

Chemistry of Reduced Graphene Oxide: Implications for the Electrophysical Properties of Segregated Graphene–Polymer Composites

 , ,
, ,  , , , , , , , and
, , , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. rGO Synthesis
2.2. Composite Fabrication
2.3. Characterization
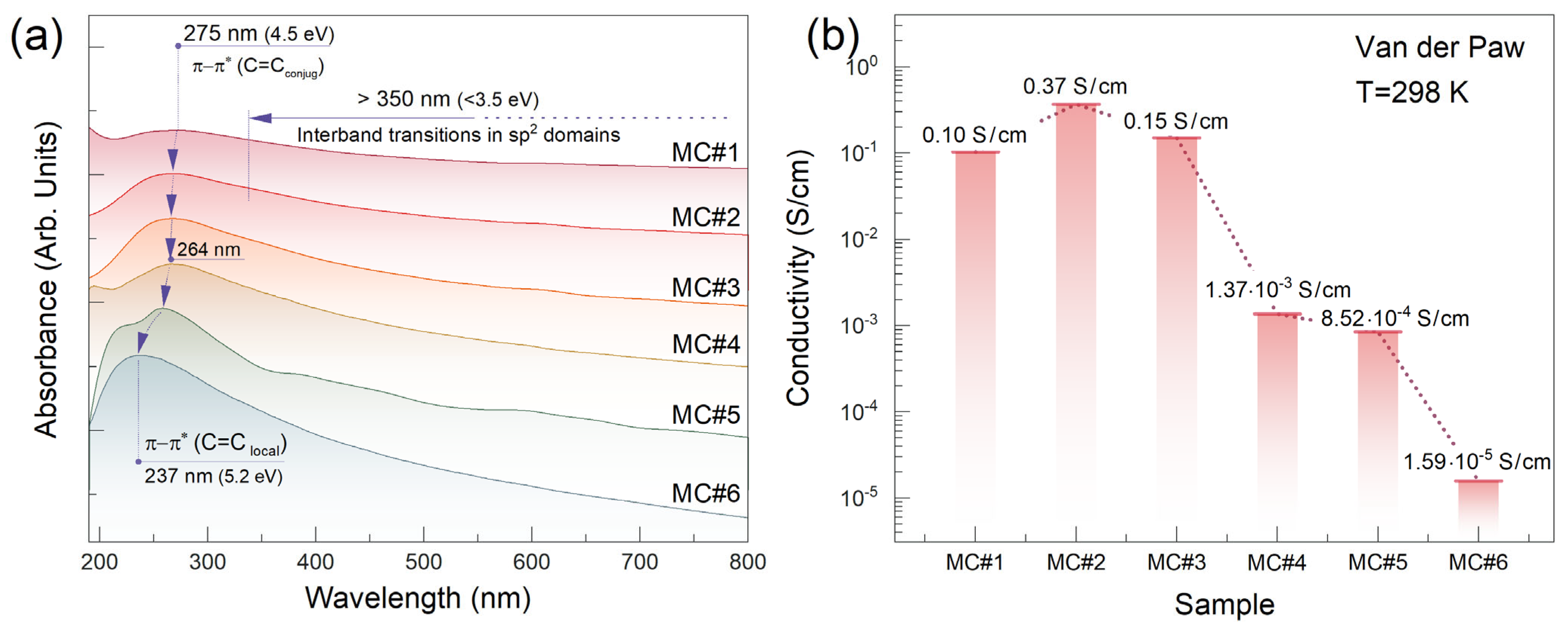
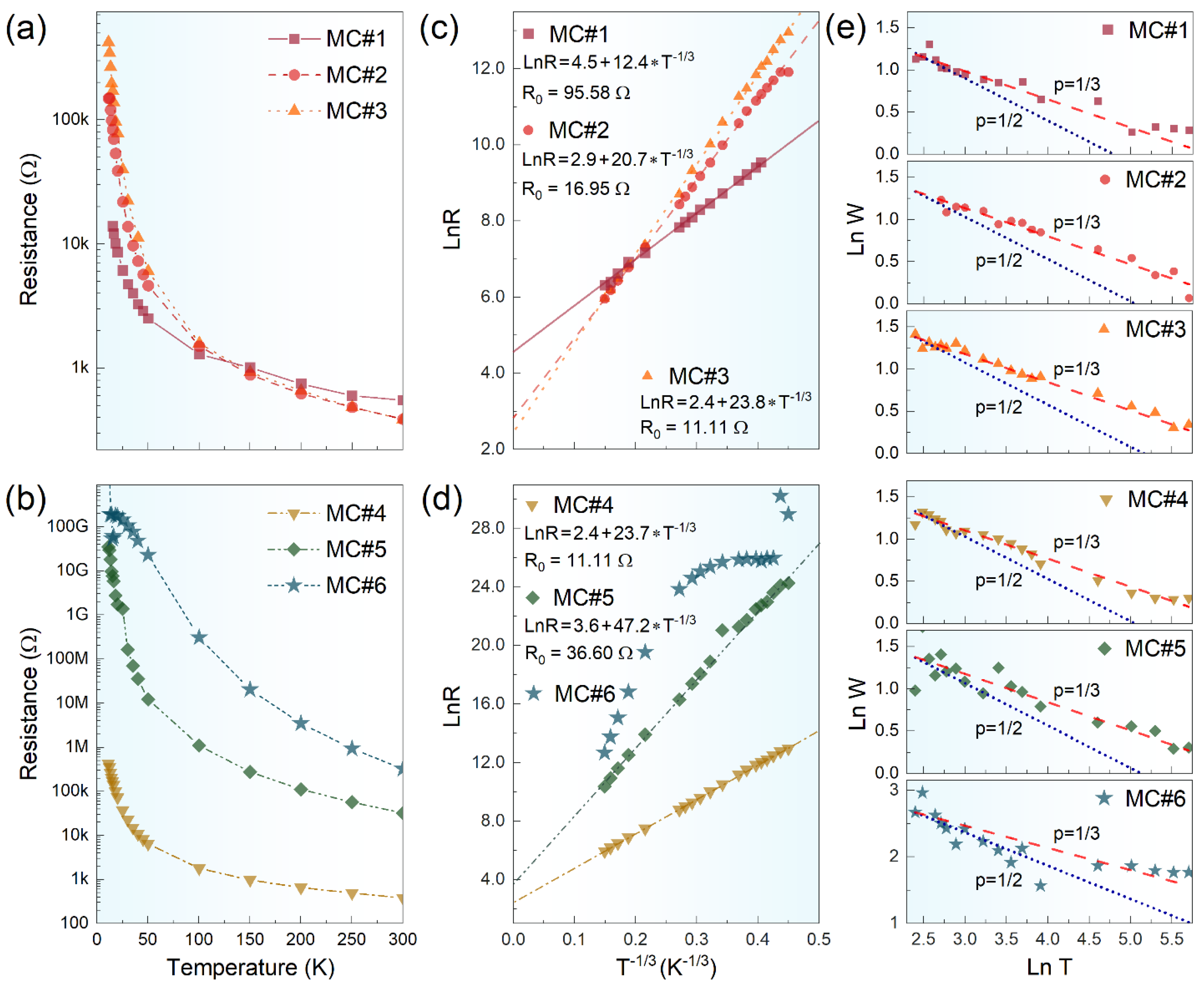
3. Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heng, W.; Solomon, S.; Gao, W. Flexible Electronics and Devices as Human–Machine Interfaces for Medical Robotics. Adv. Mater. 2022, 34, 2107902. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Li, Y.; Chen, D.; Serikawa, S.; Guizani, M.; Lv, Z. A Survey on 5G/6G, AI, and Robotics. Comput. Electr. Eng. 2021, 95, 107372. [Google Scholar] [CrossRef]
- Liu, H.; Li, Q.; Zhang, S.; Yin, R.; Liu, X.; He, Y.; Dai, K.; Shan, C.; Guo, J.; Liu, C.; et al. Electrically Conductive Polymer Composites for Smart Flexible Strain Sensors: A Critical Review. J. Mater. Chem. C 2018, 6, 12121–12141. [Google Scholar] [CrossRef]
- Tadesse, M.G.; Ahmmed, A.S.; Lübben, J.F. Review on Conductive Polymer Composites for Supercapacitor Applications. J. Compos. Sci. 2024, 8, 53. [Google Scholar] [CrossRef]
- Li, J.; Liu, X.; Feng, Y.; Yin, J. Recent Progress in Polymer/Two-Dimensional Nanosheets Composites with Novel Performances. Prog. Polym. Sci. 2022, 126, 101505. [Google Scholar] [CrossRef]
- Grunlan, J.C.; Mehrabi, A.R.; Bannon, M.V.; Bahr, J.L. Water-Based Single-Walled-Nanotube-Filled Polymer Composite with an Exceptionally Low Percolation Threshold. Adv. Mater. 2004, 16, 150–153. [Google Scholar] [CrossRef]
- Zhang, C.; Ma, C.A.; Wang, P.; Sumita, M. Temperature Dependence of Electrical Resistivity for Carbon Black Filled Ultra-High Molecular Weight Polyethylene Composites Prepared by Hot Compaction. Carbon 2005, 43, 2544–2553. [Google Scholar] [CrossRef]
