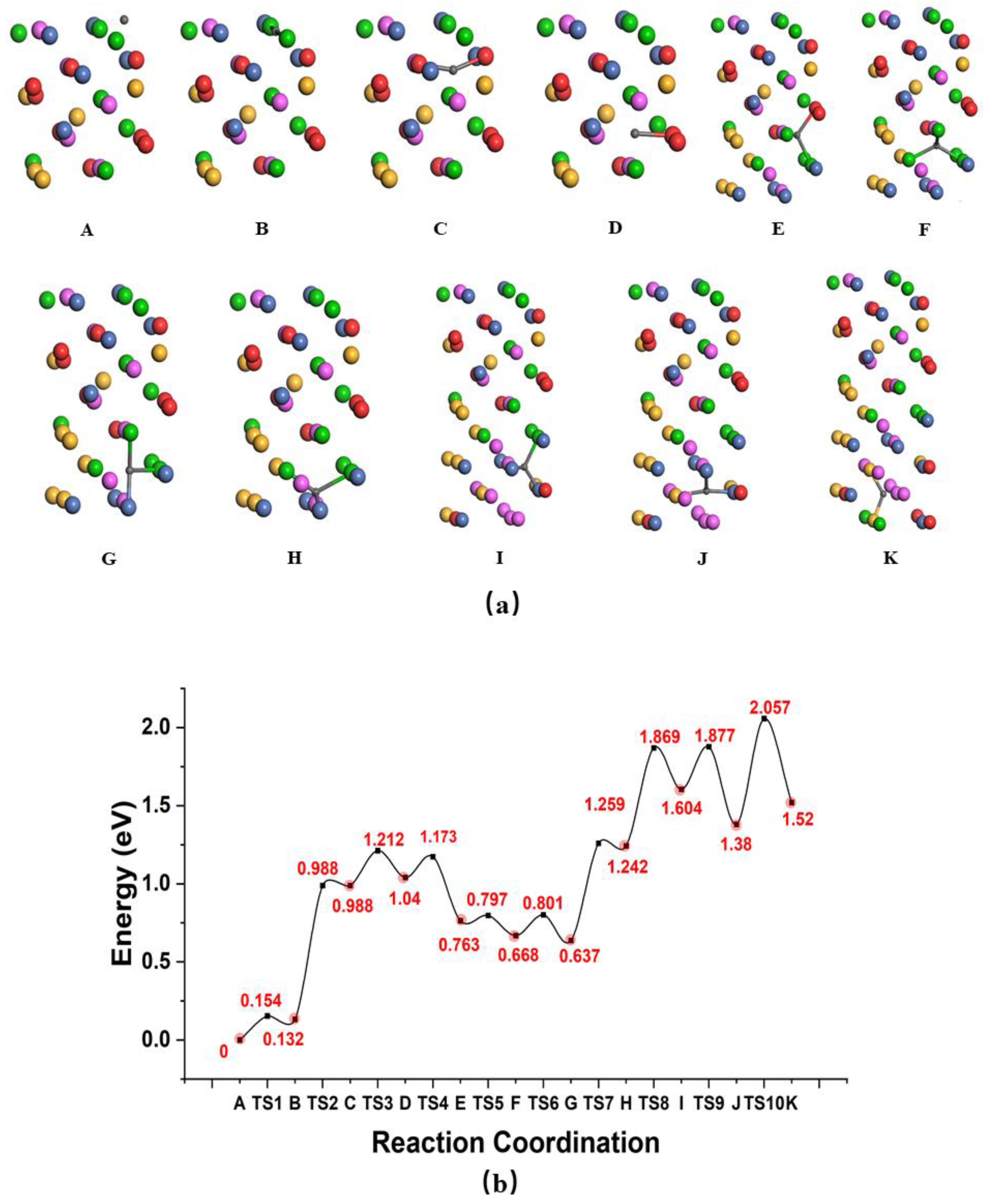
3.3. Diffusion of Atomic H on the Surface and Penetration into the Bulk
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wilder, A.B.; Dana, A.W.; Shortsleeve, F.J.; Troiano ARCastro, R.J.; Tricot, R. Relation of flake formation in steel to hydrogen, microstructure, and stress. Trans. Am. Inst. Min. Metall. Eng. 1955, 206, 1404–1405. [Google Scholar]
- Beachem, C.D. A new model for hydrogen-assisted cracking (hydrogen “embrittlement”). Metall. Mater. Trans. B 1972, 3, 441–455. [Google Scholar] [CrossRef]
- Jia, G.W.; Lei, M.Y.; Li, M.Y.; Xu, W.Q.; Li, R.; Lu, Y.H.; Cai, M.L. Hydrogen embrittlement in hydrogen-blended natural gas transportation systems: A review. Int. J. Hydrogen Energy 2023, 48, 32137–32157. [Google Scholar] [CrossRef]
- Okonkwo, P.C.; Barhoumi, E.; Ben Belgacem, I.; Mansir, I.B.; Aliyu, M.; Emori, W.; Uzoma, P.C.; Beitelmal, W.H.; Akyüz, E.; Eadwan, A.B.; et al. A focused review of the hydrogen storage tank embrittlement mechanism process. Int. J. Hydrogen Energy 2023, 48, 12935–12948. [Google Scholar] [CrossRef]
- Levchuk, D.; Koch, F.; Maier, H.; Bolt, H. Gas-driven deuterium permeation through Al2O3 coated samples. Phys. Scr. 2004, T108, 119–123. [Google Scholar] [CrossRef]
