Abstract
Changes in the work function provide a fingerprint to characterize analyte binding in charge transfer-based sensor devices. Hence, a rational sensor design requires a fundamental understanding of the microscopic factors controlling the modification of the work function. In the current investigation, we address the mechanisms behind the work function change (WFC) for the adsorption of four common volatile organic compounds (toluene, ethanol, 2-Furfurylthiol, and guaiacol) on different nitrogen-doped graphene-based 2D materials using density functional theory. We show that competition between the surface dipole moment change induced by spatial charge redistribution, the one induced by the pure adsorbate, and the one caused by the surface deformation can quantitatively predict the work function change. Furthermore, we also show this competition can explain the non-growing work function change behavior in the increasing concentrations of nitrogen-doped graphenes. Finally, we propose possible design principles for WFC of VOCs interacting with N-doped graphene materials.
1. Introduction
Gas sensing technologies are critical for tracking environmental contaminants, maintaining safety in industrial settings, and protecting public health [1]. Volatile organic compounds (VOCs) represent a key target for these technologies, given their pervasive nature and hazards. VOCs are emitted from a variety of sources, including industrial activities, consumer products, and vehicle emissions, and their long-term exposure is linked to severe health and environmental consequences, such as respiratory disorders, damage to organs, and cancer [2,3].
The performance of gas sensors for detecting volatile organic compounds (VOCs) is primarily determined by the materials used in their design. Advanced materials, particularly state-of-the-art 2D materials, are of great interest for enhancing both the sensitivity, which allows for detection of low VOC concentrations, and the selectivity, which enables differentiation among various VOC types [4,5].
An important factor in evaluating the performance of these sensors is the work function (WF) (specifically for WF-based sensors), which characterizes the interaction between the sensor material and the target VOC molecule. The work function refers to the minimum energy needed for an electron to escape from the surface of a material, and the change in work function (WFC) before and after gas molecule adsorption is widely measured as an important indicator or even direct electronic response for sensors (e.g., Schottky sensors) [6,7,8,9,10,11,12]. Computational methods such as density functional theory (DFT) are effective methods for analyzing the WFC of 2D materials upon the adsorption of VOC molecules. Numerous computational studies have been carried out to evaluate the potential sensing performance by WFC of a variety of materials upon adsorption of VOCs [13,14,15,16,17,18,19,20]. However, despite the observed WFC values reported in these studies, there is a significant lack of understanding regarding the underlying mechanisms driving these changes.