- Pang, H.; Xu, L.; Yan, D.X.; Li, Z.M. Conductive Polymer Composites with Segregated Structures. Prog. Polym. Sci. 2014, 39, 1908–1933. [Google Scholar] [CrossRef]
- Hsissou, R.; Seghiri, R.; Benzekri, Z.; Hilali, M.; Rafik, M.; Elharfi, A. Polymer Composite Materials: A Comprehensive Review. Compos. Struct. 2021, 262, 113640. [Google Scholar] [CrossRef]
- Wang, F.; Fang, W.; Ming, X.; Liu, Y.; Xu, Z.; Gao, C. A Review on Graphene Oxide: 2D Colloidal Molecule, Fluid Physics, and Macroscopic Materials. Appl. Phys. Rev. 2023, 10, 011311. [Google Scholar] [CrossRef]
- Tarcan, R.; Todor-Boer, O.; Petrovai, I.; Leordean, C.; Astilean, S.; Botiz, I. Reduced Graphene Oxide Today. J. Mater. Chem. C 2020, 8, 1198–1224. [Google Scholar] [CrossRef]
- Pang, H.; Chen, T.; Zhang, G.M.; Zeng, B.Q.; Li, Z.M. An Electrically Conducting Polymer/Graphene Composite with a Very Low Percolation Threshold. Mater. Lett. 2010, 64, 2226–2229. [Google Scholar] [CrossRef]
- Du, J.H.; Zhao, L.; Zeng, Y.; Zhang, L.L.; Li, F.; Liu, P.; Liu, C. Comparison of Electrical Properties between Multi-Walled Carbon Nanotube and Graphene Nanosheet/High Density Polyethylene Composites with a Segregated Network Structure. Carbon 2011, 49, 1094–1100. [Google Scholar] [CrossRef]
- Li, M.; Gao, C.; Hu, H.; Zhao, Z. Electrical Conductivity of Thermally Reduced Graphene Oxide/Polymer Composites with a Segregated Structure. Carbon 2013, 65, 371–373. [Google Scholar] [CrossRef]
- Shiyanova, K.A.; Gudkov, M.V.; Gorenberg, A.Y.; Rabchinskii, M.K.; Smirnov, D.A.; Shapetina, M.A.; Gurinovich, T.D.; Goncharuk, G.P.; Bazhenov, S.L.; Melnikov, V.P. Segregated Polymer Composites with High Electrical Conductivity and Well Mechanical Properties Based on PVC, P(VDF-TFE), UHMWPE and rGO for Energy Application. ACS Omega 2020, 5, 25148–25155. [Google Scholar] [CrossRef]
