
Templating Iron(III) Oxides on DNA Molecules

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Preparation of FeOOH/DNA
2.3. Preparation of Fe2O3/DNA
2.4. Preparation of Non-Templated FeOOH Nanoparticles
2.5. Preparation of Non-Templated Fe2O3
2.6. Si Substrate Preparation
2.7. Optical Absorption Spectroscopy
2.8. FTIR
2.9. Photoelectron Spectroscopy (XPS)
2.10. Powder X-ray Diffraction
2.11. Atomic Force and Scanned Conductance Microscopy
2.12. Electron Microscopy
3. Results and Discussion
3.1. Chemical Characterisation
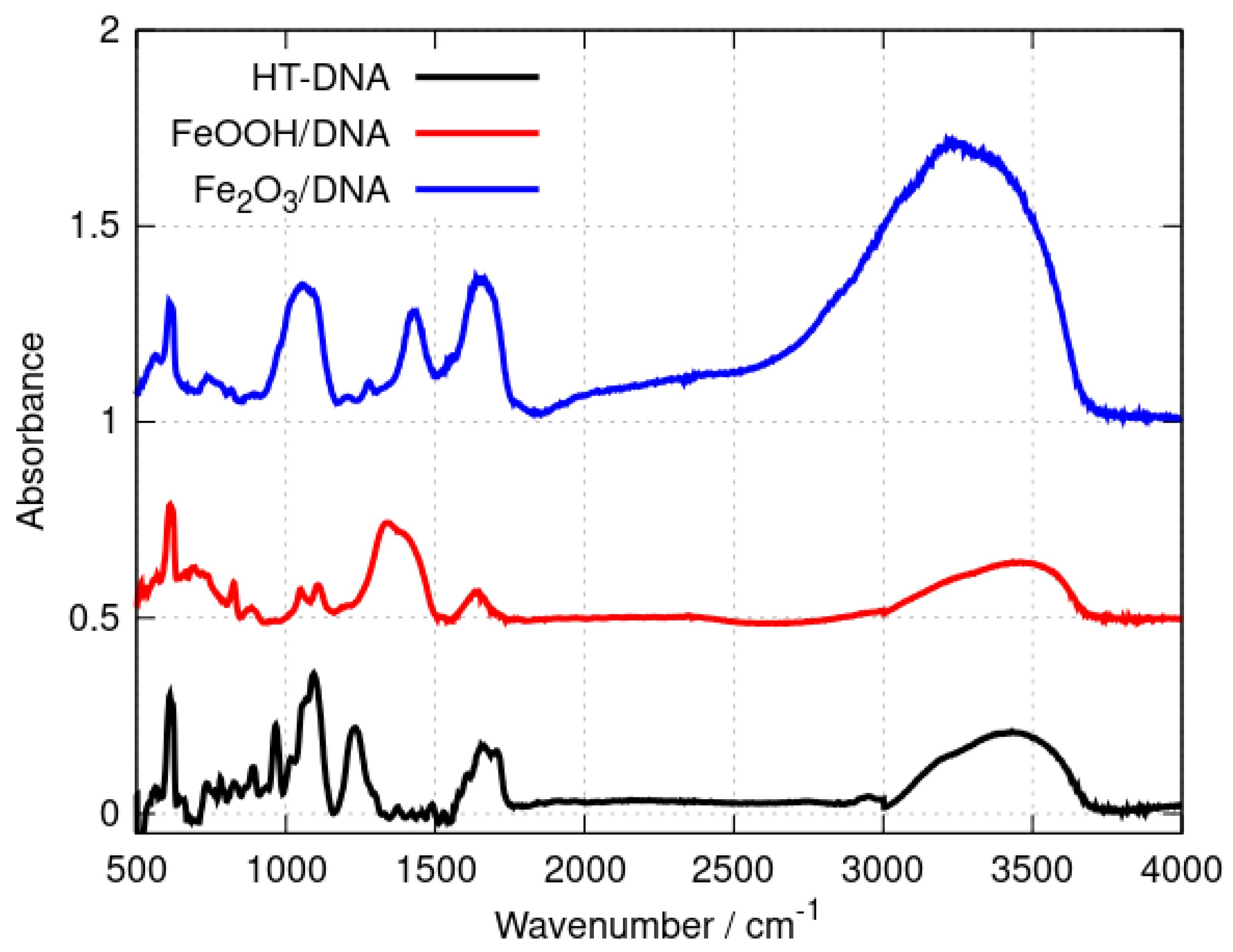
3.1.1. Fourier Transform Infrared Spectroscopy
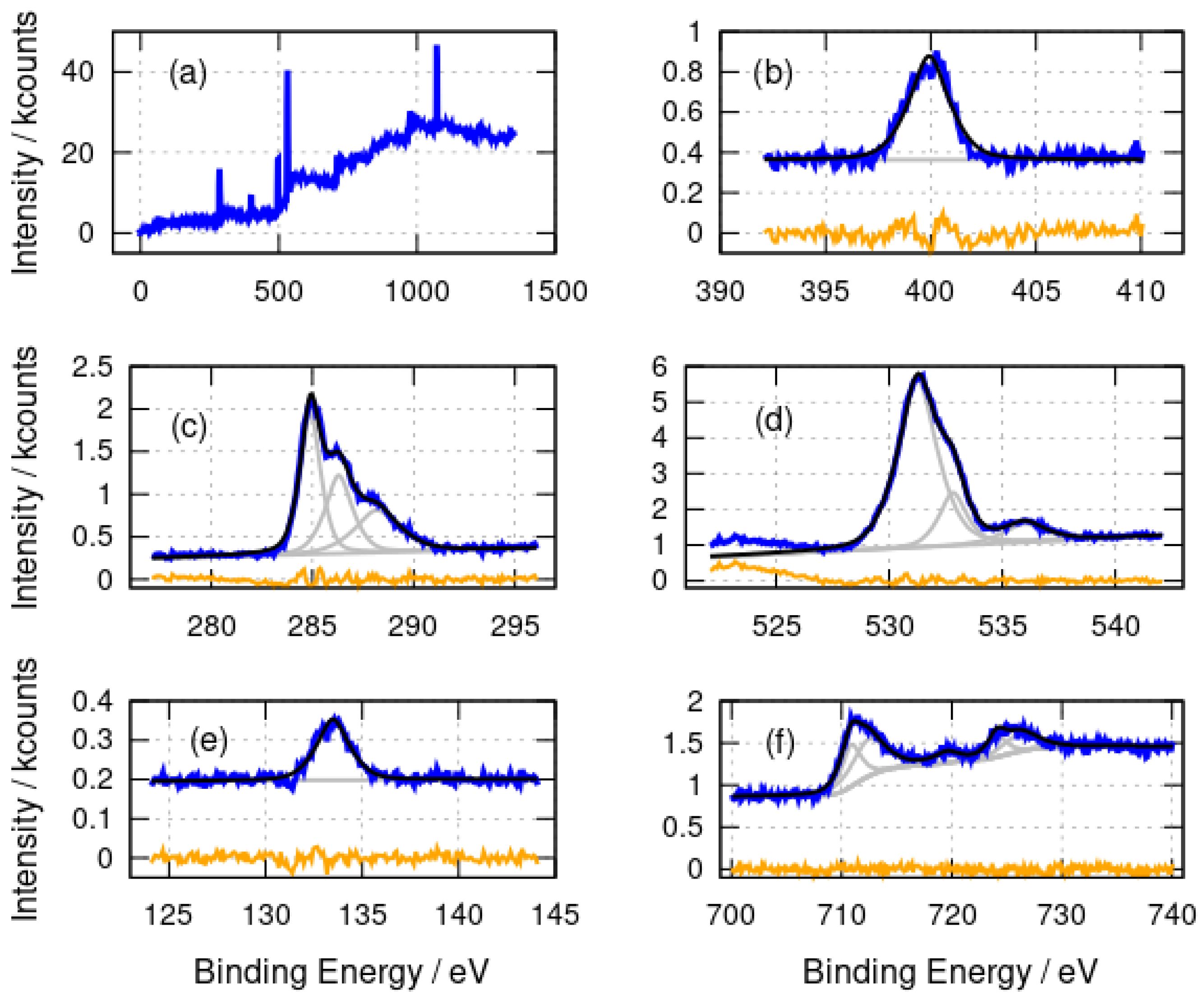
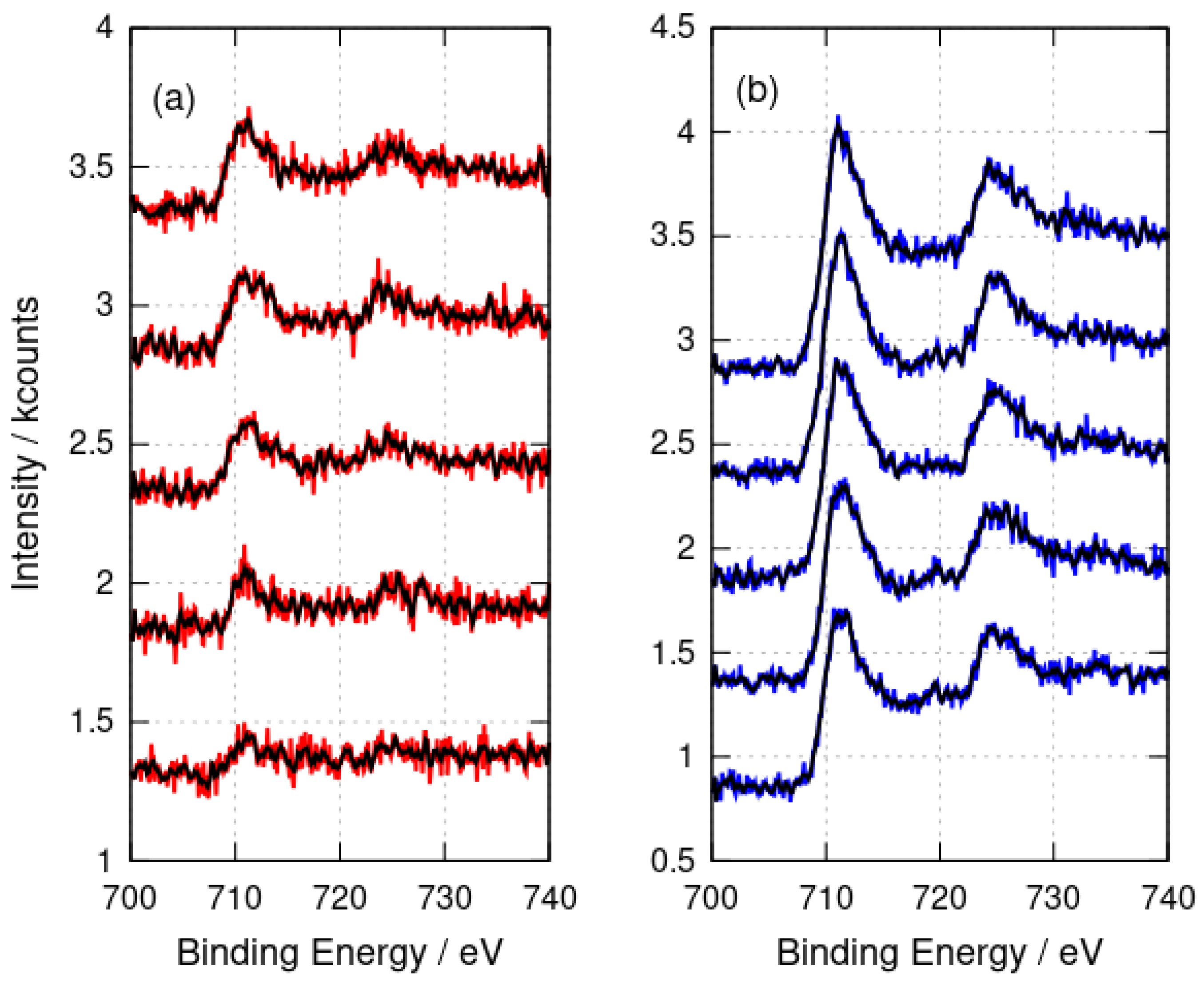
3.1.2. X-ray Photoelectron Spectroscopy
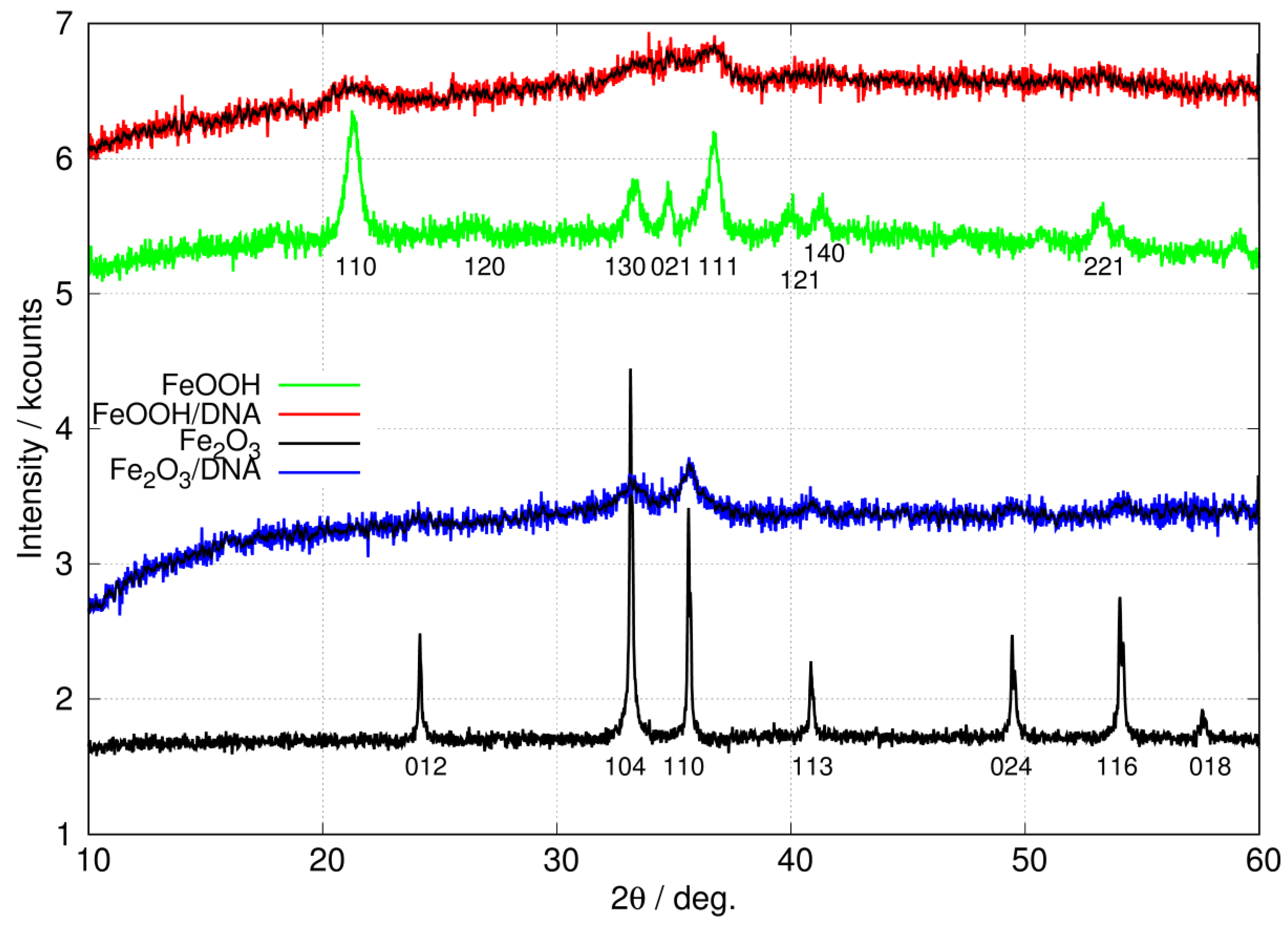
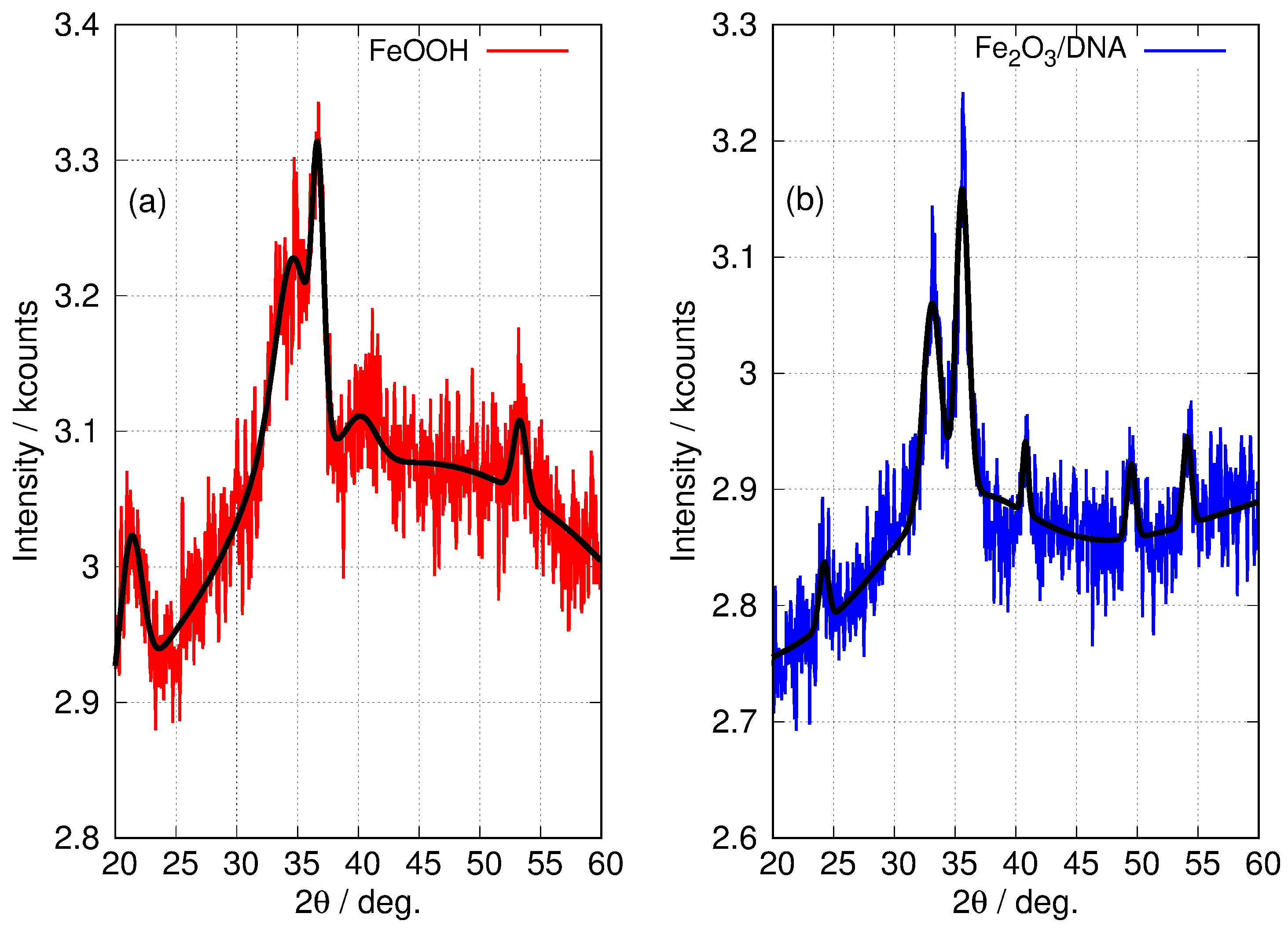
3.1.3. Powder X-ray Diffraction
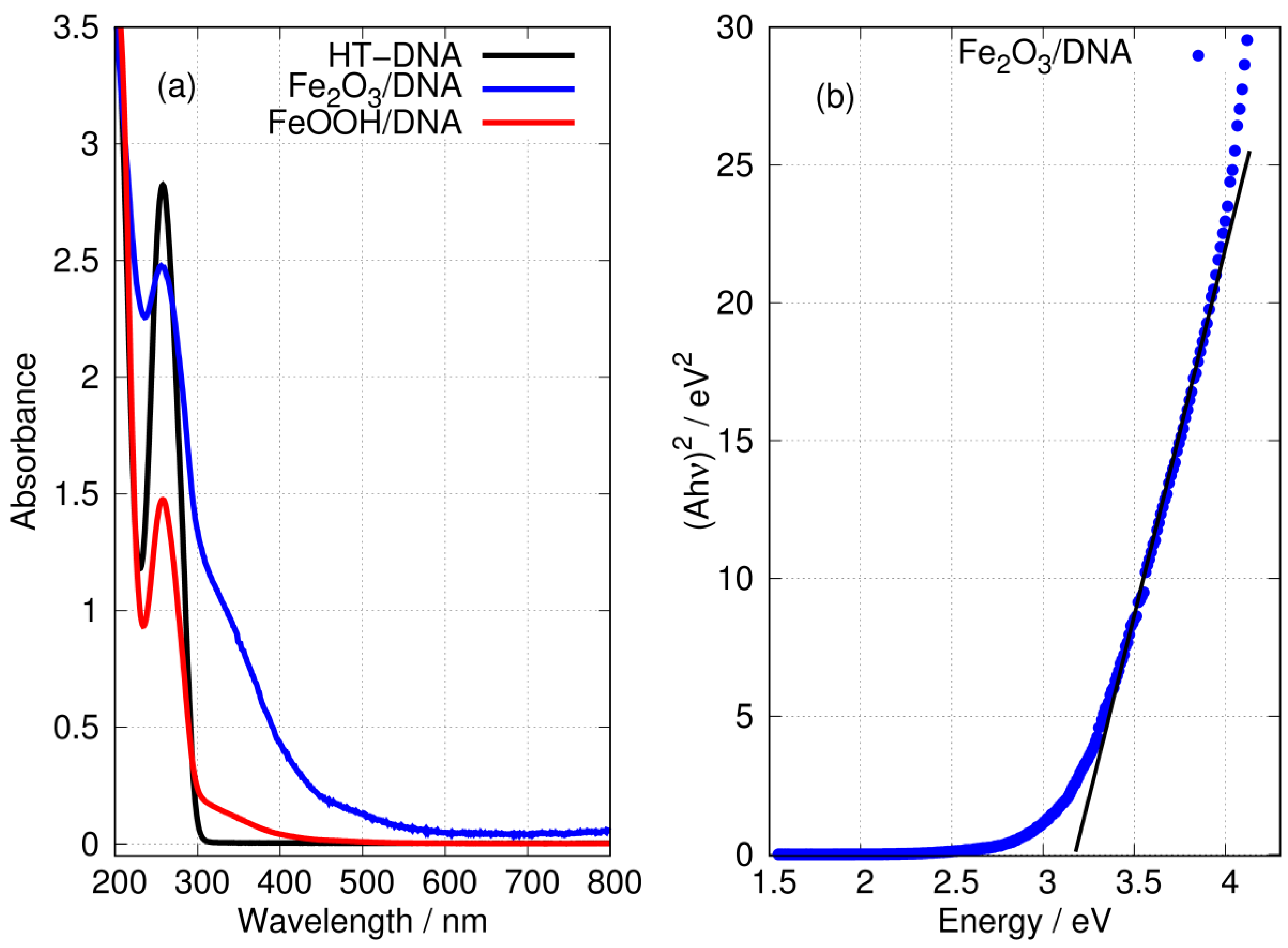
3.2. Optical Spectra and the Bandgap of DNA-Templated FeOOH and Fe2O3
Ultra-Violet Absorption Spectroscopy
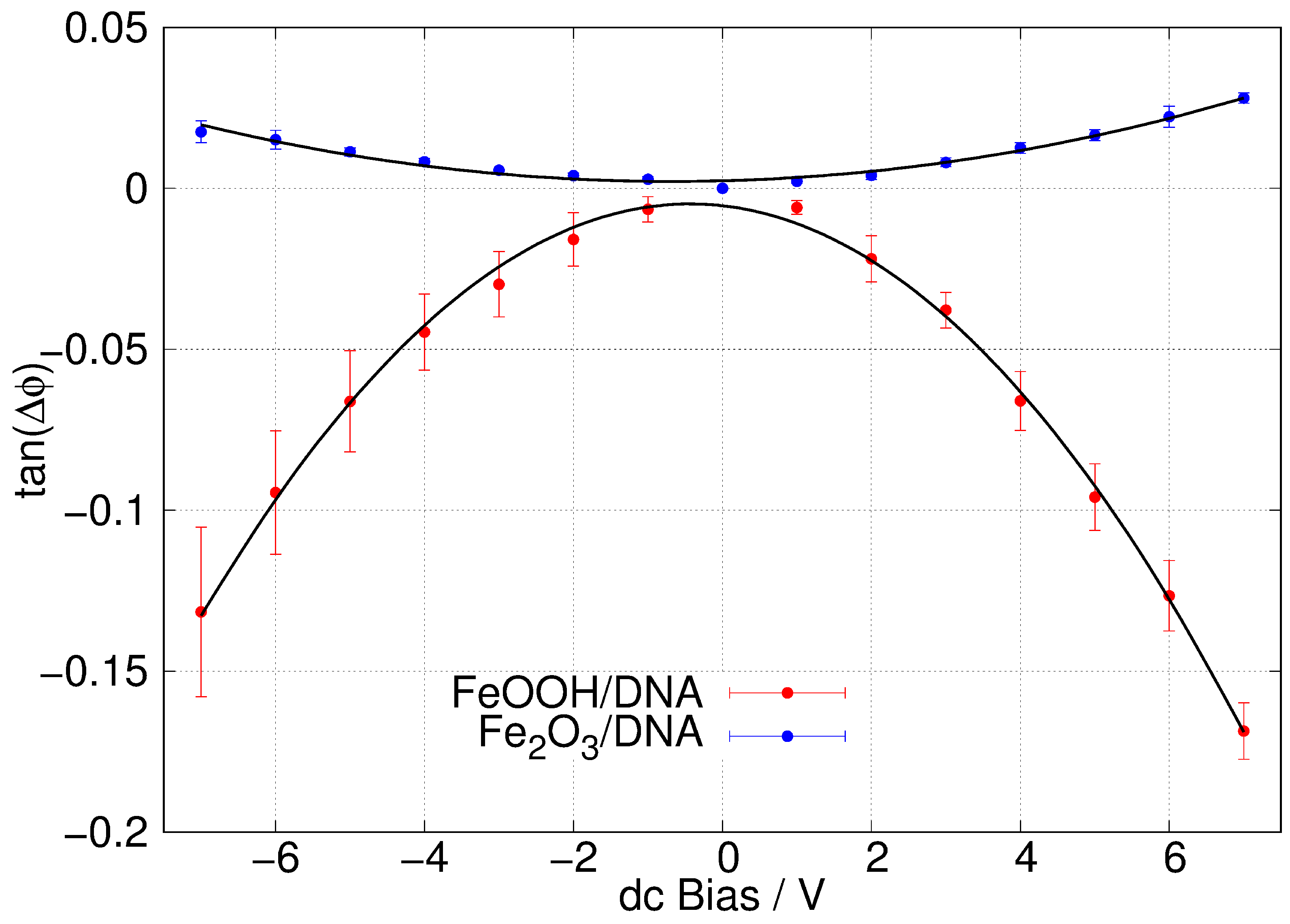
3.3. Morphology and Conductivity of DNA-Templated FeOOH and Fe2O3
Transmission Electron Microscopy
3.4. Atomic Force and Scanned Conductance Microscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, Y.; Jiang, G.; Zhang, H.; Sun, Z.; Gao, Y.; Chen, X.; Liu, H.; Tian, H.; Lai, Q.; Fan, M.; et al. α-Fe2O3, a cost effective and environmentally friendly catalyst for the generation of NH 3—A future fuel—Using a new Al2O3-looping based technology. Chem. Commun. 2017, 53, 10664–10667. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, D.M.S.N.; Mantilaka, M.M.M.G.P.G.; Palihawadana, T.C.; Chandrakumara, G.T.D.; De Silva, R.T.; Pitawala, H.M.T.G.A.; Nalin De Silva, K.M.; Amaratunga, G.A.J. Facile and low-cost synthesis of pure hematite (α-Fe2O3) nanoparticles from naturally occurring laterites and their superior adsorption capability towards acid-dyes. RSC Adv. 2019, 9, 21249–21257. [Google Scholar] [CrossRef] [PubMed]
- Hardee, K.L.; Bard, A.J. Semiconductor Electrodes: V. The Application of Chemically Vapor Deposited Iron Oxide Films to Photosensitized Electrolysis. J. Electrochem. Soc. 1976, 123, 1024–1026. [Google Scholar] [CrossRef]
- Kment, S.; Riboni, F.; Pausova, S.; Wang, L.; Wang, L.; Han, H.; Hubicka, Z.; Krysa, J.; Schmuki, P.; Zboril, R. Photoanodes based on TiO2 and α-Fe2O3 for solar water splitting—superior role of 1D nanoarchitectures and of combined heterostructures. Chem. Soc. Rev. 2017, 46, 3716–3769. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Li, C.; Jia, X.; Du, B.; Li, R.; Wang, B. Enabling high low-bias performance of Fe2O3 photoanode for photoelectrochemical water splitting. J. Colloid Interface Sci. 2023, 633, 555–565. [Google Scholar] [CrossRef]