- Li, Q.; Mo, L.B.; Wang, J.; Yan, K.; Tang, T.; Rao, Y.C.; Yao, W.Q.; Cao, J.L. Performances of Cr2O3-hydrogen isotopes permeation barriers. Int. J. Hydrogen Energy 2015, 40, 6459–6464. [Google Scholar] [CrossRef]
- Liu, L.L.; Ruan, Q.D.; Xiao, S.; Meng, X.Y.; Huang, C.; Wu, Y.Z.; Fu, R.K.Y.; Chu, P.K. Fabrication and hydrogen permeation resistance of dense CrN coatings. Surf. Coat. Technol. 2022, 437, 128326. [Google Scholar] [CrossRef]
- Nishikiori, T.; Nohira, T.; Ito, Y. Hydrogen impermeability of TiN films and its dependence on nitrogen concentration at high temperatures. J. Electrochem. Soc. 2001, 148, E52–E59. [Google Scholar] [CrossRef]
- Shan, C.Q.; Wu, A.J.; Li, Y.J.; Zhao, Z.Q.; Chen, Q.W.; Huang, Q.R.; Shi, S.L. The behavior of diffusion and permeation of tritium through 316L stainless-steel with coating of TiC and TiN + TiC. J. Nucl. Mater. 1992, 191, 221–225. [Google Scholar] [CrossRef]
- Isobe, K.; Yamanishi, T.; Konishi, S. Tritium permeation behavior in SiC/SiC composites. Fusion. Eng. Des. 2009, 85, 1012–1015. [Google Scholar] [CrossRef]
- Nakamura, H.; Isobe, K.; Nakamichi, M.; Yamanishi, T. Evaluation of deuterium permeation reduction factor of various coatings deposited on ferritic/martensitic steels for development of tritium permeation barrier. Fusion Eng. Des. 2010, 85, 1531–1536. [Google Scholar] [CrossRef]