- Hummers, W.S.; Offeman, R.E. Preparation of Graphitic Oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved Synthesis of Graphene Oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef]
- Rosillo-Lopez, M.; Salzmann, C.G. Detailed Investigation into the Preparation of Graphene Oxide by Dichromate Oxidation. ChemistrySelect 2018, 3, 6972–6978. [Google Scholar] [CrossRef]
- Gutiérrez-Cruz, A.; Ruiz-Hernández, A.R.; Vega-Clemente, J.F.; Luna-Gazcón, D.G.; Campos-Delgado, J. A Review of Top-Down and Bottom-Up Synthesis Methods for the Production of Graphene, Graphene Oxide and Reduced Graphene Oxide. J. Mater. Sci. 2022, 57, 14543–14578. [Google Scholar] [CrossRef]
- Brisebois, P.P.; Siaj, M. Harvesting Graphene Oxide – Years 1859 to 2019: A Review of Its Structure, Synthesis, Properties and Exfoliation. J. Mater. Chem. C 2020, 8, 1517–1547. [Google Scholar] [CrossRef]
- Shiyanova, K.A.; Gudkov, M.V.; Rabchinskii, M.K.; Sokura, L.A.; Stolyarova, D.Y.; Baidakova, M.V.; Shashkin, D.P.; Trofimuk, A.D.; Smirnov, D.A.; Komarov, I.A.; et al. Graphene oxide chemistry management via the use of KMnO4/K2Cr2O7 oxidizing agents. Nanomaterials 2021, 11, 915. [Google Scholar] [CrossRef] [PubMed]
- Rabchinskii, M.K.; Sysoev, V.V.; Varezhnikov, A.S.; Solomatin, M.A.; Struchkov, N.S.; Stolyarova, D.Y.; Ryzhkov, S.A.; Antonov, G.A.; Gabrelian, V.S.; Cherviakova, P.D.; et al. Toward On-Chip Multisensor Arrays for alcohols sensing: Capitalizing the graphene carbonylation. ACS Appl. Mater. Interfaces 2023, 15, 28370–28386. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, A.; Sharma, S.; Papakonstantinou, P.; Hamilton, J. Probing the Thermal Deoxygenation of Graphene Oxide Using High-Resolution In Situ X-ray-Based Spectroscopies. J. Phys. Chem. C 2011, 115, 17009–17019. [Google Scholar] [CrossRef]
