Coexistence of the Band Filling Effect and Trap-State Filling in the Size-Dependent Photoluminescence Blue Shift of MAPbBr3 Nanoparticles



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of MAPbBr3 Nanoparticles (MNPs)
2.2. Characterization
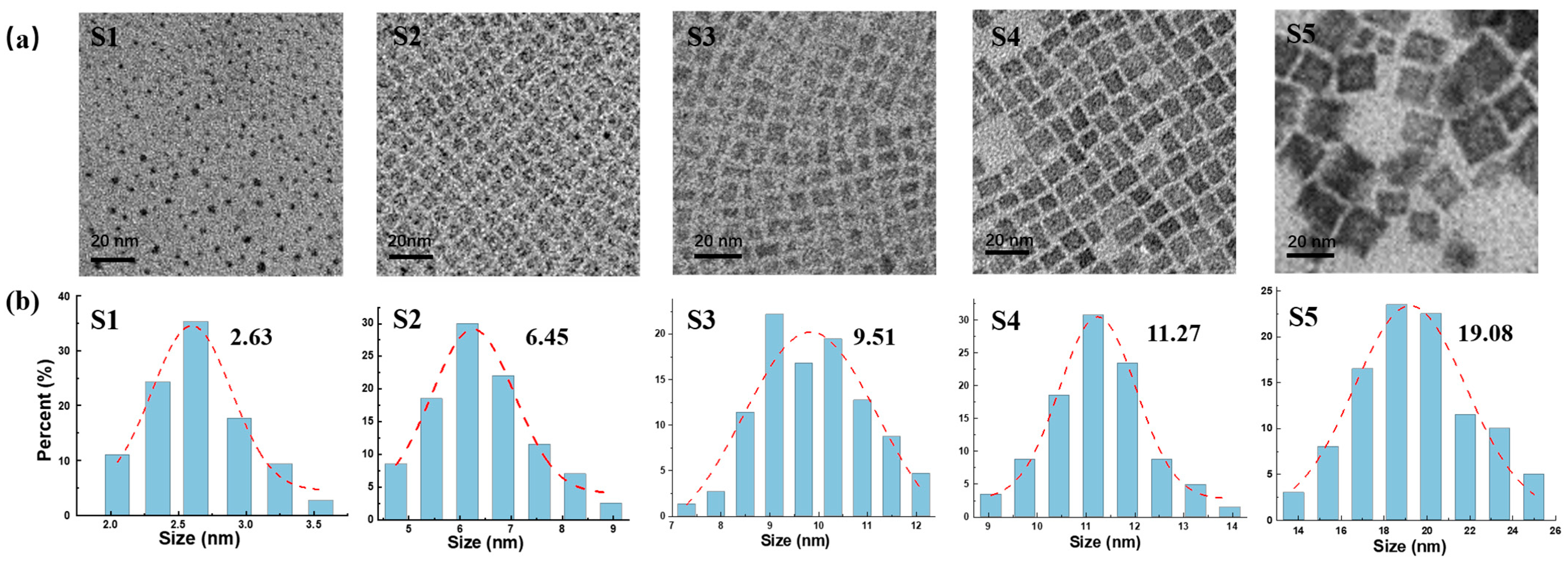
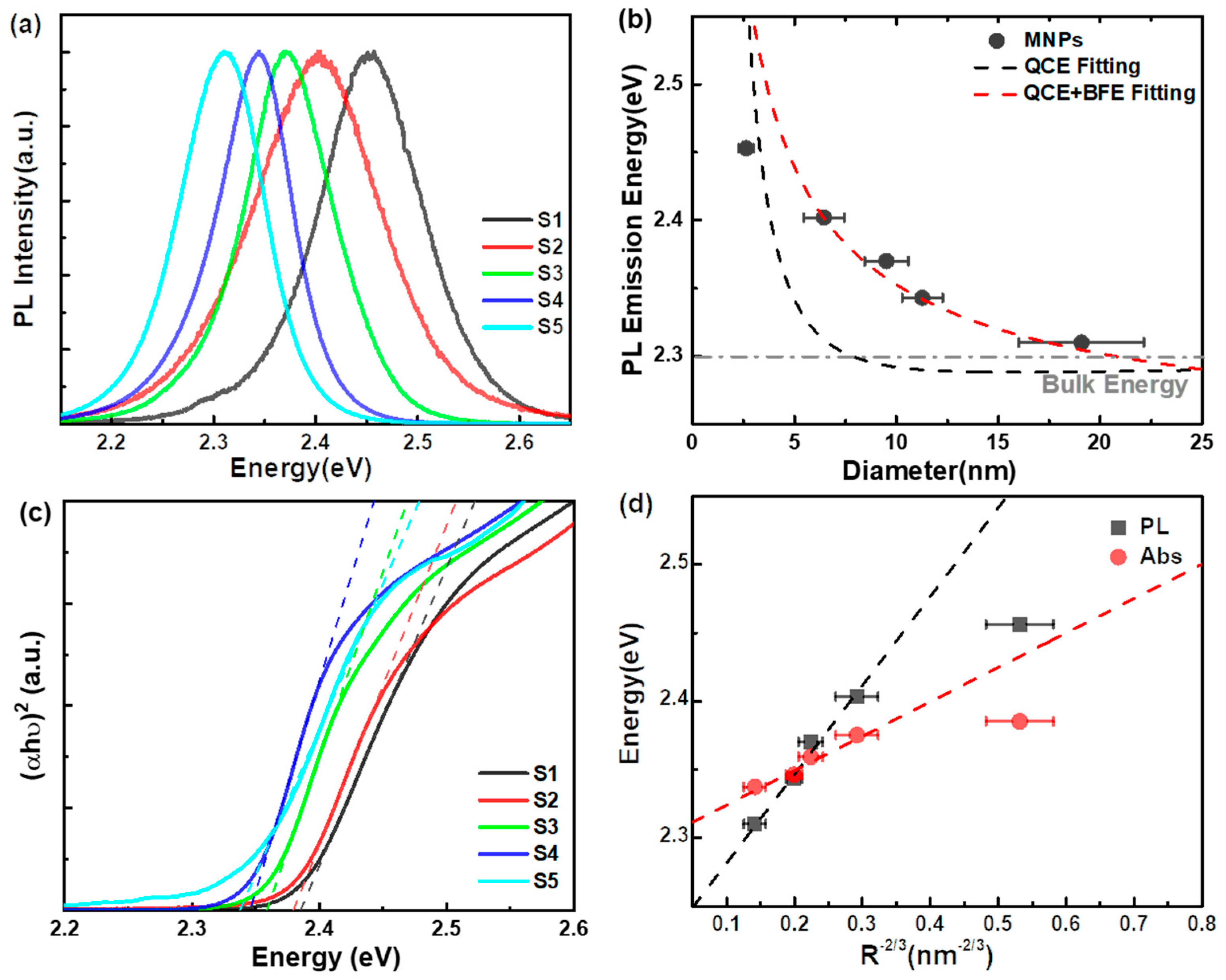
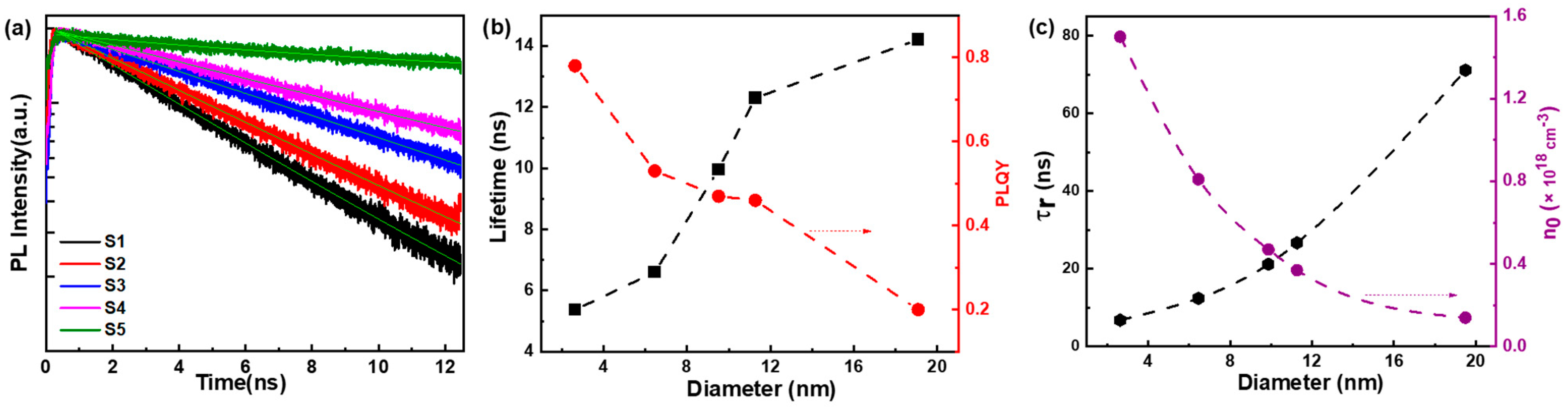
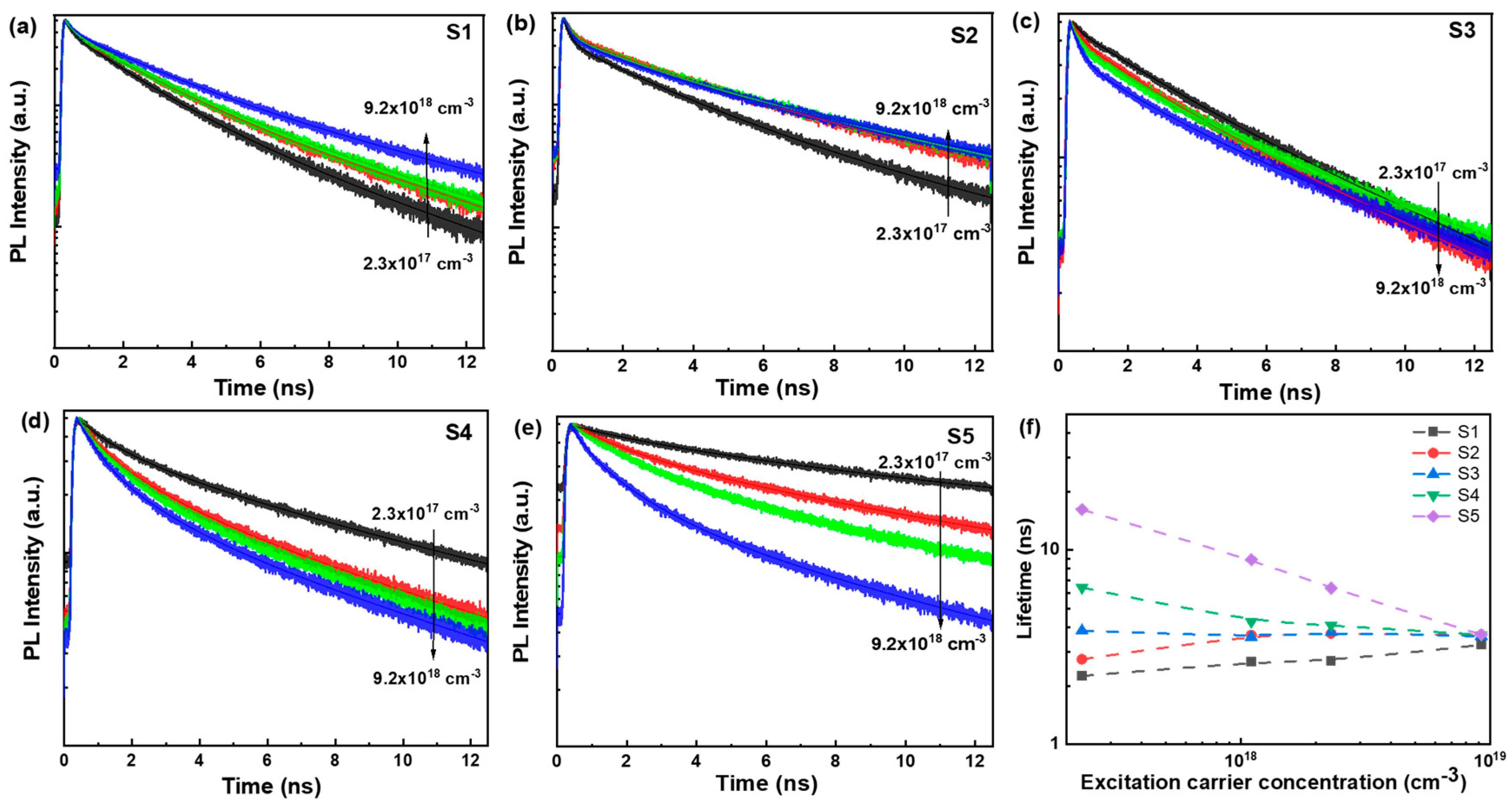
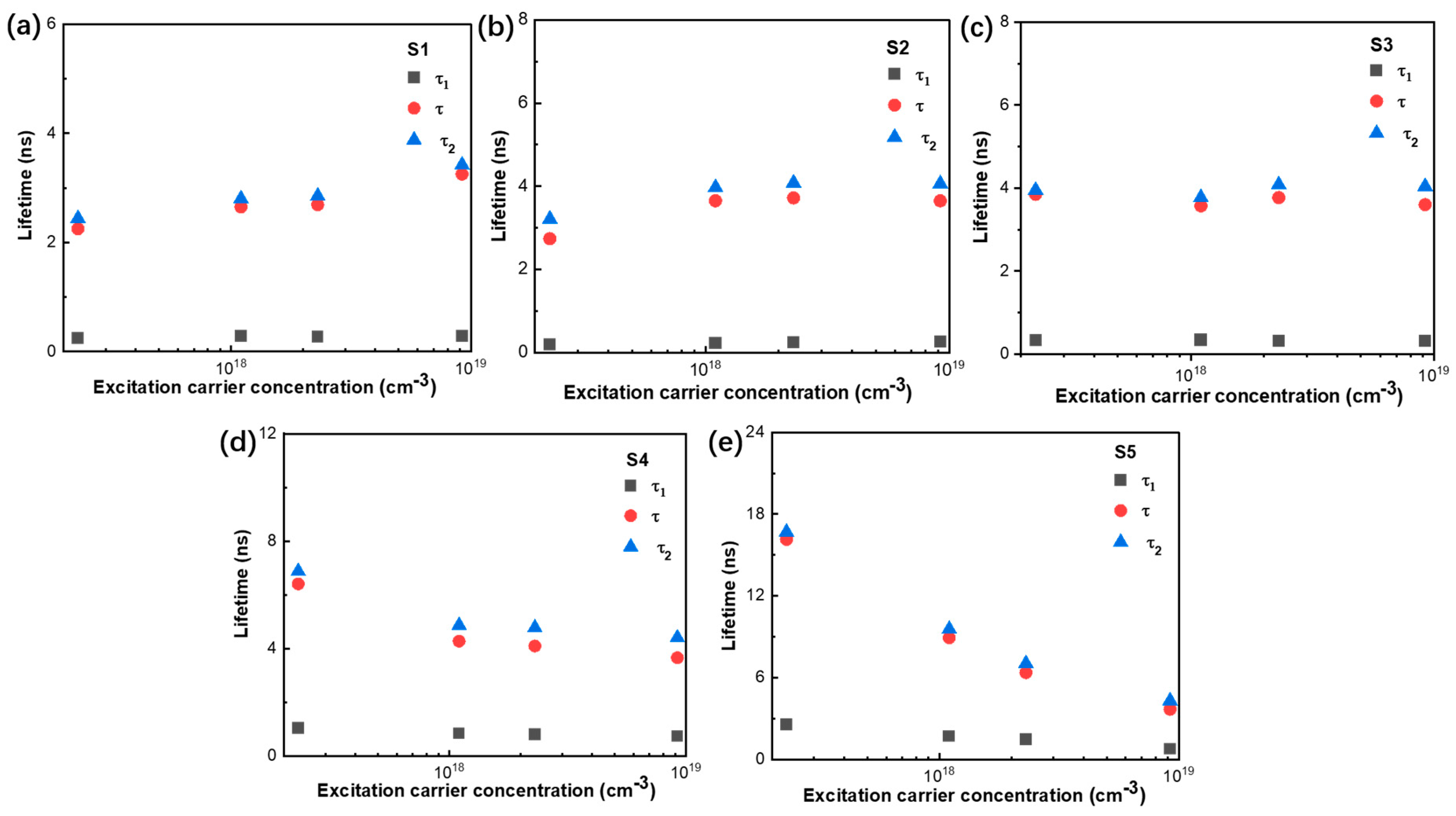
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Fu, Y.; Zhu, H.; Chen, J.; Hautzinger, M.P.; Zhu, X.Y.; Jin, S. Metal Halide Perovskite Nanostructures for Optoelectronic Applications and the Study of Physical Properties. Nat. Rev. Mater. 2019, 4, 169–188. [Google Scholar] [CrossRef]
- Quan, L.; Rand, B.P.; Friend, R.H.; Mhaisalkar, S.G.; Lee, T.W.; Sargent, E.H. Perovskites for Next-Generation Optical Sources. Chem. Rev. 2019, 119, 7444–7477. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Zhang, Y.; Wang, S.; Guo, J.; Yu, W.W.; Rogach, A.L. Metal Hailide Perovskites Light-Emitting Devices: Promising Technology for Next-Generation Displays. Adv. Funct. Mater. 2019, 29, 1902008. [Google Scholar] [CrossRef]
- Wang, Y.; Song, L.; Chen, Y.; Huang, W. Emerging New-Generation Photodetectors Based on Low-Dimensional Halide Perovskites. ACS Photonics 2020, 7, 10–28. [Google Scholar] [CrossRef]
- Shamsi, J.; Urban, A.S.; Imran, M.; De Trizio, L.; Manna, L. Metal Halide Perovskite Nanocrystals: Synthesis, Post-Synthesis Modifications, and Their Optical Properties. Chem. Rev. 2019, 119, 3296–3348. [Google Scholar] [CrossRef] [PubMed]
- Sichert, J.A.; Tong, Y.; Mutz, N.; Vollmer, M.; Fischer, S.; Milowska, K.Z.; García Cortadella, R.; Nickel, B.; Cardenas-Daw, C.; Stolarczyk, J.K.; et al. Quantum Size Effect in Organometal Halide Perovskite Nanoplatelets. Nano Lett. 2015, 15, 6521–6527. [Google Scholar] [CrossRef]