- Kumar, P.; Khanduri, H.; Pathak, S.; Singh, A.; Basheed, G.A.; Pant, R.P. Temperature selectivity for single phase hydrothermal synthesis of PEG-400 coated magnetite nanoparticles. Dalton Trans. 2020, 49, 8672–8683. [Google Scholar] [CrossRef]
- Kumar, P.; Pathak, S.; Jain, K.; Singh, A.; Kuldeep; Basheed, G.; Pant, R. Low-temperature large-scale hydrothermal synthesis of optically active PEG-200 capped single domain MnFe2O4 nanoparticles. J. Alloys Compd. 2022, 904, 163992. [Google Scholar] [CrossRef]
- Gubin, S.P.; Koksharov, Y.A.; Khomutov, G.B.; Yurkov, G.Y. Magnetic nanoparticles: Preparation, structure and properties. Russ. Chem. Rev. 2005, 74, 489–520. [Google Scholar] [CrossRef]
- Pankhurst, Q.A.; Connolly, J.; Jones, S.K.; Dobson, J. Applications of magnetic nanoparticles in biomedicine. J. Phys. D Appl. Phys. 2003, 36, R167–R181. [Google Scholar] [CrossRef]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides: Structure, Properties, Reactions, Occurences and Uses, 1st ed.; Wiley: Hoboken, NJ, USA, 2003. [Google Scholar] [CrossRef]
- Iandolo, B.; Wickman, B.; Zorić, I.; Hellman, A. The rise of hematite: Origin and strategies to reduce the high onset potential for the oxygen evolution reaction. J. Mater. Chem. A 2015, 3, 16896–16912. [Google Scholar] [CrossRef]
- Sivula, K.; Le Formal, F.; Grätzel, M. Solar Water Splitting: Progress Using Hematite (α-Fe2O3) Photoelectrodes. ChemSusChem 2011, 4, 432–449. [Google Scholar] [CrossRef] [PubMed]
- Bora, D.K.; Braun, A.; Constable, E.C. “In rust we trust”. Hematite—The prospective inorganic backbone for artificial photosynthesis. Energy Environ. Sci. 2013, 6, 407–425. [Google Scholar] [CrossRef]
- Wheeler, D.A.; Wang, G.; Ling, Y.; Li, Y.; Zhang, J.Z. Nanostructured hematite: Synthesis, characterization, charge carrier dynamics, and photoelectrochemical properties. Energy Environ. Sci. 2012, 5, 6682. [Google Scholar] [CrossRef]
- Jing, Z.; Wu, S. Preparation and magnetic properties of spherical α-Fe2O3 nanoparticles via a non-aqueous medium. Mater. Chem. Phys. 2005, 92, 600–603. [Google Scholar] [CrossRef]
- Townsend, T.K.; Sabio, E.M.; Browning, N.D.; Osterloh, F.E. Photocatalytic water oxidation with suspended alpha-Fe2O3 particles-effects of nanoscaling. Energy Environ. Sci. 2011, 4, 4270. [Google Scholar] [CrossRef]
- Wang, R.; Chen, Y.; Fu, Y.; Zhang, H.; Kisielowski, C. Bicrystalline Hematite Nanowires. J. Phys. Chem. B 2005, 109, 12245–12249. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Q. Microwave assisted hydrothermal synthesis and magnetic property of hematite nanorods. Mater. Lett. 2008, 62, 988–990. [Google Scholar] [CrossRef]