- Rabchinskii, M.K.; Shnitov, V.V.; Brzhezinskaya, M.; Baidakova, M.V.; Stolyarova, D.Y.; Ryzhkov, S.A.; Saveliev, S.D.; Shvidchenko, A.V.; Nefedov, D.Y.; Antonenko, A.O.; et al. Manifesting Epoxide and Hydroxyl Groups in XPS Spectra and Valence Band of Graphene Derivatives. Nanomaterials 2023, 13, 23. [Google Scholar] [CrossRef]
- Schultz, B.J.; Dennis, R.V.; Aldinger, J.P.; Jaye, C.; Wang, X.; Fischer, D.A.; Cartwright, A.N.; Banerjee, S. X-ray absorption spectroscopy studies of electronic structure recovery and nitrogen local structure upon thermal reduction of graphene oxide in an ammonia environment. RSC Adv. 2014, 4, 634–644. [Google Scholar] [CrossRef]
- Aguilar-Bolados, H.; Vargas-Astudillo, D.; Yazdani-Pedram, M.; Acosta-Villavicencio, G.; Fuentealba, P.; Contreras-Cid, A.; Verdejo, R.; López-Manchado, M.A. Facile and Scalable One-Step Method for Amination of Graphene Using Leuckart Reaction. Chem. Mater. 2017, 29, 6698–6705. [Google Scholar] [CrossRef]
- Rabchinskii, M.K.; Sysoev, V.V.; Glukhova, O.E.; Brzhezinskaya, M.; Stolyarova, D.Y.; Varezhnikov, A.S.; Solomatin, M.A.; Barkov, P.V.; Kirilenko, D.A.; Pavlov, S.I.; et al. Guding graphene Derivatization for the On-Chip Multisensor Arrays: From the Synthesis to the Theoretical Background. Adv. Mater. Technol. 2022, 7, 2101250. [Google Scholar] [CrossRef]
- Vesel, A.; Zaplotnik, R.; Primc, G.; Mozetič, M. A Review of Strategies for the Synthesis of N-Doped Graphene-Like Materials. Nanomaterials 2020, 10, 2286. [Google Scholar] [CrossRef]
- Gueorguiev, G.K.; Neidhardt, J.; Stafström, S.; Hultman, L. First-principles calculations on the curvature evolution and cross-linkage in carbon nitride. Chem. Phys. Lett. 2005, 410, 228–234. [Google Scholar] [CrossRef]