- Hollenberg, G.W.; Simonen, E.P.; Kalinin, G.; Terlain, A. Tritium/hydrogen barrier development. Fusion Eng. Des. 1995, 28, 190–208. [Google Scholar] [CrossRef]
- George, E.P.; Raabe, D.; Ritchie, R.O. High-entropy alloys. Nat. Rev. Mater. 2019, 4, 515–534. [Google Scholar] [CrossRef]
- Tsai, M.H.; Yeh, J.W. High-entropy alloys: A critical review. Mater. Res. Lett. 2014, 2, 107–123. [Google Scholar] [CrossRef]
- Tamura, M. Hydrogen permeation characteristics of TiN-Coated Stainless Steels. J. Mater. Sci. Eng. 2015, A5, 197–201. [Google Scholar]
- Alat, E.; Motta, A.T.; Comstock, R.J.; Partezana, J.M.; Wolfe, D.E. Multilayer (TiN, TiAlN) ceramic coatings for nuclear fuel cladding. Appl. Surf. Sci. 2018, 432, 207–213. [Google Scholar] [CrossRef]
- Tamura, M.; Takizawa, H. TiAlN/TiMoN Coatings as Hydrogen Barriers. J. Mater. Sci. Eng. 2019, A9, 1–7. [Google Scholar]
- Liu, Y.; Huang, S.S.; Ding, J.H.; Yang, Y.C.; Zhao, J.J. Vanadium carbide coating as hydrogen permeation barrier: A DFT study. Int. J. Hydrogen Energy 2019, 44, 6093–6102. [Google Scholar] [CrossRef]
- Di Stefano, D.; Mrovec, M.; Elsässer, C. First-principles investigation of quantum mechanical effects on the diffusion of hydrogen in iron and nickel. Phys. Rev. B 2015, 92, 224301. [Google Scholar] [CrossRef]