- Li, Z.; Lai, X.; Wang, H.; Mao, D.; Xing, C.; Wang, D. Direct hydrothermal synthesis of single-crystalline hematite nanorods assisted by 1,2-propanediamine. Nanotechnology 2009, 20, 245603. [Google Scholar] [CrossRef]
- Kim, H.J.; Choi, K.I.; Pan, A.; Kim, I.D.; Kim, H.R.; Kim, K.M.; Na, C.W.; Cao, G.; Lee, J.H. Template-free solvothermal synthesis of hollow hematite spheres and their applications in gas sensors and Li-ion batteries. J. Mater. Chem. 2011, 21, 6549. [Google Scholar] [CrossRef]
- Martina, M.R.; Zoli, L.; Sani, E. Synthesis and characterization of goethite (α-FeOOH) magnetic nanofluids. Int. J. Thermofluids 2022, 15, 100169. [Google Scholar] [CrossRef]
- Owusu, K.A.; Qu, L.; Li, J.; Wang, Z.; Zhao, K.; Yang, C.; Hercule, K.M.; Lin, C.; Shi, C.; Wei, Q.; et al. Low-crystalline iron oxide hydroxide nanoparticle anode for high-performance supercapacitors. Nat. Commun. 2017, 8, 14264. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Chen, J.S.; Lou, X.W.D. Glucose-Assisted One-Pot Synthesis of FeOOH Nanorods and Their Transformation to Fe3O4 @Carbon Nanorods for Application in Lithium Ion Batteries. J. Phys. Chem. C 2011, 115, 9814–9820. [Google Scholar] [CrossRef]
- Chen, X.; Zeng, Y.; Chen, Z.; Wang, S.; Xin, C.; Wang, L.; Shi, C.; Lu, L.; Zhang, C. Synthesis and Electrochemical Property of FeOOH/Graphene Oxide Composites. Front. Chem. 2020, 8, 328. [Google Scholar] [CrossRef] [PubMed]
- Amani-Ghadim, A.R.; Alizadeh, S.; Khodam, F.; Rezvani, Z. Synthesis of rod-like α-FeOOH nanoparticles and its photocatalytic activity in degradation of an azo dye: Empirical kinetic model development. J. Mol. Catal. A Chem. 2015, 408, 60–68. [Google Scholar] [CrossRef]
- Ali, F.M.; Hmadeh, M.; O’Brien, P.G.; Perovic, D.D.; Ozin, G.A. Photocatalytic Properties of All Four Polymorphs of Nanostructured Iron Oxyhydroxides. ChemNanoMat 2016, 2, 1047–1054. [Google Scholar] [CrossRef]
- Huang, Z.; Han, F.; Li, M.; Zhou, Z.; Guan, X.; Guo, L. Which phase of iron oxyhydroxides (FeOOH) is more competent in overall water splitting as a photocatalyst, goethite, akaganeite or lepidocrocite? A DFT-based investigation. Comput. Mater. Sci. 2019, 169, 109110. [Google Scholar] [CrossRef]
- Zhang, H.; Bayne, M.; Fernando, S.; Legg, B.; Zhu, M.; Penn, R.L.; Banfield, J.F. Size-Dependent Bandgap of Nanogoethite. J. Phys. Chem. C 2011, 115, 17704–17710. [Google Scholar] [CrossRef]
