

Optimization of Crystal Structures in Polylithionite Concentrate: A Molecular Dynamics Approach to Lithium Extraction Efficiency

 ,
,  ,
,  , ,
, ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. X-Ray Diffraction Analysis
2.2.2. Crystal Builder
2.2.3. Geometry Optimization
2.2.4. Structural Validation
2.2.5. Miscibility
3. Results
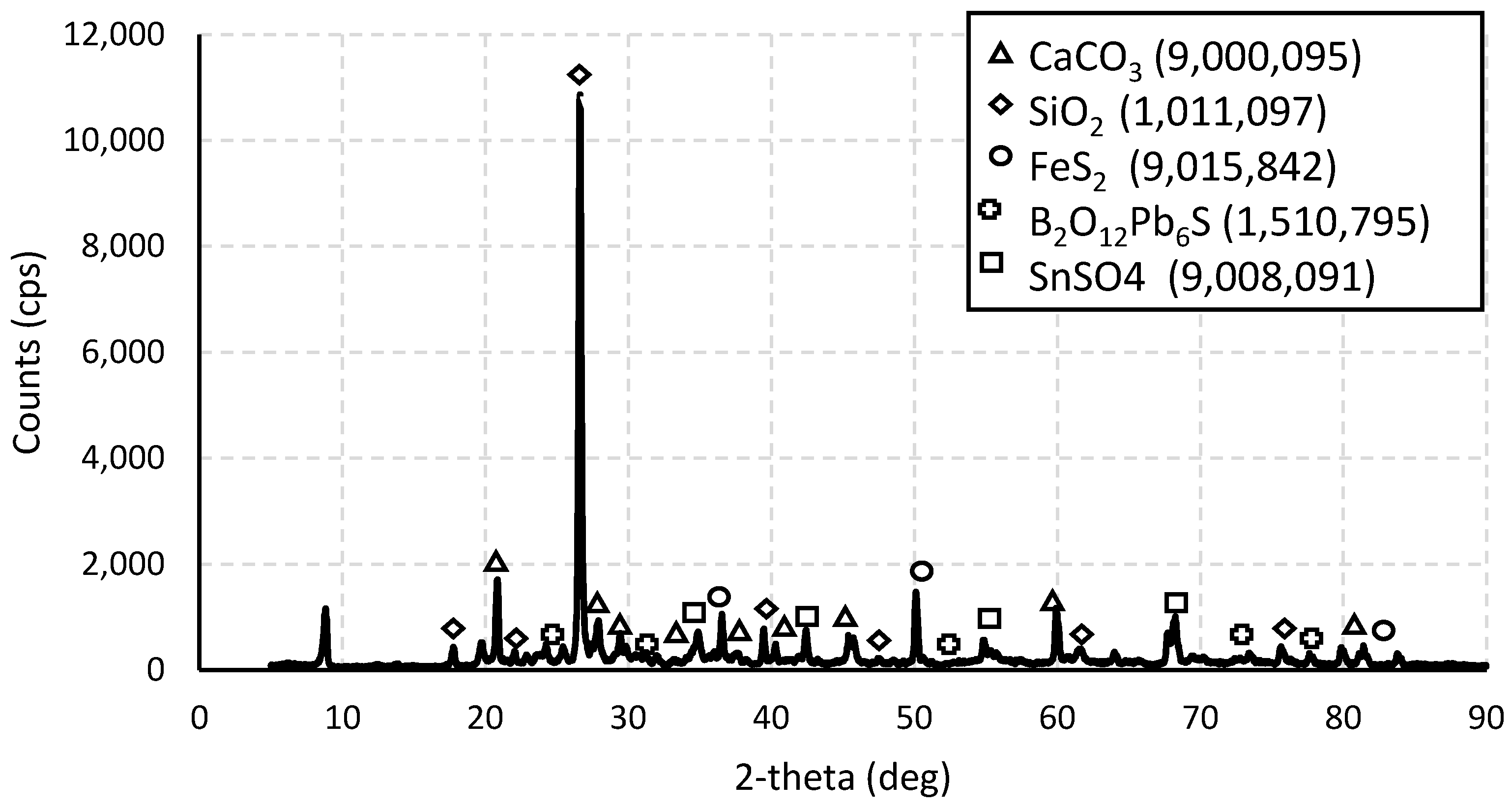
3.1. X-Ray Diffraction Analysis
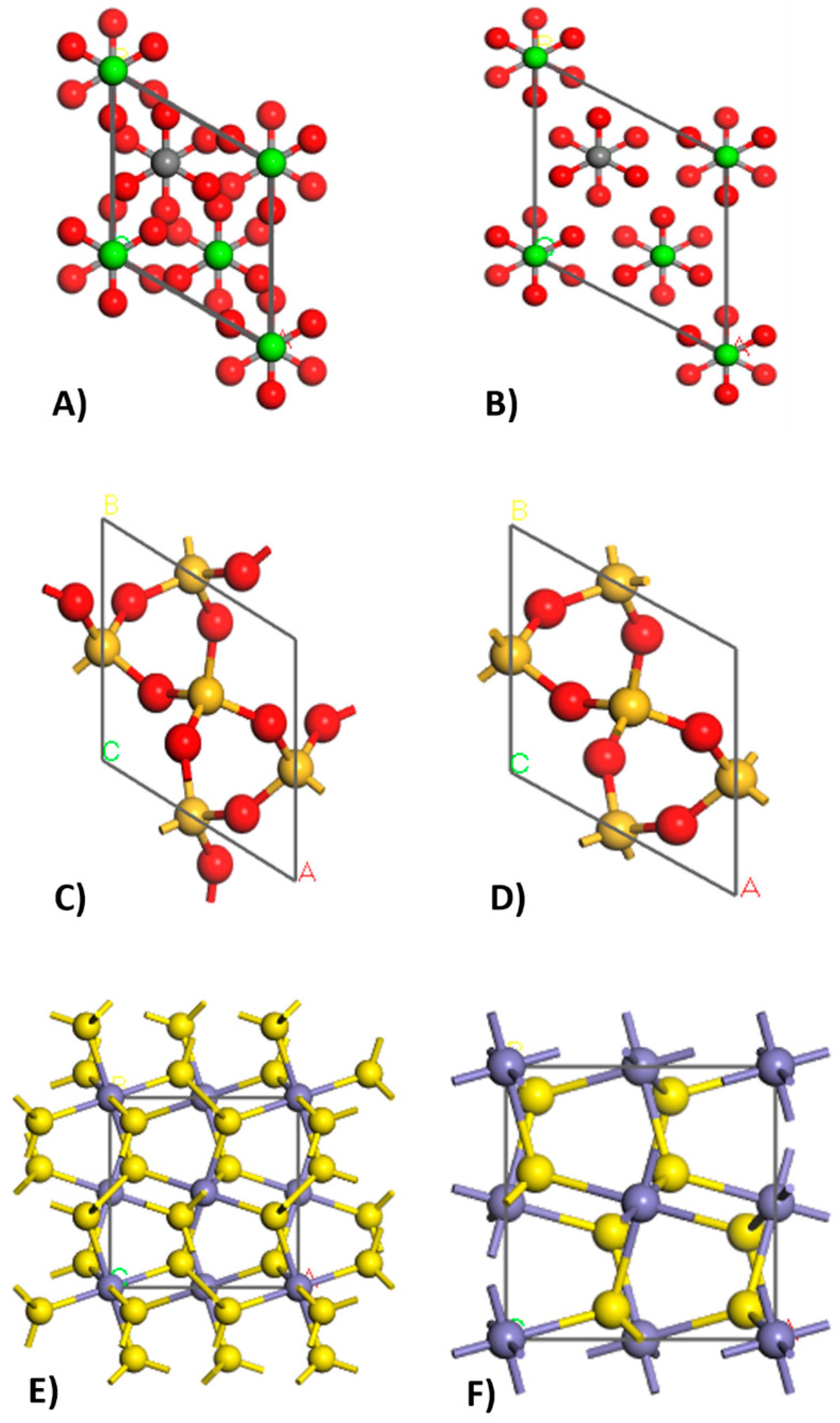
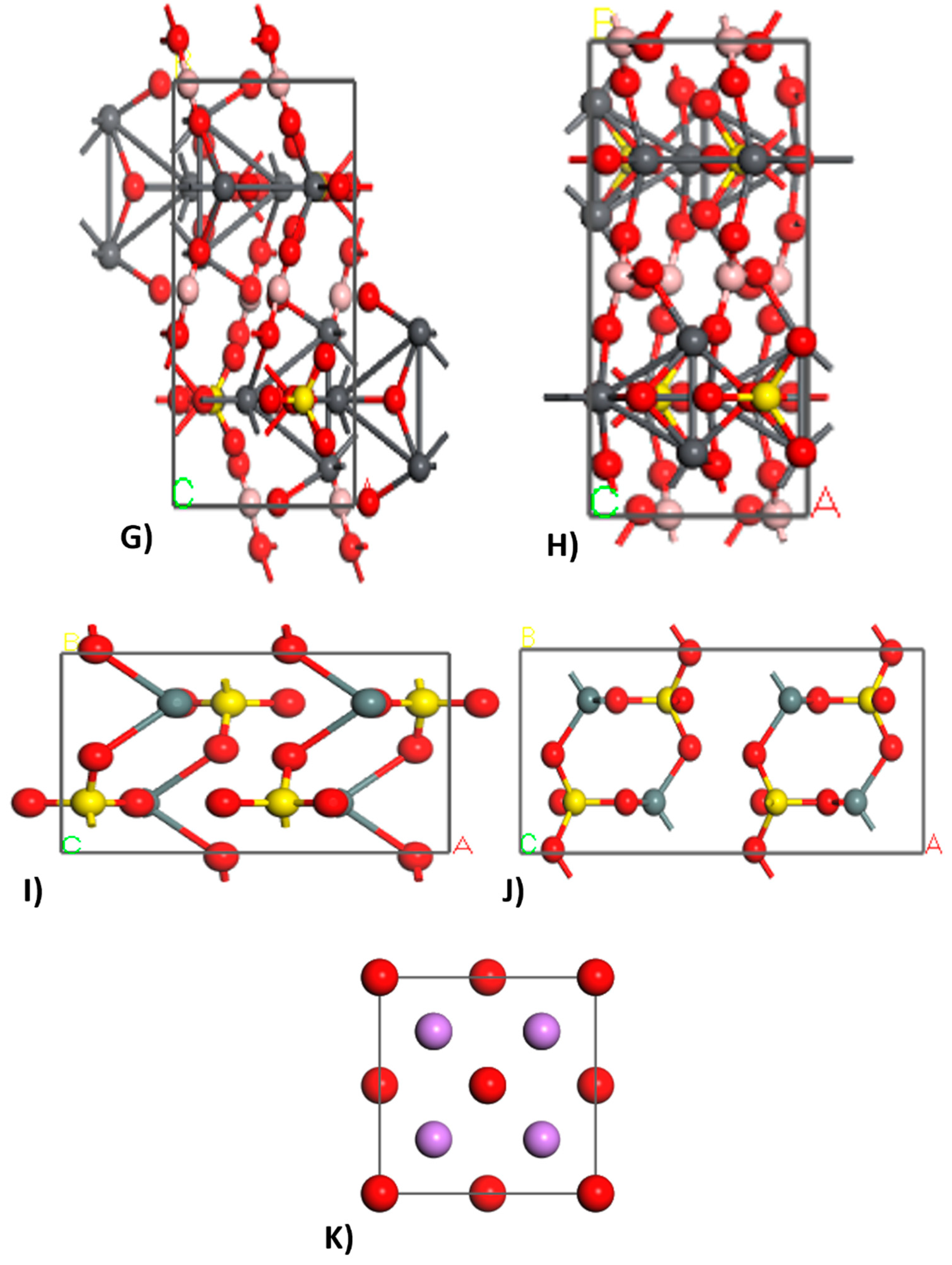
3.2. Crystal Builder
3.3. Geometry Optimization
3.4. Structural Validation
3.5. Interaction Parameter
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hollingsworth, S.A.; Dror, R.O. O. Molecular dynamics simulation for all. Neuron. 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Kozak, F.; Kurzbach, D. How to assess the structural dynamics of transcription factors by integrating sparse NMR and EPR constraints with molecular dynamics simulations. Comput. Struct. Biotechnol. J. 2021, 19, 2097–2105. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Saburi, H.; Tanaka, T.; Matsuura, Y.; Naitow, H.; Shimozono, R.; Yamamoto, N.; Inoue, H.; Nakamura, N.; Yoshizawa, Y.; et al. Multiple binding modes of a small molecule to human Keap1 revealed by X-ray crystallography and molecular dynamics simulation. FEBS Open Bio. 2015, 5, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Rumyantsev, A.V.; Borgardt, N.I.; Prikhodko, A.S.; Chaplygin, Y.A. Characterizing interface structure between crystalline and ion bombarded silicon by transmission electron microscopy and molecular dynamics simulations. Appl. Surf. Sci. 2021, 540, 148278. [Google Scholar] [CrossRef]
- Waller, C.; Marzinek, J.K.; McBurnie, E.; Bond, P.J.; Williamson, P.T.F.; Khalid, S. Impact on S. aureus and E. coli membranes of treatment with chlorhexidine and alcohol solutions: Insights from molecular simulations and nuclear magnetic resonance. J. Mol. Biol. 2023, 435, 167953. [Google Scholar] [CrossRef]
- Liu, X.; Lu, X.; Wang, R.; Zhou, H.; Xu, S. Speciation of gold in hydrosulphide-rich ore-forming fluids: Insights from first-principles molecular dynamics simulations. Geochim. Et Cosmochim. Acta 2011, 75, 185–194. [Google Scholar] [CrossRef]
- Pagliai, M.; Muniz-Miranda, M.; Cardini, G.; Schettino, V. Raman and infrared spectra of minerals from ab initio molecular dynamics simulations: The spodumene crystal. J. Mol. Struct. 2011, 993, 151–154. [Google Scholar] [CrossRef]
- Xiao, F.; Lin, W.; Cheng, Q. Ab-initio calculations and molecular dynamics simulations of In, Ag, and Cu replacing Zn in sphalerite: Implications for critical metal mineralization. Ore Geol. Rev. 2023, 163, 105699. [Google Scholar] [CrossRef]
- Vargas, K.I.; Medrano, H.; Reyes, D. Comparative study of chemical process and biotechnological process for the removal of bismuth from mining concentrates. Rev. Mex. De. Ing. Química. 2021, 20, Proc2215. [Google Scholar] [CrossRef]
- Li, S.; Hu, M.; Li, Y.; Gong, C. Fractional-order modeling and soc estimation of lithium-ion battery considering capacity loss. Int. J. Energy Res. 2018, 43, 417–429. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, C.; Dai, B.; Li, Z. Dynamic simulation of permanent magnet synchronous motor (pmsm) electric vehicle based on simulink. Energies 2022, 15, 1134. [Google Scholar] [CrossRef]
- Zhang, K.; Zhou, G.; Fang, T.; Jiang, K.; Liu, X. Structural reorganization of ionic liquid electrolyte by a rapid charge/discharge circle. J. Phys. Chem. Lett. 2021, 12, 2273–2278. [Google Scholar] [CrossRef] [PubMed]
- Alonso, E.; Álvarez, E.; Blanco, F.; Espi, J.A.; Herrera, J.; Larrea, M.; López, C.; Larrea, M.; Marqués, A.L.; De la Torre, L. Mineral Raw Materials in the Energy Transition and Digitalization. The Role of Mining and Metallurgy. Available online: https://www.raing.es (accessed on 2 June 2024).
- González, E. Bioreduction of Soluble and Insoluble Iron. Available online: https://eprints.ucm.es/id/eprint/34148/1/T36640 (accessed on 12 May 2024).