- Broitman, E.; Gueorguiev, G.K.; Furlan, A.; Son, N.T.; Gellman, A.J.; Stafström, S.; Hultman, L. Water adsorption on fullerene-like carbon nitride overcoats. Thin Solid Films 2008, 517, 1106–1110. [Google Scholar] [CrossRef]
- Deng, D.; Pan, X.; Yu, L.; Cui, Y.; Jiang, Y.; Qi, J.; Li, W.X.; Fu, Q.; Ma, X.; Xue, Q.; et al. Toward N-Doped Graphene via Solvothermal Synthesis. Chem. Mater. 2011, 23, 1188. [Google Scholar] [CrossRef]
- Mirzaei, A.; Bharath, S.P.; Kim, J.-Y.; Pawar, K.K.; Kim, H.W.; Kim, S.S. N-Doped Graphene and Its Derivatives as Resistive Gas Sensors: An Overview. Chemosensors 2023, 11, 334. [Google Scholar] [CrossRef]
- Darmstadt, H.; Roy, C. Surface Spectroscopic Study of Basic Sites on Carbon Blacks. Carbon 2003, 41, 2662–2665. [Google Scholar] [CrossRef]
- Shnitov, V.V.; Rabchinskii, M.K.; Brzhezinskaya, M.; Stolyarova, D.Y.; Pavlov, S.V.; Baidakova, M.V.; Shvidchenko, A.V.; Kislenko, V.A.; Kislenko, S.A.; Brunkov, P.N. Valence Band Structure Engineering in Graphene Derivatives. Small 2021, 17, 2104316. [Google Scholar] [CrossRef]
- Eda, G.; Chhowalla, M. Chemically derived graphene oxide: Towards large-area thin-film electronics and optoelectronics. Adv. Mater. 2010, 22, 2392–2415. [Google Scholar] [CrossRef]
- Kumar, P.V.; Bardhan, N.M.; Tongay, S.; Wu, J.; Belcher, A.M.; Grossman, J.C. Scalable enhancement of graphene oxide properties by thermally driven phase transformation. Nat. Chem. 2014, 6, 151–158. [Google Scholar] [CrossRef]
- Chien, C.-T.; Li, S.-S.; Lai, W.-J.; Yeh, Y.-C.; Chen, H.-A.; Chen, I.-S.; Chen, L.-C.; Chen, K.-H.; Nemoto, T.; Isoda, S.; et al. Tunable photoluminescence from graphene oxide. Angew. Chem. Int. Ed. 2012, 51, 6662–6666. [Google Scholar] [CrossRef]