- Braun, E.; Eichen, Y.; Sivan, U.; Ben-Yoseph, G. DNA-templated assembly and electrode attachment of a conducting silver wire. Nature 1998, 391, 775–778. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, C.; Cao, F.; Ren, J.; Qu, X. DNA metallization: Principles, methods, structures, and applications. Chem. Soc. Rev. 2018, 47, 4017–4072. [Google Scholar] [CrossRef]
- Dai, X.; Li, Q.; Aldalbahi, A.; Wang, L.; Fan, C.; Liu, X. DNA-Based Fabrication for Nanoelectronics. Nano Lett. 2020, 20, 5604–5615. [Google Scholar] [CrossRef]
- Pu, S.; Zinchenko, A.; Murata, S. Conformational behavior of DNA-templated CdS inorganic nanowire. Nanotechnology 2011, 22, 375604. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, H.D.A.; Watson, S.M.D.; Horrocks, B.R.; Houlton, A. Magnetic and conductive magnetite nanowires by DNA-templating. Nanoscale 2012, 4, 5936. [Google Scholar] [CrossRef]
- Watson, S.M.D.; Pike, A.R.; Pate, J.; Houlton, A.; Horrocks, B.R. DNA-templated nanowires: Morphology and electrical conductivity. Nanoscale 2014, 6, 4027–4037. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, J.; Zhang, G.; He, H. Polyaniline Nanowires on Si Surfaces Fabricated with DNA Templates. J. Am. Chem. Soc. 2004, 126, 7097–7101. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Schuster, G.B. Precise Sequence Control in Linear and Cyclic Copolymers of 2,5-Bis(2-thienyl)pyrrole and Aniline by DNA-Programmed Assembly. J. Am. Chem. Soc. 2013, 135, 4438–4449. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.M.D.; Galindo, M.A.; Horrocks, B.R.; Houlton, A. Mechanism of Formation of Supramolecular DNA-Templated Polymer Nanowires. J. Am. Chem. Soc. 2014, 136, 6649–6655. [Google Scholar] [CrossRef] [PubMed]
- Madsen, M.; Gothelf, K.V. Chemistries for DNA Nanotechnology. Chem. Rev. 2019, 119, 6384–6458. [Google Scholar] [CrossRef]
- Zhou, L.; Li, W.; Chen, Z.; Ju, E.; Ren, J.; Qu, X. Growth of Hydrophilic CuS Nanowires via DNA-Mediated Self-Assembly Process and Their Use in Fabricating Smart Hybrid Films for Adjustable Chemical Release. Chem.-Eur. J. 2015, 21, 2930–2935. [Google Scholar] [CrossRef]
- Nurdillayeva, R.N.; Oshido, A.B.; Bamford, T.A.; El-Zubir, O.; Houlton, A.; Hedley, J.; Pike, A.R.; Horrocks, B.R. Inkjet printing and electrical characterisation of DNA-templated cadmium sulphide nanowires. Nanotechnology 2018, 29, 135704. [Google Scholar] [CrossRef]
- Aldana, A.; Houlton, A.; Horrocks, B. Structure and optical properties of ZnxCd1−xS and Cu:ZnxCd1−xS templated on DNA molecules. Nano Express 2024, 5, 035016. [Google Scholar] [CrossRef]
- Meyer, T.A.; Zhang, C.; Bao, G.; Ke, Y. Programmable Assembly of Iron Oxide Nanoparticles Using DNA Origami. Nano Lett. 2020, 20, 2799–2805. [Google Scholar] [CrossRef] [PubMed]