- Jpseph, S.M.; Prashant, V.K. Band Filling with Free Charge Carriers in Organometal Halide Perovskites. Nat. Photon. 2014, 8, 737–747. [Google Scholar]
- Katan, C.; Mercier, N.; Even, J. Quantum Confinement and Dielectric Confinement Effects in Lower-Dimensional Hybrid Perovskite Semiconductors. Chem. Rev. 2019, 119, 3140–3192. [Google Scholar] [CrossRef]
- Akkerman, Q.A.; Motti, S.G.; Kandada, A.R.S.; Mosconi, E.; D’Innocenzo, V.; Bertoni, G.; Marras, S.; Kamino, B.A.; Miranda, L.; De Angelis, F.; et al. Solution Synthesis Approach to Colloidal Cesium Lead Halide Perovskite Nanoplatelets with Monolayer-Level Thickness Control. J. Am. Chem. Soc. 2016, 138, 1010–1016. [Google Scholar] [CrossRef]
- Butkus, J.; Vashishtha, P.; Chen, K.; Gallaher, J.K.; Prasad, S.K.K.; Metin, D.Z.; Laufersky, G.; Gaston, N.; Halpert, J.E.; Hodgkiss, J.M. The Evolution of Quantum Confinement in CsPbBr3 Perovskite Nanocrystals. Chem. Mater. 2017, 29, 3644–3652. [Google Scholar] [CrossRef]
- Protesescu, L.; Yakunin, S.; Bodnarchuk, M.I.; Krieg, F.; Caputo, R.; Hendon, C.H.; Yang, R.X.; Walsh, A.; Kovalenko, M.V. Nanocrystals of Cesium Lead Halide Perovskites (CsPbX3, X = Cl, Br, and I): Novel Optoelectronic Materials Showing Bright Emission with Wide Color Gamut. Nano Lett. 2015, 15, 3692–3696. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Gomez, L.; De Weerd, C.; Fujiwara, Y.; Gregorkiewicz, T.; Suenaga, K. Direct Observation of Band Structure Modifications in Nanocrystals of CsPbBr3 Perovskite. Nano Lett. 2016, 16, 7198–7202. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.F.; Zhang, Q.; Yip, J.N.; Xiong, Q.H.; Sum, T.C. Wavelength Tunable Single Nanowire Lasers Based on Surface Plasmon Polariton Enhanced Burstein-Moss Effect. Nano Lett. 2013, 13, 5336–5343. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Liao, Z.M.; Bie, Y.Q.; Wu, H.C.; Zhou, Y.B.; Fu, X.W.; Yu, D.P. Luminescence Blue-Shift of CdSe Nanowires Beyond the Quantum Confinement Regime. Appl. Phys. Lett. 2011, 99, 103103. [Google Scholar] [CrossRef]