- Zhang, G.K.; Wang, X.L.; Xiong, Y.F.; Shi, Y.; Song, J.F.; Luo, D.L. Mechanism for adsorption, dissociation and diffusion of hydrogen in hydrogen permeation barrier of α-Al2O3: A density functional theory study. Int. J. Hydrogen Energy 2013, 38, 1157–1165. [Google Scholar] [CrossRef]
- Ke, X.Z.; Kramer, G.J. Absorption and diffusion of hydrogen in palladium-silver alloys by density functional theory. Phys. Rev. B 2002, 66, 184304. [Google Scholar] [CrossRef]
- Hansen, N.F.; Andersen, H.C. Properties of quantum transition state theory and its corrections. J. Phys. Chem. 1996, 100, 1137–1143. [Google Scholar] [CrossRef]
- Kresse, G.; Furthermuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthermuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Hafner, J.; Kresse, G. The Vienna Ab-initio Simulation Program VASP: An efficient and versatile tool for studying the structural, dynamic, and electronic properties of materials. In Properties of Complex Inorganic Solids; Gonis, A., Meike, A., Turchi, P.E.A., Eds.; Springer: Boston, MA, USA, 1997; pp. 69–82. [Google Scholar]
- Blöchl, P.E. Project augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1966, 77, 3865–3868. [Google Scholar] [CrossRef]
- White, J.A.; Bird, D.M. Implementation of gradient-corrected exchange-correlation potentials in Car-Parrinello total-energy calculations. Phys. Rev. B 1994, 50, 4954–4957. [Google Scholar] [CrossRef]