- Khomutov, G.B. Interfacially formed organized planar inorganic, polymeric and composite nanostructures. Adv. Colloid Interface Sci. 2004, 111, 79–116. [Google Scholar] [CrossRef] [PubMed]
- Khomutov, G.B. DNA-Based Synthesis and Assembly of Organized Iron Oxide Nanostructures. In Nanomaterials for Application in Medicine and Biology; Giersig, M., Khomutov, G.B., Eds.; Springer: Dordrecht, The Netherlands, 2008; pp. 39–57. [Google Scholar] [CrossRef]
- Sanger, F.; Coulson, A.R.; Hong, G.F.; Hill, D.F.; Petersen, G.B. Nucleotide sequence of bacteriophage lambda DNA. J. Mol. Biol. 1982, 162, 729–773. [Google Scholar] [CrossRef] [PubMed]
- Bensimon, D.; Simon, A.J.; Croquette, V.; Bensimon, A. Stretching DNA with a Receding Meniscus: Experiments and Models. Phys. Rev. Lett. 1995, 74, 4754–4757. [Google Scholar] [CrossRef] [PubMed]
- Biesinger, M.C. Accessing the robustness of adventitious carbon for charge referencing (correction) purposes in XPS analysis: Insights from a multi-user facility data review. Appl. Surf. Sci. 2022, 597, 153681. [Google Scholar] [CrossRef]
- Shirley, D.A. High-Resolution X-ray Photoemission Spectrum of the Valence Bands of Gold. Phys. Rev. B 1972, 5, 4709–4714. [Google Scholar] [CrossRef]
- Nečas, D.; Klapetek, P. Gwyddion: An open-source software for SPM data analysis. Cent. Eur. J. Phys. 2012, 10, 181–188. [Google Scholar] [CrossRef]
- Ouameur, A.A.; Tajmir-Riahi, H.A. Structural Analysis of DNA Interactions with Biogenic Polyamines and cobalt(III)hexamine Studied by Fourier Transform Infrared and Capillary Electrophoresis. J. Biol. Chem. 2004, 279, 42041–42054. [Google Scholar] [CrossRef]
- Alex, S.; Dupuis, P. FT-IR and Raman investigation of cadmium binding by DNA. Inorg. Chim. Acta 1989, 157, 271–281. [Google Scholar] [CrossRef]
- Dovbeshko, G.I.; Gridina, N.Y.; Kruglova, E.B.; Pashchuk, O.P. FTIR Spectroscopy Studies of Nucleic Acid Damage. Talanta 2000, 53, 233–246. [Google Scholar] [CrossRef]
- Mateo-Martí, E.; Briones, C.; Pradier, C.; Martín-Gago, J. A DNA biosensor based on peptide nucleic acids on gold surfaces. Biosens. Bioelectron. 2007, 22, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, D.N.; Hollander, J.M.; Jolly, W.L. Nitrogen ls electron binding energies. Correlations with molecular orbital calculated nitrogen charges. Inorg. Chem. 1969, 8, 2642–2647. [Google Scholar] [CrossRef]
- Lo, P.; Tsai, W.; Lee, J.; Hung, M. The Electrochemical Behavior of Electroless Plated Ni-P Alloys in Concentrated NaOH Solution. J. Electrochem. Soc. 1995, 142, 91–96. [Google Scholar] [CrossRef]