- Chen, C.W.; Chen, K.H.; Shen, C.H.; Ganguly, A.; Chen, L.C.; Wu, J.J.; Wen, H.; Pong, W.F. Anomalous Blueshift in Emission Spectra of ZnO Nanorods with Sizes Beyond Quantum Confinement Regime. Appl. Phys. Lett. 2006, 88, 241905. [Google Scholar] [CrossRef]
- Yang, Y.H.; Chen, X.Y.; Feng, Y.; Yang, G.W. Physical Mechanism of Blue-Shift of UV Luminescence of a Single Pencil-like ZnO Nanowire. Nano Lett. 2017, 7, 3879–3883. [Google Scholar] [CrossRef]
- Wang, W.; Li, Y.; Wang, X.; Lv, Y.; Wang, S.; Wang, K.; Shi, W.Y.; Xiao, L.; Chen, Z.; Gong, Q. Density-Dependent Dynamical Coexistence of Excitons and Free Carriers in the Organolead Perovskite CH3NH3PbI3. Phys. Rev. B. 2016, 94, 140302. [Google Scholar] [CrossRef]
- Cho, J.; Choi, Y.H.; O’Loughlin, T.E.; De Jesus, L.; Banerjee, S. Ligand-Mediated Modulation of Layer Thicknesses of Perovskite Methylammonium Lead Bromide Nanoplatelets. Chem. Mater. 2016, 28, 6909–6916. [Google Scholar] [CrossRef]
- Efros, A.L.; Nesbitt, D.J. Origin and Control of Blinking in Quantum Dots. Nat. Nanotechnol. 2016, 11, 661–671. [Google Scholar] [CrossRef]
- Zheng, K.; Zhu, Q.S.; Abdellah, M.; Messing, M.E.; Zhang, W.; Generalov, A.; Niu, Y.; Ribaud, L.; Canton, S.E.; Pullerits, T. Exciton Binding Energy and the Nature of Emission States in Organometal Halide Perovskites. J. phys. Chem. Lett. 2015, 6, 2969–2975. [Google Scholar] [CrossRef]