- Jiang, S.; Shao, L.; Fan, T.W.; Duan, J.M.; Chen, X.T.; Tang, B.Y. Elastic and thermodynamic properties of high entropy carbide (HfTaZrTi)C and (HfTaZrNb)C from ab initio investigation. Ceram. Int. 2020, 46, 15104–15112. [Google Scholar] [CrossRef]
- Van de Walle, A.; Ceder, G. Automating first-principles phase diagram calculations. J. Phase Equilib. 2002, 23, 348–359. [Google Scholar] [CrossRef]
- Wang, V.; Xu, N.; Liu, J.C.; Tang, G.; Geng, W.T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Kehr, K.W. Theory of the diffusion of hydrogen in metals. In Hydrogen in Metals I: Basic Properties; Alefeld, G., Völkl, J., Eds.; Springer: Berlin, Germany, 1978; pp. 197–226. [Google Scholar]
- Guo, L.Z.; Peano, V.; Marthaler, M.; Dykman, M.I. Quantum critical temperature of a modulated oscillator. Phys. Rev. A 2013, 87, 062117. [Google Scholar] [CrossRef]
- Jónsson, H.; Mills, G.; Jacobsen, K.W. Nudged elastic band method for finding minimum energy paths of transitions. In Classical and Quantum Dynamics in Condensed Phase Simulations; Berne, B.J., Ciccotti, G., Coker, D.F., Eds.; Hong Kong World Scientific: Hong Kong, China, 1998; pp. 385–403. [Google Scholar]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Erikat, I.A.; Hamad, B.A.; Khalifeh, J.M. A density functional study on adsorption and dissociation of O2 on Ir (100) surface. Chem. Phys. 2011, 385, 35–40. [Google Scholar] [CrossRef]