- Alatorre, A.E.; & Santillán, Y. Lithium Deposits. Available online: https://www.revistageomimet.mx/wp-content/uploads/2020/12/G_348.pdf. (accessed on 9 May 2024).
- Oliveira, A.; Barbosa, M.; Netz, P. Interplay between structure and density anomaly for an isotropic core-softened ramp-like potential. Phys. A Stat. Mech. Its Appl. 2007, 386, 744–747. [Google Scholar] [CrossRef]
- Errington, J.; Debenedetti, P. Relationship between structural order and the anomalies of liquid water. Nature 2001, 409, 318–321. [Google Scholar] [CrossRef]
- Svärd, M.; Rasmuson, Å.C. Force fields and point charges for crystal structure modeling. Ind. Eng. Chem. Res. 2009, 48, 2899–2912. [Google Scholar] [CrossRef]
- Nam, K.H.; Choi, H.K.; Yeo, H.; You, N.H.; Ku, B.C.; Yu, J. Molecular design and property prediction of sterically confined polyimides for thermally stable and transparent materials. Polymers 2018, 10, 630. [Google Scholar] [CrossRef]
- Fereidoon, A.; Aleaghaee, S.; Taraghi, I. Mechanical properties of hybrid graphene/TiO2 (rutile) nanocomposite: A molecular dynamics simulation. Comput. Mater. Sci. 2015, 102, 220–227. [Google Scholar] [CrossRef]
- De la Torre, A.; Reyes, D.; Medrano, H. Molecular Dynamics: An Alternative for the Prediction of Affinity Between Mining Compounds. Rev. Electrónica ANFEI Digit. 2021, 13. Available online: http://www.anfei.mx/revista/index.php/revista/article/view/721 (accessed on 20 September 2024).
- Gupta, J.; Nunes, C.; Vyas, S.; Jonnalagadda, S. Prediction of solubility parameters and miscibility of pharmaceutical compounds by molecular dynamics simulations. J. Phys. Chem. B 2011, 115, 2014–2023. [Google Scholar] [CrossRef]
- Valavi, M.; Ukrainczyk, M.; Dehghani, M.R. Prediction of solubility of active pharmaceutical ingredients by semi-predictive Flory Huggins/Hansen model. J. Mol. Liquids. 2017, 246, 166–172. [Google Scholar] [CrossRef]
- Fuh, A.Y.G.; Huang, K.Y.C. Solubilization of functionalized (5, 5) single-walled carbon nanotubes in 5CB nematic liquid crystals: Simulation using Flory–Huggins theory. Model. Simul. Mater. Sci. Eng. 2011, 19, 025006. [Google Scholar] [CrossRef]
- Anamu, U.S.; Ayodele, O.O.; Olorundaisi, E.; Babalola, B.J.; Odetola, P.I.; Ogunmefun, A.; Ukoba, K.; Jen, T.C.; Olubambi, P.A. Fundamental design strategies for advancing the development of high entropy alloys for thermo-mechanical application: A critical review. J. Mater. Res. Technol. 2023, 27, 4833–4860. [Google Scholar] [CrossRef]
- Cardenal, A.D.; Ramadhar, T.R. The crystalline sponge method: Quantum chemical in silico derivation and analysis of guest binding energies. CrystEngComm 2021, 23, 7570–7575. [Google Scholar] [CrossRef]
- The Materials Project. “Materials Data on Li2O by Materials Project”. United States. 2020. Available online: https://next-gen.materialsproject.org/materials/mp-1960 (accessed on 20 September 2024).