- Lee, S.; Park, J.H.; Lee, B.R.; Jung, E.D.; Yu, J.C.; Di Nuzzo, D.; Friend, R.H.; Song, M.H. Amine-Based Passivating Materials for Enhanced Optical Properties and Performance of Organic-Inorganic Perovskites in Light-Emitting Diodes. J. Phys. Chem. Lett. 2017, 8, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Bhaumik, S.; Veldhuis, S.A.; Ng, Y.F.; Li, M.; Muduli, S.K.; Sum, T.C.; Damodaran, B.; Mhaisalkar, S.; Mathews, N. Highly Stable, Luminescent Core-Shell Type Methylammonium-Octylammonium Lead Bromide Layered Perovskite Nanoparticles. Chem. Commun. 2016, 52, 7118. [Google Scholar] [CrossRef] [PubMed]
- Kirakosyan, A.; Kim, J.; Lee, S.W.; Swathi, I.; Yoon, S.-G.; Choi, J. Optical Properties of Colloidal CH3NH3PbBr3 Nanocrystals by Controlled Growth of Lateral Dimension. Cryst. Groth. Des. 2017, 17, 794–799. [Google Scholar] [CrossRef]
- Ahmed, G.H.; Yin, J.; Bose, R.; Sinatra, L.; Alarousu, E.; Yengel, E.; AlYami, N.M.; Saidaminov, M.I.; Zhang, Y.; Hedhili, M.N.; et al. Pyridine-Induced Dimensionality Change in Hybrid Perovskite Nanocrystals. Chem. Mater. 2017, 29, 4393–4400. [Google Scholar] [CrossRef]
- Debellis, D.; Gigli, G.; Brinck, S.T.; Infant, I.; Giansante, C. Quantum-Confined and Enhanced Optical Absorption of Colloidal PbS Quantum Dots at Wavelengths with Expected Bulk Behavior. Nano Lett. 2017, 17, 1248–1254. [Google Scholar] [CrossRef]
- Ning, J.Q.; Zheng, C.C.; Zhang, X.H.; Xu, S.J. Strong Quantum Confinement Effect and Reduced Frohlich Exciton-Phonon Coupling an ZnO Quantum Dots Embedded Inside a SiO2 Matrix. Nanoscale 2015, 7, 17482. [Google Scholar] [CrossRef]
- Brus, L. Electronic Wave Functions in Semiconductor Clusters: Experiment and Theory. J. Phys. Chem. 1986, 90, 2555–2560. [Google Scholar] [CrossRef]
- Galkowski, K.; Mitioglu, A.; Miyata, A.; Plochocha, P.; Portugall, O.; Eperon, G.E.; Wang, J.T.-W.; Stergiopoulos, T.; Stranks, S.D.; Snaith, H.J.; et al. Determination of the Exciton Binding Energy and Effective Masses for Methylammonium and Formamidinium Lead Tri-Halide Perovskite Semiconductors. Energy Environ. Sci. 2016, 9, 962–970. [Google Scholar] [CrossRef]