- Aduru, S.; Contarini, S.; Rabalais, J.W. Electron-, X-ray-, and ion-stimulated decomposition of nitrate salts. J. Phys. Chem. 1986, 90, 1683–1688. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, H.T.; Yoo, K.O. Effect of Ferric Oxide on the High-Temperature Removal of Hydrogen Sulfide over ZnO-Fe2O3 Mixed Metal Oxide Sorbent. Ind. Eng. Chem. Res. 1995, 34, 1181–1188. [Google Scholar] [CrossRef]
- Tan, B.J.; Klabunde, K.J.; Sherwood, P.M.A. X-ray photoelectron spectroscopy studies of solvated metal atom dispersed catalysts. Monometallic iron and bimetallic iron-cobalt particles on alumina. Chem. Mater. 1990, 2, 186–191. [Google Scholar] [CrossRef]
- Schulze, P.D.; Shaffer, S.L.; Hance, R.L.; Utley, D.L. Adsorption of water on rhenium studied by XPS. J. Vac. Sci. Technol. A Vac. Surfaces Film. 1983, 1, 97–99. [Google Scholar] [CrossRef]
- Petrovykh, D.Y.; Kimura-Suda, H.; Whitman, L.J.; Tarlov, M.J. Quantitative Analysis and Characterization of DNA Immobilized on Gold. J. Am. Chem. Soc. 2003, 125, 5219–5226. [Google Scholar] [CrossRef]
- Wandelt, K. Photoemission studies of adsorbed oxygen and oxide layers. Surf. Sci. Rep. 1982, 2, 1–121. [Google Scholar] [CrossRef]
- Welsh, I.D.; Sherwood, P.M.A. Photoemission and electronic structure of FeOOH: Distinguishing between oxide and oxyhydroxide. Phys. Rev. B 1989, 40, 6386–6392. [Google Scholar] [CrossRef]
- Fujii, T.; De Groot, F.M.F.; Sawatzky, G.A.; Voogt, F.C.; Hibma, T.; Okada, K. In situ XPS analysis of various iron oxide films grown by NO2 -assisted molecular-beam epitaxy. Phys. Rev. B 1999, 59, 3195–3202. [Google Scholar] [CrossRef]
- Li, Y.; Lv, Z.; Zhang, S.; Zhang, Y.; Wu, S.; Liu, R. Controlled Fabrication and Characterization of α-FeOOH Nanorods. J. Inorg. Organomet. Polym. Mater. 2022, 32, 1400–1408. [Google Scholar] [CrossRef]
- Vernekar, D.; Jagadeesan, D. Tunable acid–base bifunctional catalytic activity of FeOOH in an orthogonal tandem reaction. Catal. Sci. Technol. 2015, 5, 4029–4038. [Google Scholar] [CrossRef]
- Lassoued, A.; Dkhil, B.; Gadri, A.; Ammar, S. Control of the shape and size of iron oxide (α-Fe2O3) nanoparticles synthesized through the chemical precipitation method. Results Phys. 2017, 7, 3007–3015. [Google Scholar] [CrossRef]
- Almeida, T.P.; Fay, M.; Zhu, Y.; Brown, P.D. Process Map for the Hydrothermal Synthesis of α-Fe2O3 Nanorods. J. Phys. Chem. C 2009, 113, 18689–18698. [Google Scholar] [CrossRef]
- Hua, J.; Gengsheng, J. Hydrothermal synthesis and characterization of monodisperse α-Fe2O3 nanoparticles. Mater. Lett. 2009, 63, 2725–2727. [Google Scholar] [CrossRef]