- Park, S.; Hu, Y.; Hwang, J.O.; Lee, E.-S.; Casabianca, L.B.; Cai, W.; Potts, J.R.; Ha, H.-W.; Chen, S.; Oh, J.; et al. Chemical Structures of Hydrazine-Treated Graphene Oxide and Generation of Aromatic Nitrogen Doping. Nat. Commun. 2012, 3, 638–645. [Google Scholar] [CrossRef]
- Joung, D.; Khondaker, S.I. Efros-Shklovskii variable-range hopping in reduced graphene oxide sheets of varying carbon sp2 fraction. Phys. Rev. B 2012, 86, 235423. [Google Scholar] [CrossRef]
- Van Lien, N. Crossovers in two-dimensional variable range hopping. Phys. Lett. A 1995, 207, 379–384. [Google Scholar] [CrossRef]
- Mott, N.F.; Pollitt, M.P.S.; Wallis, S.; Adkins, C. The Anderson transition. Proc. R. Soc. Lond. A 1975, 345, 169. [Google Scholar] [CrossRef]
- Khondaker, S.I.; Shlimak, I.S.; Nicholls, J.T.; Pepper, M.; Ritchie, D.A. Coulomb Blockade in a GaAs Quantum Dot. Phys. Rev. B 1991, 59, 4580. [Google Scholar] [CrossRef]
- Lee, Y.-C.; Liu, C.-I.; Yang, Y.; Elmquist, R.E.; Liang, C.-T. Crossover from Efros–Shklovskii to Mott Variable Range Hopping in Monolayer Epitaxial Graphene Grown on SiC. Chin. J. Phys. 2017, 55, 1235–1241. [Google Scholar] [CrossRef]
- Bennaceur, K.; Jacques, P.; Portier, F.; Roche, P.; Glattli, D.C. Unveiling Quantum Hall Transport by Efros-Shklovskii to Mott Variable-Range Hopping Transition in Graphene. Phys. Rev. B 2012, 86, 085433. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Saito, S. Formation, Stabilities, and Electronic Properties of Nitrogen Defects in Graphene. Phys. Rev. B 2011, 84, 245446. [Google Scholar] [CrossRef]
- Carva, K.; Sanyal, B.; Fransson, J.; Eriksson, O. Defect-Controlled Electronic Transport in Single, Bilayer, and N-Doped Graphene: Theory. Phys. Rev. B 2010, 81, 245405. [Google Scholar] [CrossRef]