- Tilchin, J.; Dirin, D.N.; Maikov, G.I.; Sashchiuk, A.; Kovalenko, M.V.; Lifshitz, E. Hydrogen-like Wannier-Mott Excitons in Single Crystal of Methylammonium Lead Bromide Perovskite. ACS Nano 2016, 10, 6363–6371. [Google Scholar] [CrossRef]
- Tanaka, K.; Takahashi, T.; Ban, T.; Kondo, T.; Uchida, K.; Miura, N. Comparative Study on the Excitons in Lead-Halide-Based Perovskite-Type Crystals CH3NH3PbBr3 CH3NH3PbI3. Solid State Commun. 2003, 127, 619–623. [Google Scholar] [CrossRef]
- Do, T.; Del Águila, A.G.; Cui, C.; Xing, J.; Ning, Z.J.; Xiong, Q. Optical Study on Intrinsic Exciton States in High-Quality CH3NH3PbBr3 Single Crystals. Phys. Rev. B 2017, 96, 075308. [Google Scholar]
- Laban, W.A.; Etgar, L. Depleted Hole Conductor-Free Lead Halide Iodide Heterojunction Solar Cells. Energy Environ. Sci. 2013, 6, 3249–3253. [Google Scholar] [CrossRef]
- Kim, J.; Lee, S.H.; Lee, J.H.; Hong, K.-H. The Role of Intrinsic Defects in Methylammonium Lead Iodide Perovskite. J. Phys. Chem. Lett. 2014, 5, 1312–1317. [Google Scholar] [CrossRef]
- Xing, G.; Mathews, N.; Lim, S.S.; Yantara, N.; Liu, X.; Sabba, D.; Gratzel, M.; Mhaisalkar, S.; Sum, T.C. Low-Temperature Solution-Processed Wavelength-Tunable Perovslites for Lasing. Nat. Mater. 2014, 13, 476–480. [Google Scholar] [CrossRef]
- Thomas, G.A.; Rice, T.M. Trions, Molecules and Excitons Above the Mott Density in Ge. Solid State Commun. 1977, 23, 359–363. [Google Scholar] [CrossRef]
- He, H.; Yu, Q.; Li, H.; Li, J.; Si, J.; Jin, Y.; Wang, N.; Wang, J.; He, J.; Wang, X.; et al. Exciton Localization in Solution-Processed Organolead Trihalide Perovskites. Nat. Comm. 2016, 7, 10896. [Google Scholar] [CrossRef]