- Lemine, O. Microstructural characterisation of nanoparticles using, XRD line profiles analysis, FE-SEM and FT-IR. Superlattices Microstruct. 2009, 45, 576–582. [Google Scholar] [CrossRef]
- Danno, T.; Nakatsuka, D.; Kusano, Y.; Asaoka, H.; Nakanishi, M.; Fujii, T.; Ikeda, Y.; Takada, J. Crystal Structure of β-Fe2O3 and Topotactic Phase Transformation to α-Fe2O3. Cryst. Growth Des. 2013, 13, 770–774. [Google Scholar] [CrossRef]
- Jia, C.J.; Sun, L.D.; Luo, F.; Han, X.D.; Heyderman, L.J.; Yan, Z.G.; Yan, C.H.; Zheng, K.; Zhang, Z.; Takano, M.; et al. Large-Scale Synthesis of Single-Crystalline Iron Oxide Magnetic Nanorings. J. Am. Chem. Soc. 2008, 130, 16968–16977. [Google Scholar] [CrossRef]
- Qu, X.F.; Zhou, G.T.; Yao, Q.Z.; Fu, S.Q. Aspartic-Acid-Assisted Hydrothermal Growth and Properties of Magnetite Octahedrons. J. Phys. Chem. C 2010, 114, 284–289. [Google Scholar] [CrossRef]
- Teng, X.; Yang, H. Effects of surfactants and synthetic conditions on the sizes and self-assembly of monodisperse iron oxide nanoparticlesElectronic supplementary information (ESI) available: XRD data of iron oxide nanoparticles. J. Mater. Chem. 2004, 14, 774. [Google Scholar] [CrossRef]
- Tian, Y.; Yu, B.; Li, X.; Li, K. Facile solvothermal synthesis of monodisperse Fe3O4 nanocrystals with precise size control of one nanometre as potential MRI contrast agents. J. Mater. Chem. 2011, 21, 2476. [Google Scholar] [CrossRef]
- Cui, H.; Liu, Y.; Ren, W. Structure switch between α-Fe2O3, γ-Fe2O3 and Fe3O4 during the large scale and low temperature sol–gel synthesis of nearly monodispersed iron oxide nanoparticles. Adv. Powder Technol. 2013, 24, 93–97. [Google Scholar] [CrossRef]
- Amini, M.; Mousazade, Y.; Zand, Z.; Bagherzadeh, M.; Najafpour, M.M. Ultra-small and highly dispersive iron oxide hydroxide as an efficient catalyst for oxidation reactions: A Swiss-army-knife catalyst. Sci. Rep. 2021, 11, 6642. [Google Scholar] [CrossRef]
- Murgueitio, E.; Cumbal, L.; Abril, M.; Izquierdo, A.; Debut, A.; Tinoco, O. Green Synthesis of Iron Nanoparticles: Application on the Removal of Petroleum Oil from Contaminated Water and Soils. J. Nanotechnol. 2018, 2018, 4184769. [Google Scholar] [CrossRef]
- Lany, S. Semiconducting transition metal oxides. J. Phys. Condens. Matter 2015, 27, 283203. [Google Scholar] [CrossRef]
- Lany, S. Band-structure calculations for the 3 d transition metal oxides in G W. Phys. Rev. B 2013, 87, 085112. [Google Scholar] [CrossRef]
- Seo, O.; Tayal, A.; Kim, J.; Song, C.; Chen, Y.; Hiroi, S.; Katsuya, Y.; Ina, T.; Sakata, O.; Ikeya, Y.; et al. Tuning of structural, optical band gap, and electrical properties of room-temperature-grown epitaxial thin films through the Fe2O3:NiO ratio. Sci. Rep. 2019, 9, 4304. [Google Scholar] [CrossRef]