- Rodríguez, D.; Pámanes, G.; Delgado, E.; Rodríguez, M.D.; Medrano, H.; Reyes, D. Extraction optimization and molecular dynamic simulation of cellulose nanocrystals obtained from bean forage. Biocatal. Agric. Biotechnol. 2022, 43, 102443. [Google Scholar] [CrossRef]
- Perrot, V.; Roussey, A.; Benayad, A.; Veillerot, M.; Mariolle, D.; Solé-Daura, A.; Jousseaume, V. Zif-8 thin films by a vapor-phase process: Limits to growth. Nanoscale 2023, 15, 7115–7125. [Google Scholar] [CrossRef] [PubMed]
- Gobato, R. Calculation by uff method of frequencies and vibrational temperatures of the unit cell of the rhodochrosite crystal. Int. J. Adv. Chem. 2019, 7, 77–81. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y.; Yang, Y.; Li, Y.; Zhao, M.; Liu, W.; Qu, Y. Efficient helium separation with two-dimensional metal–organic framework Fe/Ni-PTC: A theoretical study. Membranes 2021, 11, 927. [Google Scholar] [CrossRef]
- Dokur, D.; Keskın, S. Effects of force field selection on the computational ranking of MOFs for CO2 separations. Ind. Eng. Chem. Res. 2018, 57, 2298–2309. [Google Scholar] [CrossRef]
- Aly, S.; Elganzory, H.; Mahross, M.; Abdalla, E. Quantum chemical studies and effect of gamma irradiation on the spectral, thermal, x-ray diffraction and DNA interaction with Pd (ii), Cu(i), and Cd (ii) of hydrazone derivatives. Appl. Organomet. Chem. 2021, 35, e6153. [Google Scholar] [CrossRef]
- Deljoo, M. The molecular dynamics study of CO2, SO2, and NO2 pollution capture from O2 environment via polyether/polyamide-Ag membranes. Preprint 2024. [Google Scholar] [CrossRef]
- Addicoat, M.; Vankova, N.; Akter, I.; Heine, T. Extension of the universal force field to metal–organic frameworks. J. Chem. Theory Computation. 2014, 10, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Jaillet, L.; Artemova, S.; Redon, S. Im-uff: Extending the universal force field for interactive molecular modeling. J. Mol. Graph. Model. 2017, 77, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Tokarský, J.; Čapková, P.; Burda, J. Structure and stability of kaolinite/TiO2 nanocomposite: DFT and MM computations. J. Mol. Model. 2011, 18, 2689–2698. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Y.; Huang, L.; Su, B.; Yang, Y. Investigating the microscopic mechanism of ultrasonic-vibration-assisted-pressing of WC-co powder by simulation. Materials 2023, 16, 5199. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Pan, Y.; Zhang, W.; Liu, X.; Li, L. The size effect to O2−–Ce4+ charge transfer emission and band gap structure of Sr2CeO4. Luminescence 2018, 33, 907–912. [Google Scholar] [CrossRef]
- Goldsby, J. Basic elastic properties predictions of cubic cerium oxide using first-principles methods. J. Ceramics. 2013, 1, 323018. [Google Scholar] [CrossRef]
- Tagne, A.; Simo-Tagne, M.; Kharchi, R.; Ndukwu, M.; Assoumani, N.; Compaoré, A.; Zoulalian, A. Numerical study of the pyrolysis of wood chips for biocharcoal production: Influence of chips geometry and initial moisture content. Energies 2022, 15, 4098. [Google Scholar] [CrossRef]