- Kawaharamura, T.; Nishinaka, H.; Kamada, Y.; Ohshima, T.; Ye, Z.Z.; Zeng, Y.J.; Zhu, L.P.; He, H.P.; Zhao, B.H. Carrier Concentration Dependence of Band Gap Shift in n-Type Zno:Al Films. J. Appl. Phys. 2007, 101, 083705. [Google Scholar]
- Chen, S.; Wen, X.; Sheng, R.; Huang, S.; Deng, X.; Green, M.A.; Ho-Baillie, A. Mobile Ion Induced Slow Carrier Dynamic in Organic-Inorganic Perovskite CH3NH3PbBr3. ACS Appl. Mater. Inter. 2016, 8, 5351–5357. [Google Scholar] [CrossRef]
- Malgras, V.; Tominaka, S.; Ryan, J.W.; Henzie, J.; Takei, T.; Ohara, K.; Yamauchi, Y. Observation Of Quantum Confinement InMonodisperse Methylammonium Lead Halide Perovskite Nanocryst-als Embedded In Mesoporous Silica. J. Am. Chem. Soc. 2016, 138, 13874–13881. [Google Scholar] [CrossRef]
- D’Innocenzo, V.; Srimath Kandada, A.R.; De Bastiani, M.; Gandini, M.; Petrozza, A. Tuning The Light Emission Properties ByBand Gap Engineering In Hybrid Lead Halide Perovskite. J. Am. Chem.Soc. 2014, 136, 17730–17733. [Google Scholar] [CrossRef]
- Swain, S.K.; Duenow, J.N.; Johnston, S.W.; Amarasinghe, M.; McCoy, J.J.; Metzger, W.K.; Lynn, K.G. Approach to defect-free lifetime and high electron density in CdTe. J. Elect. Mater. 2019, 48, 7. [Google Scholar] [CrossRef]
- Yu, Z. The Rashab Effect and Indirect Electron-Hole Recombination in Hybrid Organic-Inorganic Perovskites. Phys. Chem. Chem. Phys. 2017, 19, 14907–14912. [Google Scholar] [CrossRef] [PubMed]
- Xing, G.C.; Wu, B.; Wu, X.Y.; Li, M.J.; Du, B.; Wei, Q.; Guo, J.; Yeow, E.K.L.; Sum, T.C.; Huang, W. Transcending the Slow Bimolecular Recombination in Lead-Halide Perovskites for Electroluminescence. Nat. Commun. 2017, 8, 14558. [Google Scholar] [CrossRef]
- Droseros, N.; Longo, G.; Brauer, J.C.; Sessolo, M.; Bolink, H.J.; Banerji, N. Origin of the Enhanced Photoluminescence Quantum Yield in MaPbBr3 Perovskite with Reduced Crystal Size. ACS Energy Lett. 2018, 3, 1458–1466. [Google Scholar] [CrossRef]