- McKenzie, R.; Zurawsky, W.; Mijovic, J. A Molecular Interpretation of Maxwell–Wagner–Sillars Processes. J. Non-Cryst. Solids 2014, 406, 11–21. [Google Scholar] [CrossRef]
- Prodromakis, T.; Papavassiliou, C. Engineering the Maxwell–Wagner Polarization Effect. Appl. Surf. Sci. 2009, 255, 6989–6994. [Google Scholar] [CrossRef]
- Yadav, R.; Tirumali, M.; Wang, X.; Naebe, M.; Kandasubramanian, B. Polymer Composite for Antistatic Application in Aerospace. Def. Technol. 2020, 16, 107–118. [Google Scholar] [CrossRef]
- Zhu, A.; Wang, H.; Sun, S.; Zhang, C. The Synthesis and Antistatic, Anticorrosive Properties of Polyaniline Composite Coating. Prog. Org. Coat. 2018, 122, 270–279. [Google Scholar] [CrossRef]
- Abbasi, H.; Antunes, M.; Velasco, J.I. Recent Advances in Carbon-Based Polymer Nanocomposites for Electromagnetic Interference Shielding. Prog. Mater. Sci. 2019, 103, 319–373. [Google Scholar] [CrossRef]
- Omana, L.; Chandran, A.; John, R.E.; Wilson, R.; George, K.C.; Unnikrishnan, N.V.; Varghese, S.S.; George, G.; Simon, S.M.; Paul, I. Recent Advances in Polymer Nanocomposites for Electromagnetic Interference Shielding: A Review. ACS Omega 2022, 7, 25921–25947. [Google Scholar] [CrossRef]
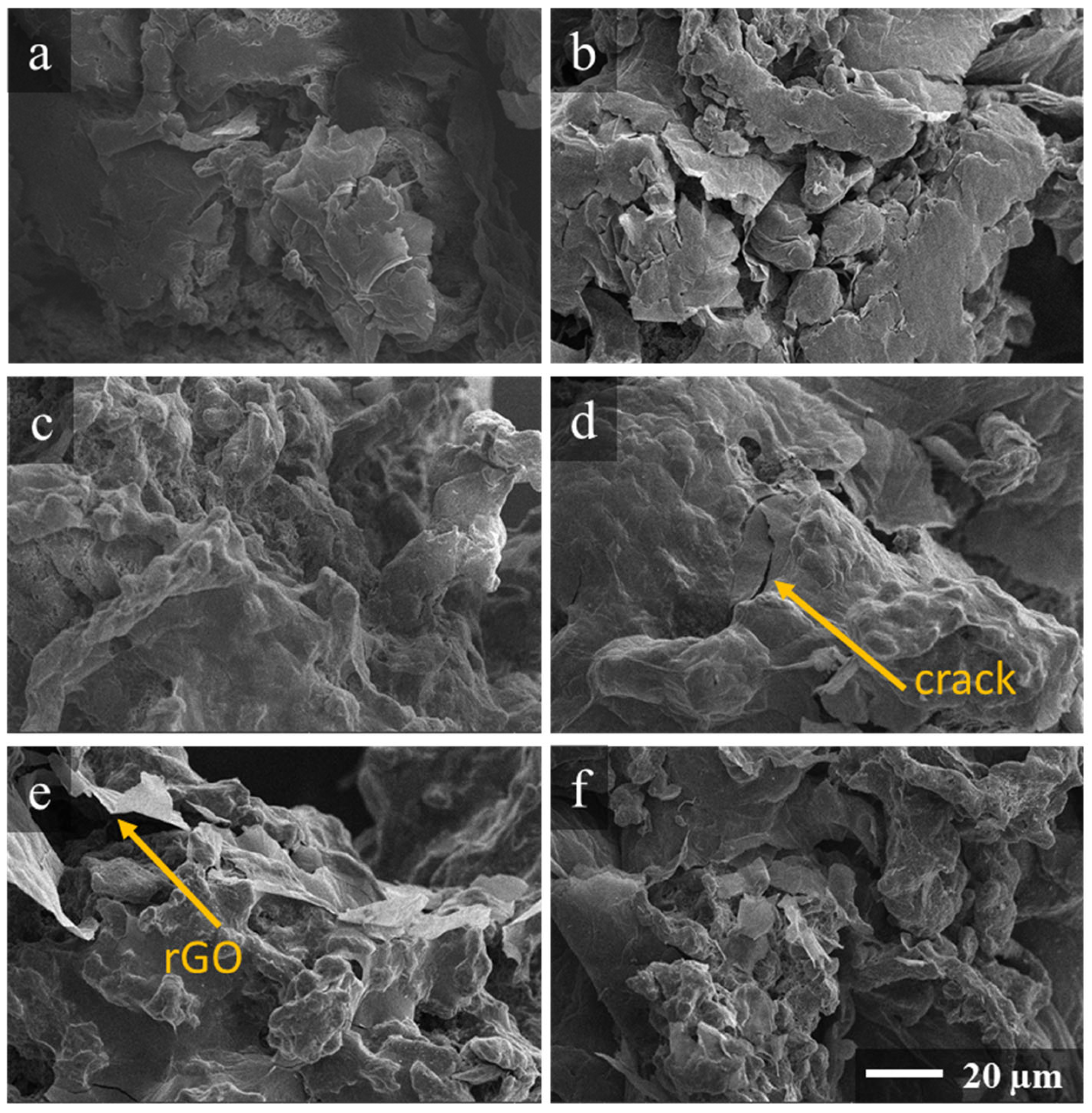
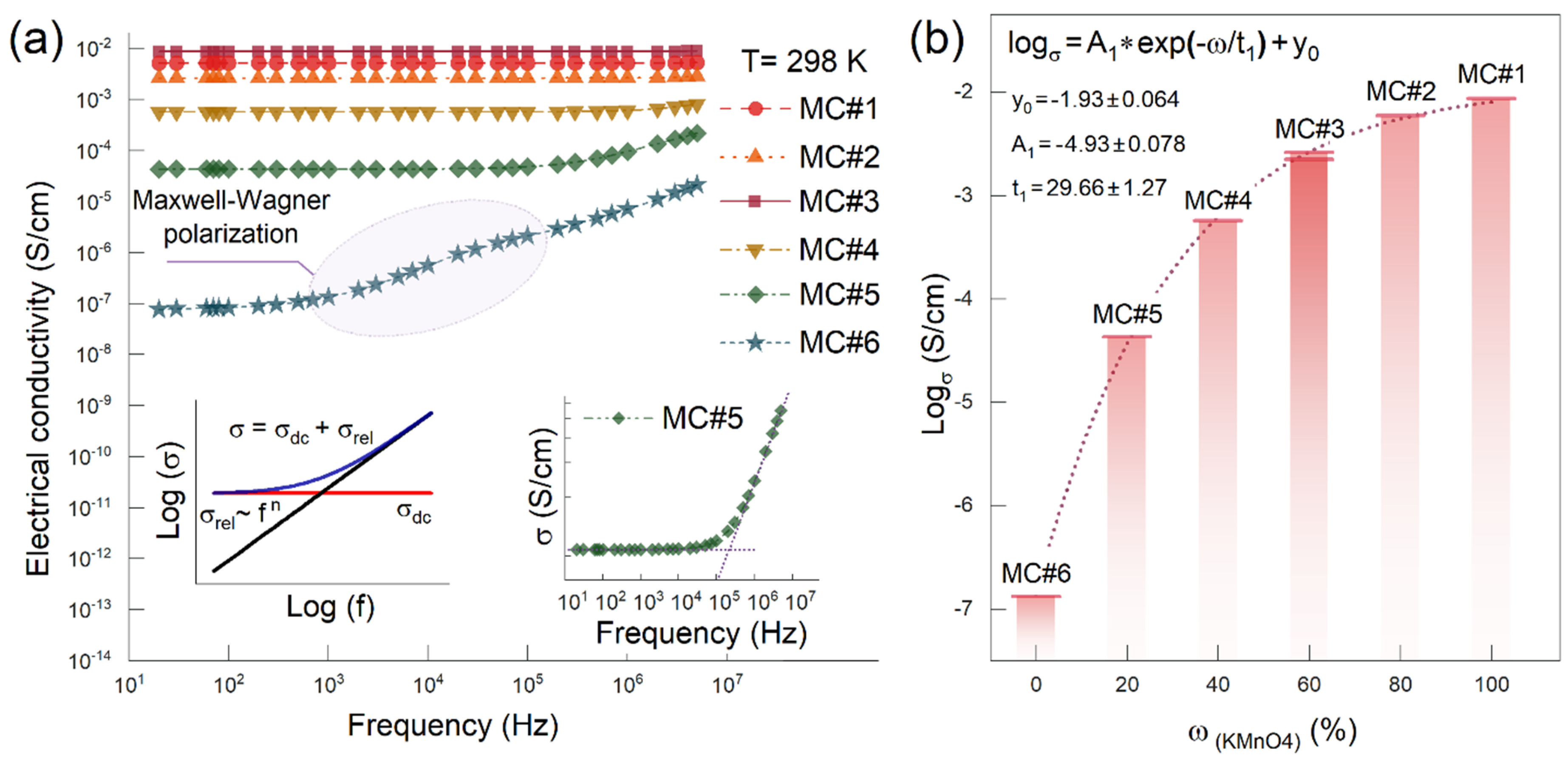

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
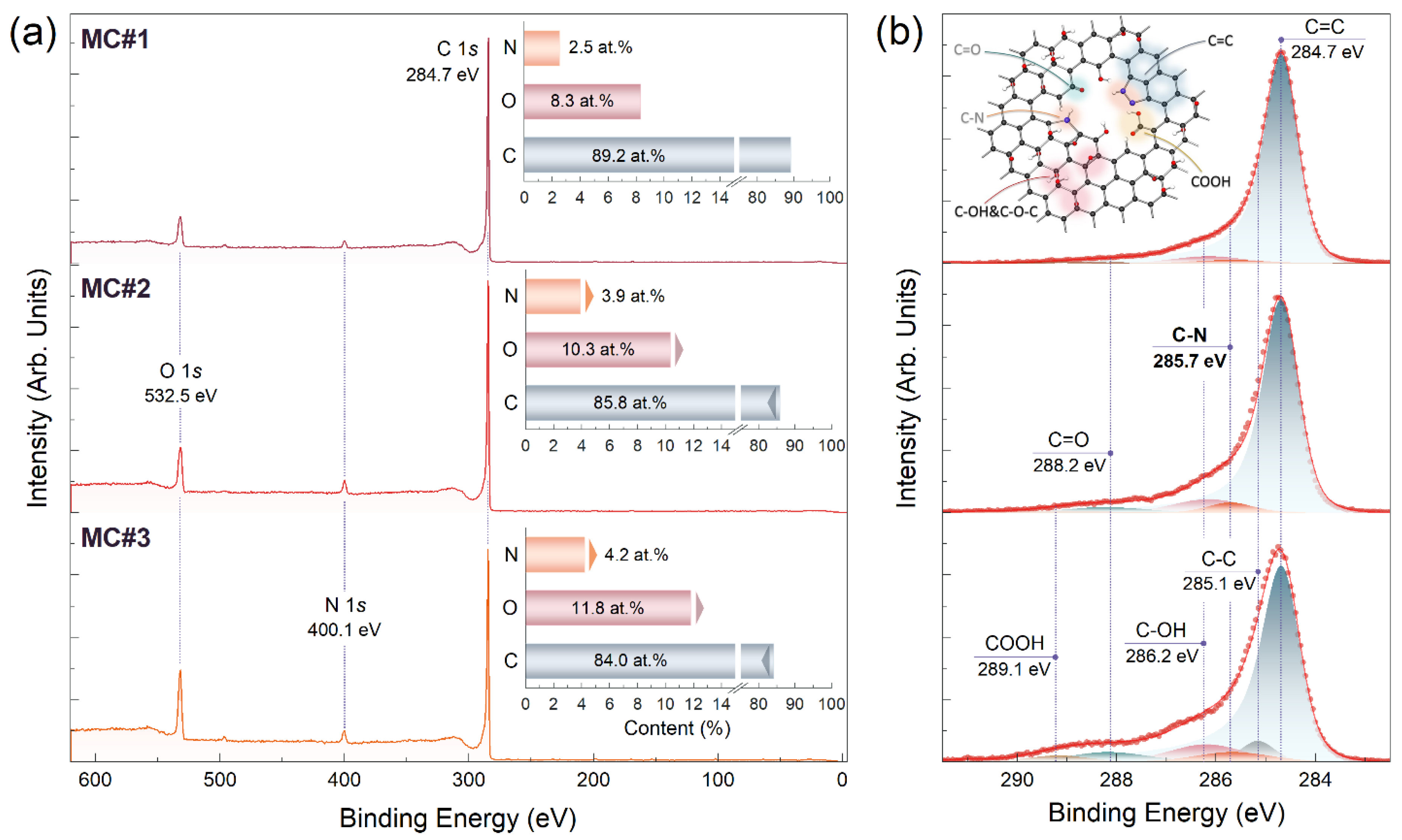
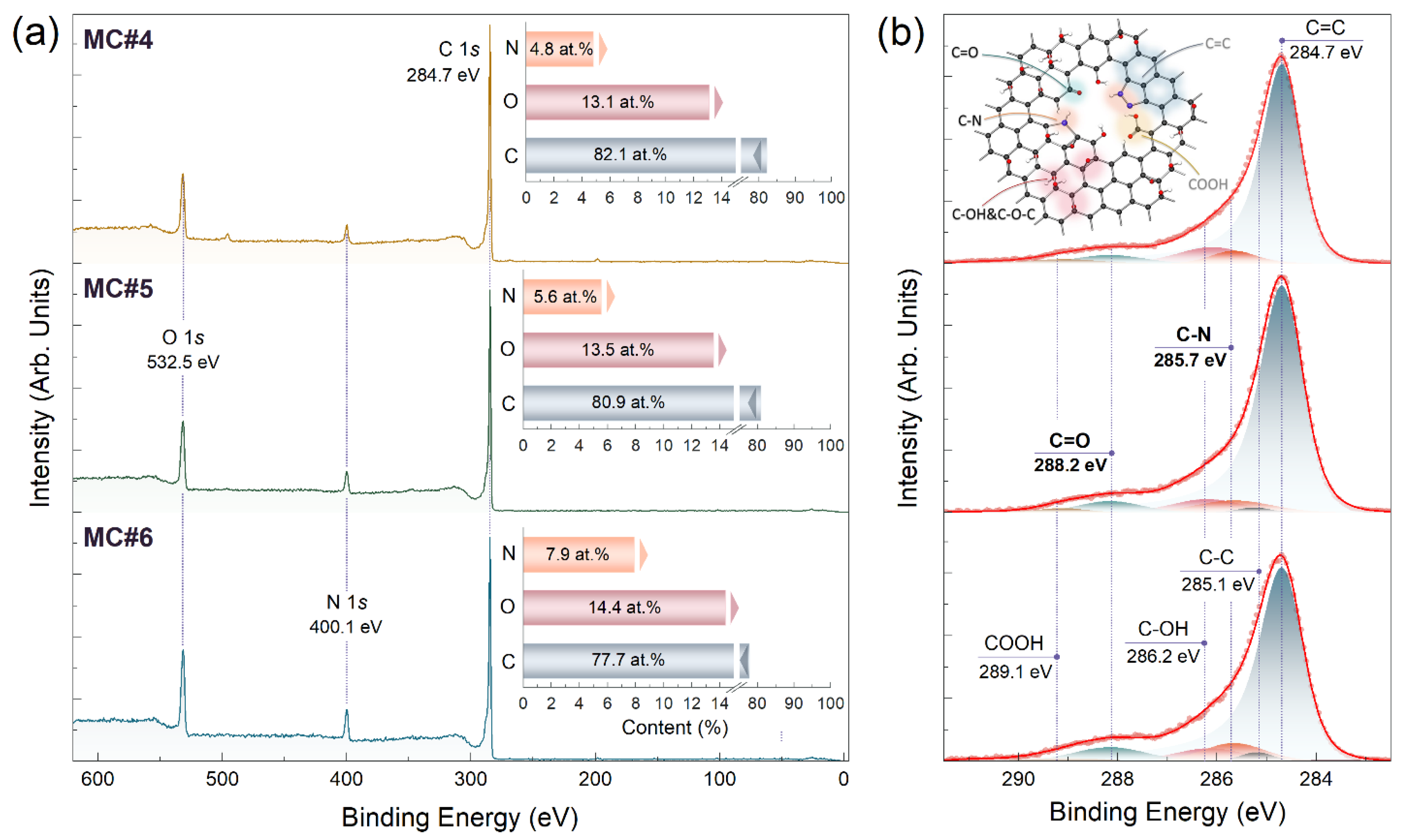
| Component | C-V | C=C | C-C | C-OH&C-O-C | C=O | COOH | C-N | C/O Ratio |
|---|---|---|---|---|---|---|---|---|
| Binding Energy [eV] | 283.7 | 284.7 | 285.1 | 286.2 | 288.2 | 289.1 | 285.7 | |
| MC#1 | <0.1 | 91.8 | <0.1 | 5.4 | 0.5 | <0.1 | 2.3 | 14.6 |
| MC#2 | <0.1 | 85.6 | <0.1 | 6.4 | 3.9 | <0.1 | 4.1 | 7.9 |
| MC#3 | <0.1 | 77.9 | 5.2 | 6.6 | 5.7 | 0.3 | 4.3 | 6.1 |
| MC#4 | <0.1 | 79.1 | 4.6 | 6.0 | 4.7 | 0.7 | 4.9 | 6.7 |
| MC#5 | <0.1 | 77.7 | 3.2 | 6.4 | 6.4 | 0.7 | 5.6 | 7.9 |