- Makuła, P.; Pacia, M.; Macyk, W. How To Correctly Determine the Band Gap Energy of Modified Semiconductor Photocatalysts Based on UV–Vis Spectra. J. Phys. Chem. Lett. 2018, 9, 6814–6817. [Google Scholar] [CrossRef]
- Bockrath, M.; Markovic, N.; Shepard, A.; Tinkham, M.; Gurevich, L.; Kouwenhoven, L.P.; Wu, M.W.; Sohn, L.L. Scanned Conductance Microscopy of Carbon Nanotubes and λ-DNA. Nano Lett. 2002, 2, 187–190. [Google Scholar] [CrossRef]
- Staii, C.; Johnson, A.T.; Pinto, N.J. Quantitative Analysis of Scanning Conductance Microscopy. Nano Lett. 2004, 4, 859–862. [Google Scholar] [CrossRef]
- Warnes, B.; Aplan, F.; Simkovich, G. Electrical conductivity and seebeck voltage of Fe2O3, pure and doped, as a function of temperature and oxygen pressure. Solid State Ion. 1984, 12, 271–276. [Google Scholar] [CrossRef]
- Zhang, X.; An, L.; Yin, J.; Xi, P.; Zheng, Z.; Du, Y. Effective Construction of High-quality Iron Oxy-hydroxides and Co-doped Iron Oxy-hydroxides Nanostructures: Towards the Promising Oxygen Evolution Reaction Application. Sci. Rep. 2017, 7, 43590. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Energies/eV | |||||
|---|---|---|---|---|---|
| Sample | Sat. | ||||
| -FeOOH/DNA | 711.1 | 712.5 | 719.7 | 724.5 | 726.3 |
| -Fe2O3/DNA | 710.9 | 712.5 | 719.6 | 724.1 | 726.3 |
| Sample | /Degrees and Assignment (hkl) | ‡ /nm | ||||||
|---|---|---|---|---|---|---|---|---|
| FeOOH | 21.3 | 33.2 | 34.7 | 36.7 | 40.1 | 41.4 | 53.3 | |
| (110) * | (130) * | (021) * | (111) * | (121) * | (140) | (221) * | ||
| Fe2O3 | 24.2 | 33.1 | 35.6 | 40.8 | 49.5 | 54.1 | 57.6 | |
| (012) * | (104) * | (110) * | (113) * | (024) * | (116) * | (018) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zubairu, S.M.; Idris, S.O.; Gimba, C.E.; Uzairu, A.; Houlton, A.; Horrocks, B.R. Templating Iron(III) Oxides on DNA Molecules. Nanomaterials 2024, 14, 1609. https://doi.org/10.3390/nano14191609
Zubairu SM, Idris SO, Gimba CE, Uzairu A, Houlton A, Horrocks BR. Templating Iron(III) Oxides on DNA Molecules. Nanomaterials. 2024; 14(19):1609. https://doi.org/10.3390/nano14191609
Chicago/Turabian StyleZubairu, Siyaka Mj, Sulaiman O. Idris, Casmir E. Gimba, Adamu Uzairu, Andrew Houlton, and Benjamin R. Horrocks. 2024. "Templating Iron(III) Oxides on DNA Molecules" Nanomaterials 14, no. 19: 1609. https://doi.org/10.3390/nano14191609
APA StyleZubairu, S. M., Idris, S. O., Gimba, C. E., Uzairu, A., Houlton, A., & Horrocks, B. R. (2024). Templating Iron(III) Oxides on DNA Molecules. Nanomaterials, 14(19), 1609. https://doi.org/10.3390/nano14191609