- Liu, W.; Butte, R.; Dussaigne, A.; Grandjean, N.; Deveaud, B.; Jacopin, G. Carrier-Density-Dependent Recombination Dynamics of Excitons and Electron-Hole Plasma in m-Plane InGaN/GaN Quantum Wells. Phys. Rev. B 2016, 94, 195411. [Google Scholar] [CrossRef]
- Krückemeier, L.; Krogmeier, B.; Liu, Z.; Rau, U.; Kirchartz, T. Understanding Transient Photoluminescence in Halide Perovskite Layer Stacks and Solar Cells. Adv. Energy Mater. 2021, 11, 2003489. [Google Scholar] [CrossRef]
- Yamada, Y.; Yamada, T.; Shimazaki, A.; Wakamiya, A.; Kanemitsu, Y. Interfacial Charge-Carrier Trapping in CH3NH3PbI3-Based Heterolayered Structures Revealed by Time-Resolved Photoluminescence Spectroscopy. J. Phys. Chem. Lett. 2016, 7, 1972–1977. [Google Scholar] [CrossRef]
- Ridley, B.K. Kinetics of Radiative Recombination in Quantum Wells. Phys. Rev. B 1990, 41, 12190. [Google Scholar] [CrossRef]
- Stranks, S.D.; Burlakov, V.M.; Leijtens, T.; Ball, J.M.; Goriely, A.; Snaith, H.J. Recombination Kinetics in Organic-Inorganic Perovskites: Excitons, Free Charge, and Subgap States. Phys. Rev. Appl. 2014, 2, 034007. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Oan, L.; Tamai, N. Effects of Size and Capping Reagents on Biexciton Auger Recombination Dynamics of CdTe Quantum Dots. J. Phys. Chem. C. 2009, 113, 11783–11789. [Google Scholar] [CrossRef]
- Staub, F.; Hempel, H.; Hebig, J.; Mock, J.; Paetzold, U.W.; Rau, U.; Unold, T.; Kirchartz, T. Beyond Bulk Lifetimes: Insights into Lead Halide Perovskite Films from Time-Resolved Photoluminescence. Phys. Rev. Appl. 2016, 6, 044017. [Google Scholar] [CrossRef]
- Kumar, P.; Muthu, C.; Vijayakumar, C.; Nair, V.; Narayan, K. Quantum Confinement Effects in Organic Lead Tribromide Perovskite Nanoparticles. J. Phys. Chem. 2016, 120, 18333–18339. [Google Scholar] [CrossRef]