| MC#6 | <0.1 | 76.3 | 2.1 | 5.6 | 7.5 | 0.7 | 7.8 | 6.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rabchinskii, M.K.; Shiyanova, K.A.; Brzhezinskaya, M.; Gudkov, M.V.; Saveliev, S.D.; Stolyarova, D.Y.; Torkunov, M.K.; Chumakov, R.G.; Vdovichenko, A.Y.; Cherviakova, P.D.; et al. Chemistry of Reduced Graphene Oxide: Implications for the Electrophysical Properties of Segregated Graphene–Polymer Composites. Nanomaterials 2024, 14, 1664. https://doi.org/10.3390/nano14201664
Rabchinskii MK, Shiyanova KA, Brzhezinskaya M, Gudkov MV, Saveliev SD, Stolyarova DY, Torkunov MK, Chumakov RG, Vdovichenko AY, Cherviakova PD, et al. Chemistry of Reduced Graphene Oxide: Implications for the Electrophysical Properties of Segregated Graphene–Polymer Composites. Nanomaterials. 2024; 14(20):1664. https://doi.org/10.3390/nano14201664
Chicago/Turabian StyleRabchinskii, Maxim K., Kseniya A. Shiyanova, Maria Brzhezinskaya, Maksim V. Gudkov, Sviatoslav D. Saveliev, Dina Yu. Stolyarova, Mikhail K. Torkunov, Ratibor G. Chumakov, Artem Yu. Vdovichenko, Polina D. Cherviakova, and et al. 2024. "Chemistry of Reduced Graphene Oxide: Implications for the Electrophysical Properties of Segregated Graphene–Polymer Composites" Nanomaterials 14, no. 20: 1664. https://doi.org/10.3390/nano14201664
APA StyleRabchinskii, M. K., Shiyanova, K. A., Brzhezinskaya, M., Gudkov, M. V., Saveliev, S. D., Stolyarova, D. Y., Torkunov, M. K., Chumakov, R. G., Vdovichenko, A. Y., Cherviakova, P. D., Novosadov, N. I., Nguen, D. Z., Ryvkina, N. G., Shvidchenko, A. V., Prasolov, N. D., & Melnikov, V. P. (2024). Chemistry of Reduced Graphene Oxide: Implications for the Electrophysical Properties of Segregated Graphene–Polymer Composites. Nanomaterials, 14(20), 1664. https://doi.org/10.3390/nano14201664