- Vegge, T. Locating the rate-limiting step for the interaction of hydrogen with Mg(0001) using density-functional theory calculations and rate theory. Phys. Rev. B 2004, 70, 035412. [Google Scholar] [CrossRef]
- Belonoshko, A.B.; Rosengren, A.; Dong, Q.; Hultquist, G.; Leygraf, C. First-principles study of hydrogen diffusion in α-Al2O3 and liquid alumina. Phys. Rev. B 2004, 69, 024302. [Google Scholar] [CrossRef]
- Jiang, D.E.; Carter, E.A. Diffusion of interstitial hydrogen into and through bcc Fe from first principle. Phys. Rev. B 2004, 70, 064102. [Google Scholar] [CrossRef]
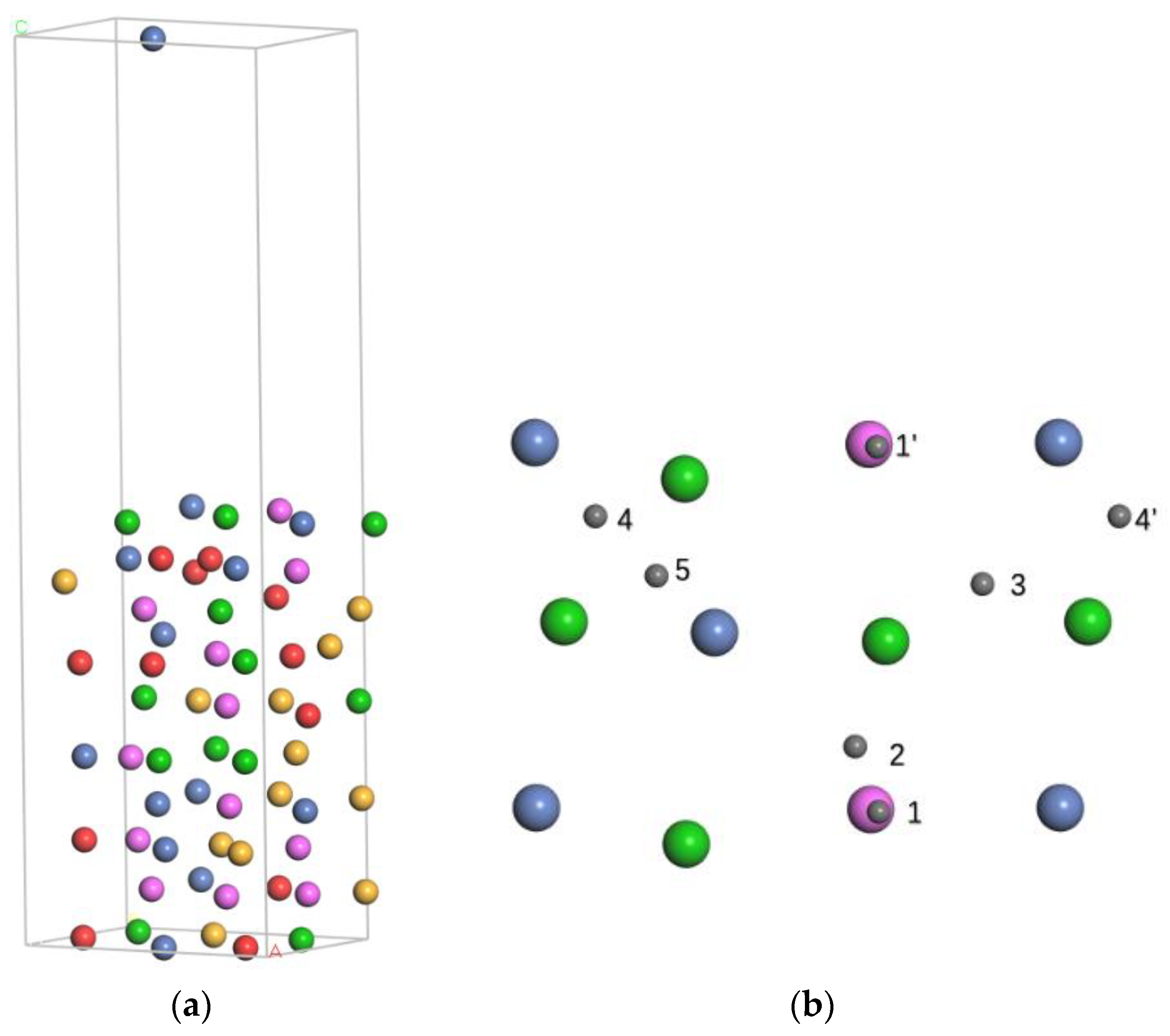
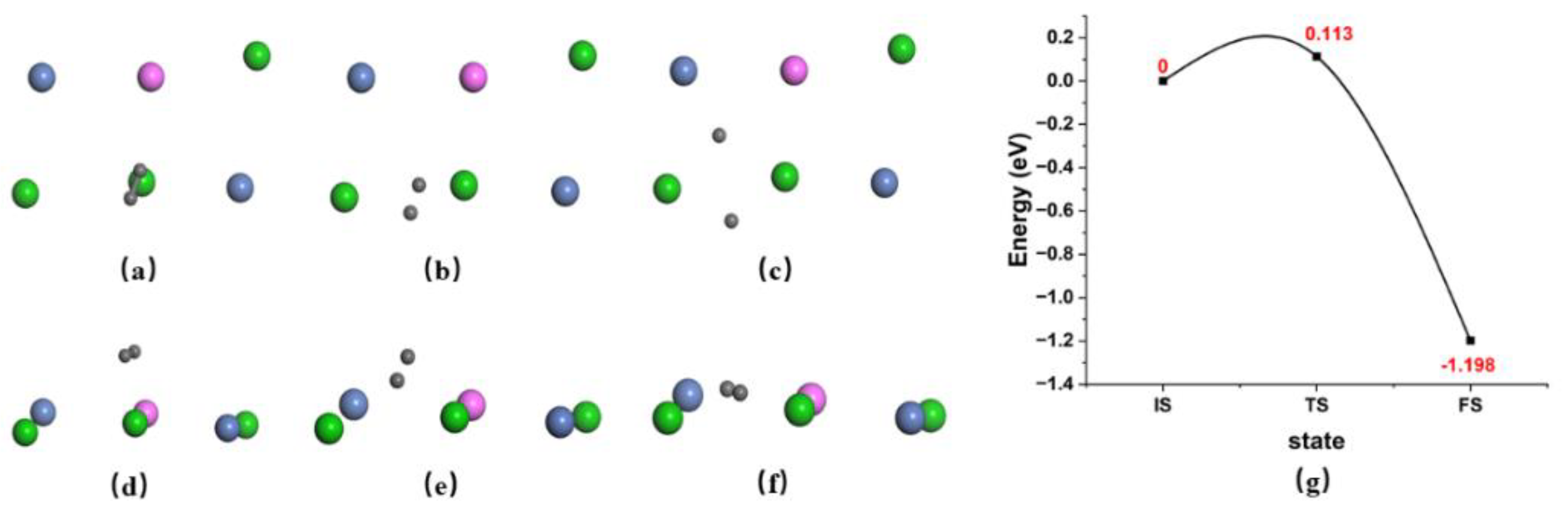
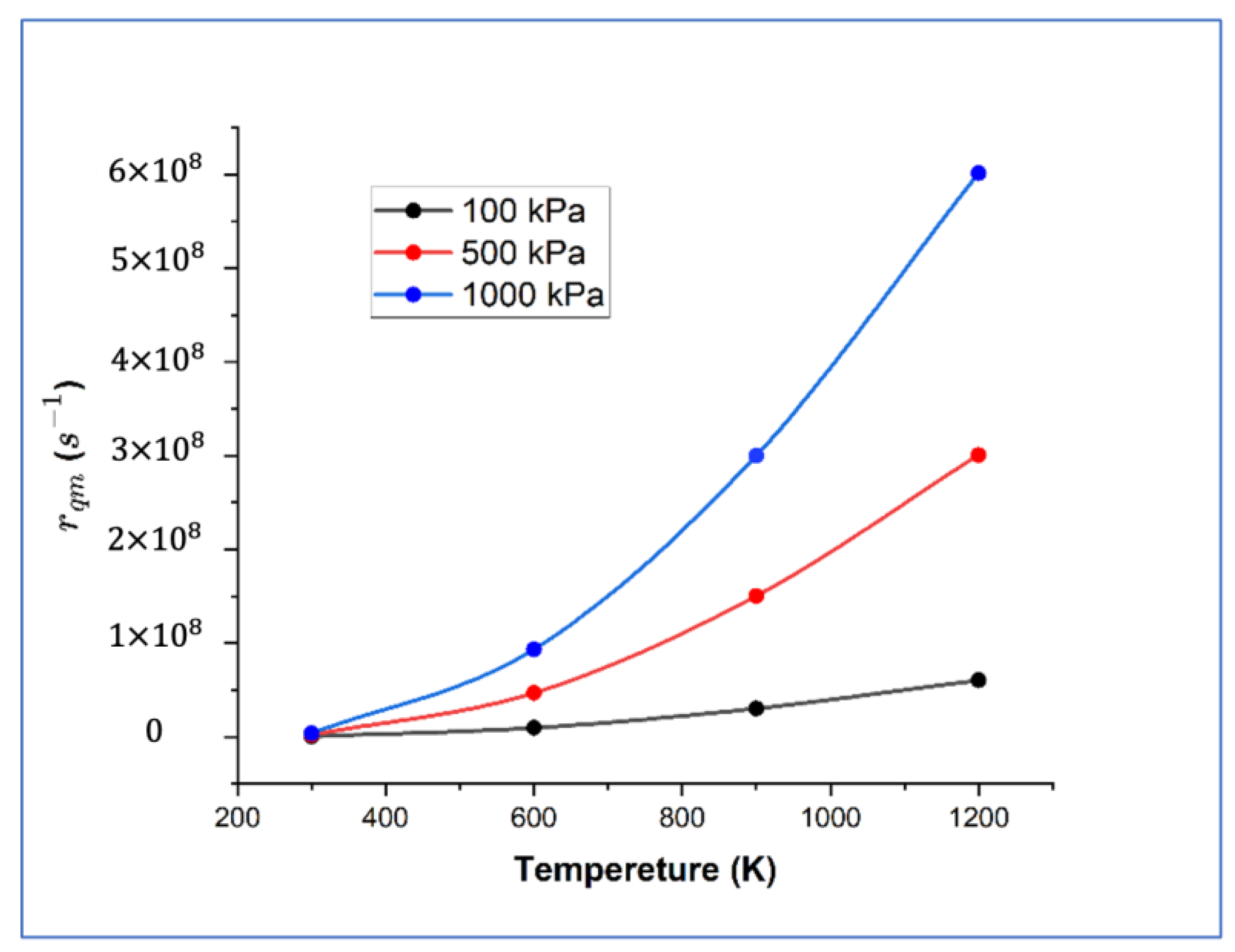
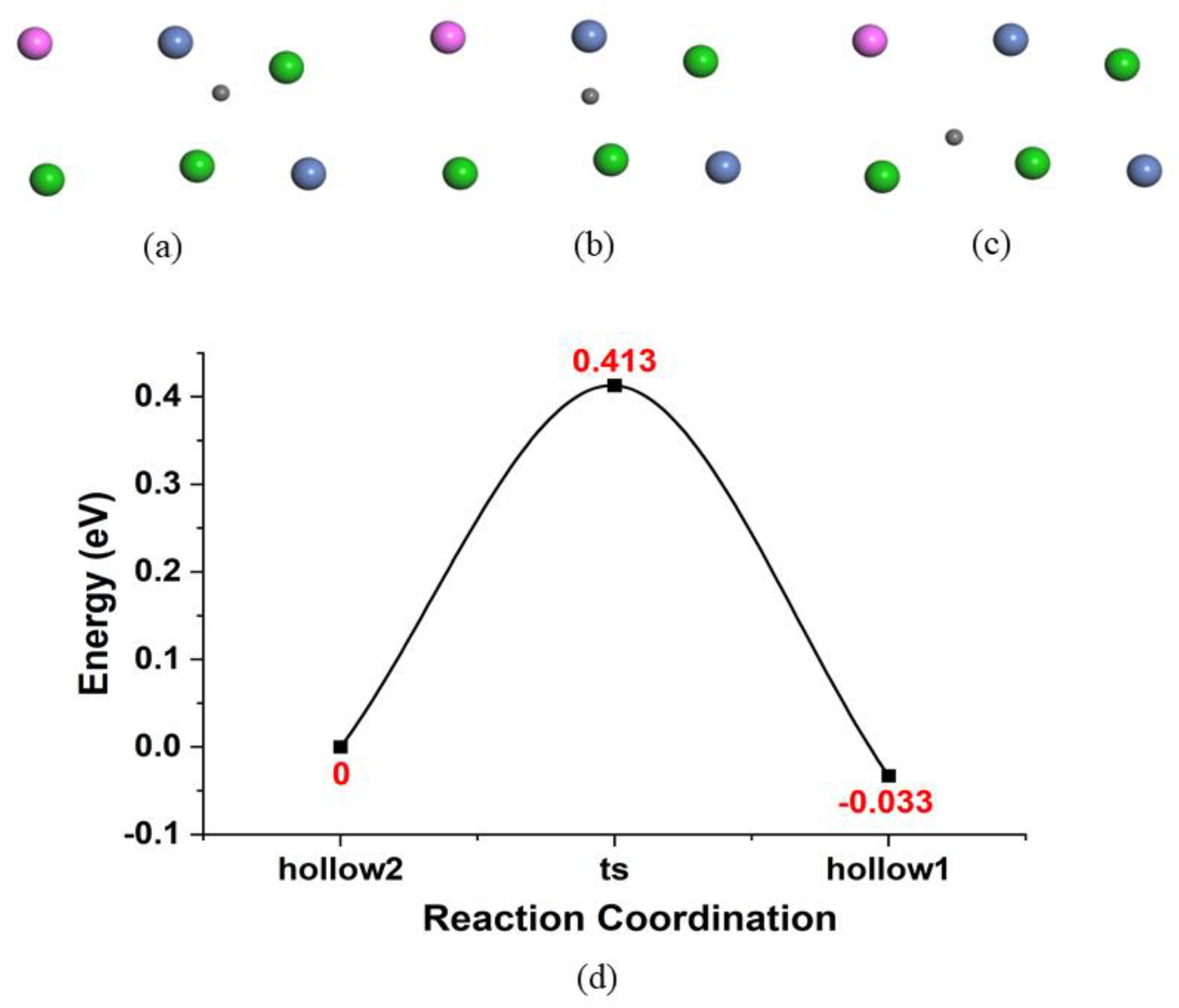

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorption Site | Distance to Surface (Å) | ||
|---|---|---|---|
| H2 molecule | Ti-Top | −0.152 | 1.994 |
| H atom | 1 (Top) | −0.123 | 1.590 |
| 2 (Bridge) | −0.463 | 1.109 | |
| 3 (Hollow) | −0.863 | 0.597 | |
| 4 (Hollow) | −0.830 | 1.035 | |
| 5 (Hollow) | −0.803 | 1.040 | |
| 2 H atoms | Hollow | −0.723 | 0.418, 0.587 |