- Gamero-Barraza, J.I.; Pámanes-Carrasco, G.A.; Delgado, E.; Cabrales-Arellano, C.P.; Medrano-Roldán, H.; Gallegos-Ibáñez, D.; Wedwitschka, H.; Reyes-Jáquez, D. Computational modelling of extrusion process temperatures on the interactions between black soldier fly larvae protein and corn flour starch. Food Chem. Molecular Sci. 2024, 8, 100202. [Google Scholar] [CrossRef]
- Zhuravlev, Y.N.; Obolonskaya, O.S. Structure, mechanical stability, and chemical bond in alkali metal oxides. J. Struct. Chem. 2010, 51, 1005–1013. [Google Scholar] [CrossRef]
- David, W.I.F.; Jones, M.O.; Gregory, D.H.; Jewell, C.M.; Johnson, S.R.; Walton, A.; Edwards, P.P. A mechanism for non-stoichiometry in the lithium amide/lithium imide hydrogen storage reaction. J. Am. Chem. Soc. 2007, 129, 1594–1601. [Google Scholar] [CrossRef]
- Annamareddy, A.; Eapen, J. Ion hopping and Constrained Li diffusion pathways in the superionic state of antifluorite Li2O. Entropy 2017, 19, 227. [Google Scholar] [CrossRef]
- Yuan, P.; Liu, B.; Sun, H. Optimization of the crystallization process of bis(2-hydroxyethyl) terephthalate. Cryst. Res. Technol. 2021, 56, 2100025. [Google Scholar] [CrossRef]
- Song, L.; Bediako, J.; Song, M.; Kim, J.; Cho, C.; Zhao, Y.; Choi, J. Effective recovery of Pt(IV) from acidic solution by a defective metal-organic frameworks using central composite design for synthesis. Sustain. Chem. Eng. 2019, 7, 7510–7518. [Google Scholar] [CrossRef]
- Moustafa, I.; Mohamed, N.; Ibrahim, S. Molecular modeling and antimicrobial screening studies on some 3-aminopyridine transition metal complexes. Open J. Inorg. Chem. 2022, 12, 39–56. [Google Scholar] [CrossRef]
- Nagappayya, S.; Gaikar, V.; Ali, S. Designing of ligands for solvent extraction of Cs+ using molecular modeling approach. Desalination Water Treat. 2012, 38, 1–7. [Google Scholar] [CrossRef]
- Orekhov, M. Coordination numbers of bivalent ions in organic solvents. Russ. J. Phys. Chem. A 2021, 95, 2059–2064. [Google Scholar] [CrossRef]
- Rodríguez, I.; Valladares, R.; Hinojosa-Romero, D.; Valladares, A. Emergence of magnetism in bulk amorphous palladium. Phys. Rev. B 2019, 100, 024422. [Google Scholar] [CrossRef]
- Liu, X.; Du, J.; Hua, L.; Liu, K. First-principles comparative study of CuFeSe2 and CuFeS2. Mater. Res. 2023, 26, e20220371. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, S.; Li, X.; Zhu, J.; Liang, H.; Zhang, X.; Shu, Q. Molecular dynamics simulations for 5,5′-bistetrazole-1,1′-diolate (TKX-50) and its PBXs. RSC Adv. 2016, 6, 20034–20041. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Formula | Space Group | Latency Parameters | |||||
|---|---|---|---|---|---|---|---|---|
| a (Å) | b (Å) | c (Å) | (°) | (°) | (°) | |||
| Calcite | CaCO3 | 167: R-3c | 4.9937 | 4.9937 | 17.0792 | 90 | 90 | 120 |
| Quartz | SiO2 | 152: P3121 | 4.9198 | 4.9198 | 5.4018 | 90 | 90 | 120 |
| Pyrite | FeS2 | 205: Pa-3 | 5.4279 | 5.4279 | 5.4279 | 90 | 90 | 90 |
| Pb6O2(BO3)2SO4 | B2O12Pb6S | 62: Pnma | 6.3849 | 11.3296 | 17.7568 | 90 | 90 | 90 |
| Tin oxide | SnSO4 | 62: Pnma | 8.6952 | 5.3834 | 6.9650 | 90 | 90 | 90 |
| Lithium Oxide * | LiO2 | Fm3̅m | 4.65 | 4.65 | 4.65 | 90 | 90 | 90 |
| Phase | Element | X | Y | X |