- Huang, H.; Zhao, F.; Liu, L.; Zhang, F.; Wu, X.; Shi, L.; Zou, B.; Pei, Q.; Zhong, H. Emulsion Synthesis of Size-Tunable CH3NH3PbBr3 Quantum Dots: An Alternative Route Toward Efficient Light-Emitting Diodes. ACS Appl. Mater. Interfaces 2015, 7, 28128–28133. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C.; Kim, J.; Lee, T. Structural and Thermal Disorder of Solution-Processed CH3NH3PbBr3 Hybrid Perovskite Thin Films. Acs Appl. Mater. Interfaces 2017, 9, 10344–10348. [Google Scholar] [CrossRef]
- Kumawat, N.K.; Dey, A.; Narasimhan, K.L.; Kabra, D. Near Infrared to Visible Electroluminescent Diodes Based on Organometallic Halide Perovskites: Structural and Optical Investigation. ACS Photon. 2015, 2, 349–354. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, J.; Chen, M.; Huang, T.; Ding, G.; Wang, Z. Coexistence of the Band Filling Effect and Trap-State Filling in the Size-Dependent Photoluminescence Blue Shift of MAPbBr3 Nanoparticles. Nanomaterials 2024, 14, 1546. https://doi.org/10.3390/nano14191546
Sun J, Chen M, Huang T, Ding G, Wang Z. Coexistence of the Band Filling Effect and Trap-State Filling in the Size-Dependent Photoluminescence Blue Shift of MAPbBr3 Nanoparticles. Nanomaterials. 2024; 14(19):1546. https://doi.org/10.3390/nano14191546
Chicago/Turabian StyleSun, Jing, Mengzhen Chen, Tao Huang, Guqiao Ding, and Zhongyang Wang. 2024. "Coexistence of the Band Filling Effect and Trap-State Filling in the Size-Dependent Photoluminescence Blue Shift of MAPbBr3 Nanoparticles" Nanomaterials 14, no. 19: 1546. https://doi.org/10.3390/nano14191546
APA StyleSun, J., Chen, M., Huang, T., Ding, G., & Wang, Z. (2024). Coexistence of the Band Filling Effect and Trap-State Filling in the Size-Dependent Photoluminescence Blue Shift of MAPbBr3 Nanoparticles. Nanomaterials, 14(19), 1546. https://doi.org/10.3390/nano14191546