| Adsorption Site | Top-Al1 | Top-Ti1 | Top-Ti2 | Top-Ti3 | Top-Ni1 | Top-Ni2 | |
|---|---|---|---|---|---|---|---|
| Energy (eV) | −421.72 | −421.30 | −421.25 | −421.33 | −421.81 | −421.54 | |
| Frequency (THz) | 1 | 51.36 | 46.10 | 44.31 | 43.86 | 52.53 | 52.37 |
| 2 | 11.74 | 6.40 | 1.12i | 8.09i | 3.89i | 2.22i | |
| 3 | 5.52 | 3.37i | 10.46i | 11.49i | 13.60i | 8.46i | |
| Band-length of H-M () | 1.59 | 1.77 | 1.78 | 1.77 | 1.53 | 1.5 | |
| Coverage (ML) | Energy (eV) | Ead (eV/atom) | EadZPE (eV/atom) |
|---|---|---|---|
| 1/6 | −422.46 | −0.86 | −0.82 |
| 1/3 | −426.49 | −0.75 | −0.72 |
| 1/2 | −430.67 | −0.77 | −0.74 |
| 2/3 | −434.48 | −0.68 | −0.65 |
| 1 | −441.87 | −0.56 | −0.53 |
| Path | (eV) | (eV) | D0qm (ps−1) | D0cl (ps−1) | D0cl (1 × 10−8 m2s−1) | TC (K) | |||
|---|---|---|---|---|---|---|---|---|---|
| T = 300 K | T = 600 K | T = 900 K | |||||||
| Diffusion on surface | 0.431 | −0.034 | 6.128 | 10.512 | 12.996 | 20.16 | 22.487 | 116 | |
| Penetration from surface | A→B | 0.148 | −0.027 | 6 | 9.83 | 11.85 | 17.07 | 3.449 | 70 |
| B→C | 0.856 | −0.011 | 6.11 | 10.45 | 12.75 | 17 | 8.169 | 61 | |
| C→D | 0.224 | −0.03 | 5.91 | 9.16 | 10.7 | 15.17 | 18.024 | 151 | |
| D→E | 0.132 | −0.003 | 5.542 | 7.721 | 8.45 | 9.363 | 4.233 | 100 | |
| E→F | 0.033 | −0.043 | 6.123 | 10.654 | 13.419 | 16.89 | 3.139 | 126 | |