|---|---|---|---|---|
| Calcite (CaCO3) | Ca | 0.000 | 0.000 | 0.000 |
| C | 0.000 | 0.000 | 0.250 | |
| O | 0.257 | 0.000 | 0.250 | |
| Quartz (SiO2) | Si | 0.465 | 0.000 | 0.333 |
| O | 0.417 | 0.278 | 0.222 | |
| Pyrite (FeS2) | Fe | 0.000 | 0.000 | 0.333 |
| S | 0.625 | 0.625 | 0.625 | |
| Pb6O2(BO3)2SO4 | O | 0.030 | 0.025 | 0.447 |
| S | 0.212 | 0.250 | 0.493 | |
| O | 0.167 | 0.650 | 0.974 | |
| O | 0.087 | 0.250 | 0.161 | |
| Pb | 0.224 | 0.250 | 0.292 | |
| Pb | 0.171 | 0.093 | 0.105 | |
| O | 0.164 | 0.250 | 0.571 | |
| Pb | 0.402 | 0.250 | 0.699 | |
| B | 0.079 | 0.508 | 0.748 | |
| O | 0.141 | 0.597 | 0.718 | |
| Pb | 0.645 | 0.590 | 0.102 | |
| O | 0.750 | 0.491 | 0.829 | |
| O | 0.281 | 0.750 | 0.355 | |
| O | 0.024 | 0.402 | 0.713 | |
| Tin sulfate (O4SSn) | Sn | 0.208 | 0.250 | 0.222 |
| S | 0.069 | 0.250 | 0.694 | |
| O | -0.083 | 0.250 | 0.597 | |
| O | 0.194 | 0.250 | 0.550 | |
| O | 0.088 | 0.020 | 0.819 | |
| Lithium Oxide (Li2O) | O | 0.000 | 0.000 | 0.000 |
| Li | 0.250 | 0.750 | 0.250 |
| Name | Formula | Density [g/cm3] | |
|---|---|---|---|
| Experimental | Simulated | ||
| Calcite | CaCO3 | 2.703 | 1.697 |
| Quartz | SiO2 | 2.643 | 2.708 |
| Pyrite | FeS2 | 4.983 | 4.594 |
| Pb6O2(BO3)2SO4 | B2O12Pb6S | 7.699 | 6.330 |
| Tin sulfate | SnSO4 | 4.375 | 3.540 |
| Lithium oxide | LiO2 | 1.960 | 1.959 |
| Base | Display | |
|---|---|---|
| Pb6O2(BO3)2SO4 | LiO2 | 67.4689 |
| Tin sulfate | LiO2 | 11.1287 |
| Quartz | LiO2 | 8.9556 |
| Calcite | LiO2 | 4.2978 |
| Pyrite | LiO2 | 0.6052 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quezada-Aldaco, M.G.; Delgado, E.; Zazueta-Álvarez, D.E.; Martínez-Gómez, V.J.; Medrano-Roldán, H.; Vázquez-Ortega, P.G.; Hernández-Rodarte, F.S.; Reyes-Jáquez, D. Optimization of Crystal Structures in Polylithionite Concentrate: A Molecular Dynamics Approach to Lithium Extraction Efficiency. Nanomaterials 2024, 14, 1713. https://doi.org/10.3390/nano14211713
Quezada-Aldaco MG, Delgado E, Zazueta-Álvarez DE, Martínez-Gómez VJ, Medrano-Roldán H, Vázquez-Ortega PG, Hernández-Rodarte FS, Reyes-Jáquez D. Optimization of Crystal Structures in Polylithionite Concentrate: A Molecular Dynamics Approach to Lithium Extraction Efficiency. Nanomaterials. 2024; 14(21):1713. https://doi.org/10.3390/nano14211713
Chicago/Turabian StyleQuezada-Aldaco, María Guadalupe, Efren Delgado, David Enrique Zazueta-Álvarez, Víctor Jesús Martínez-Gómez, Hiram Medrano-Roldán, Perla Guadalupe Vázquez-Ortega, Felipe Samuel Hernández-Rodarte, and Damián Reyes-Jáquez. 2024. "Optimization of Crystal Structures in Polylithionite Concentrate: A Molecular Dynamics Approach to Lithium Extraction Efficiency" Nanomaterials 14, no. 21: 1713. https://doi.org/10.3390/nano14211713
APA StyleQuezada-Aldaco, M. G., Delgado, E., Zazueta-Álvarez, D. E., Martínez-Gómez, V. J., Medrano-Roldán, H., Vázquez-Ortega, P. G., Hernández-Rodarte, F. S., & Reyes-Jáquez, D. (2024). Optimization of Crystal Structures in Polylithionite Concentrate: A Molecular Dynamics Approach to Lithium Extraction Efficiency. Nanomaterials, 14(21), 1713. https://doi.org/10.3390/nano14211713