| F→G | 0.132 | −0.018 | 6.157 | 10.688 | 13.148 | 18.343 | 4.235 | 139 | |
| G→H | 0.621 | 0.016 | 6.07 | 10.04 | 11.81 | 12.721 | 4.853 | 159 | |
| H→I | 0.625 | −0.001 | 6.118 | 10.424 | 12.627 | 15.729 | 3.632 | 251 | |
| I→J | 0.271 | −0.03 | 6.223 | 11.515 | 14.981 | 24.466 | 7.267 | 236 | |
| J→K | 0.6 | 0.056 | 6.027 | 9.354 | 10.239 | 7.856 | 4.433 | 231 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, W.; Wu, L.; Shuai, Q.; Li, Z.; Wang, H.; Fu, W.; Jiang, Z.; Zhao, C.; Hua, Q. Mechanism for Adsorption, Dissociation, and Diffusion of Hydrogen in High-Entropy Alloy AlCrTiNiV: First-Principles Calculation. Nanomaterials 2024, 14, 1391. https://doi.org/10.3390/nano14171391
Zheng W, Wu L, Shuai Q, Li Z, Wang H, Fu W, Jiang Z, Zhao C, Hua Q. Mechanism for Adsorption, Dissociation, and Diffusion of Hydrogen in High-Entropy Alloy AlCrTiNiV: First-Principles Calculation. Nanomaterials. 2024; 14(17):1391. https://doi.org/10.3390/nano14171391
Chicago/Turabian StyleZheng, Weilong, Liangliang Wu, Qilin Shuai, Zhaoqiang Li, Haoqi Wang, Wei Fu, Zhenxiong Jiang, Chuang Zhao, and Qingsong Hua. 2024. "Mechanism for Adsorption, Dissociation, and Diffusion of Hydrogen in High-Entropy Alloy AlCrTiNiV: First-Principles Calculation" Nanomaterials 14, no. 17: 1391. https://doi.org/10.3390/nano14171391