To interpret the origin of the work function change, Leung et al. [21] developed the surface dipole moment model and revealed the work function change mechanism for single-atom adsorption on W surfaces. Subsequently this model has been applied to many systems such as single-atom adsorption [22], surface functionalization [23], and interfaces [24,25,26,27] to gain insights into the property/structure–property relationships for their systems of interest. Herein, we validate a model for physisorption of VOCs on typical 2D sensing materials. This facilitates a better rational design of sensing materials for WF-based sensors.
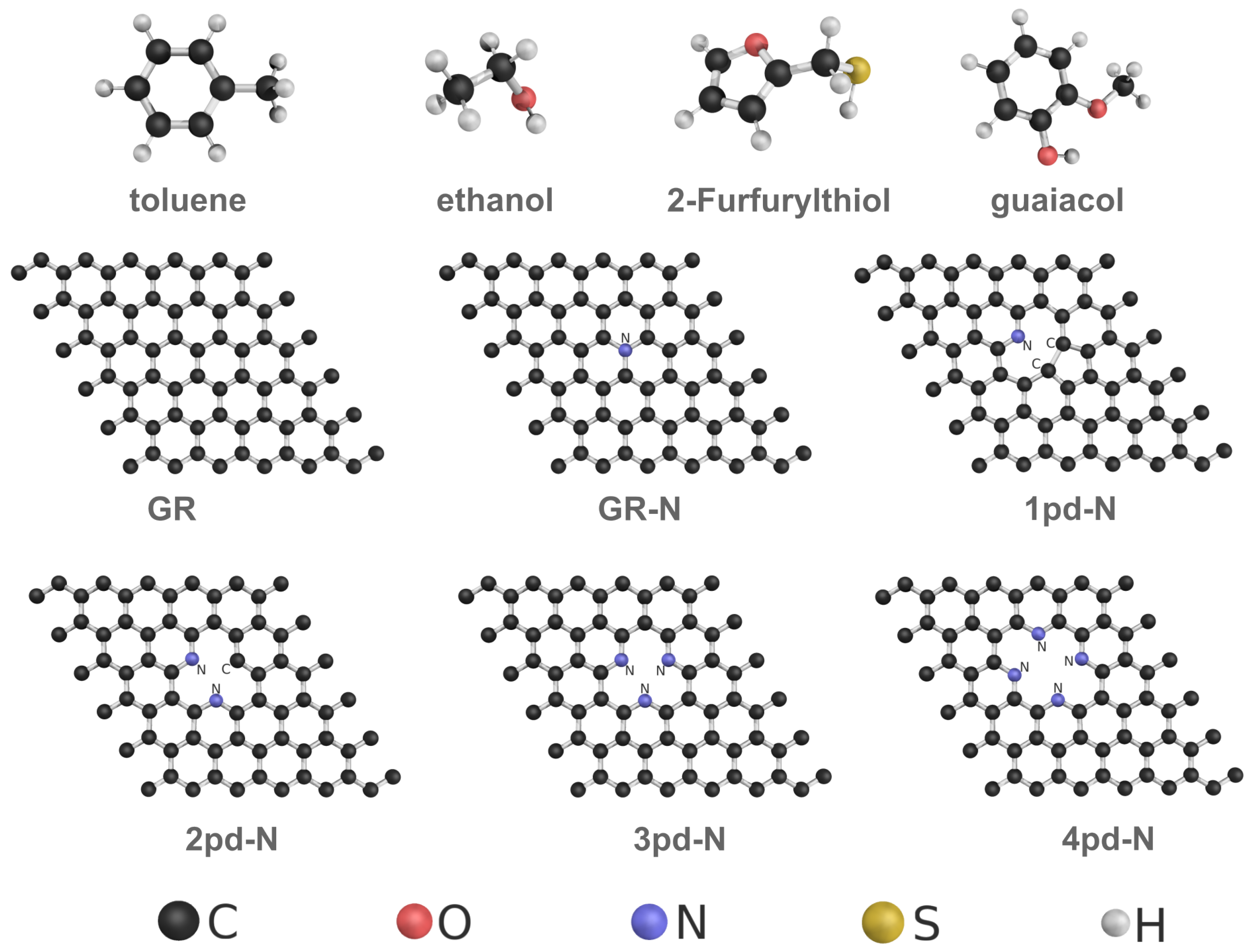
In this work, we investigate the WFC as an electronic response on different 2D N-doped graphenes upon adsorption of four common VOC molecules (toluene [28], ethanol [28], 2-Furfurylthiol [29], and guaiacol [29]). The substrates encompass pristine graphene (GR) and graphene substrates doped by single graphitic-N (GR-N) and pyridinic-N (1pd-N), as shown in Figure 1. Pyrrolic-N (pr-N) has been excluded, as it is not stable and converts to 1pd-N in all simulations. Furthermore, to probe the influence of local nitrogen and point defect concentration, we expanded from 1pd-N up to 4pd-N, expecting to increase the WF value [30,31]. Next, we validated the linear relationship of the WFC and the surface dipole moment change , and the observed behaviors of the WFC were quantitatively analyzed based on the components of the surface dipole moment change. Finally, we propose possible design principles for the WFC of VOCs by sensing for 2D materials.
Figure 1.
Ball-and-stick representation of VOC molecules and graphene-based substrates.
2. Computational Methods
The Quantum ESPRESSO package [32,33,34] (version 7.0) was used to perform periodic DFT calculations with the Perdew–Burke–Ernzerhof (PBE) version of the generalized gradient approximation (GGA) exchange–correlation functional [35]. DFT-D3 was involved in all simulations to describe the van der Waals (vdW) interactions [36]. The cutoff energy of planar basis sets generated by the projector augmented wave (PAW) method was set to 60 Ry, while the charge density cut-off was 450 Ry. The Gaussian smearing spreading was employed with a width of 0.01 Ry. A (6 × 6) hexagonal supercell composed of 72 carbon atoms was modeled with a fixed vacuum thickness of 30.68 Å, ensuring negligible interaction of periodic images of isolated gas molecules. The atomic positions and lattice parameters (at the fixed z length) were optimized by constraining the cell length along the c direction to be constant until the total energies and the imposed force on the atoms became less than 10−6 Ry/cell and 10−4 Ry·Bohr−1, with <0.01 kbar of residual stress. A (2 × 2 × 1) Monkhorst–Pack grid was sampled in the first Brillouin zone for the k-points meshes. In addition, the coulomb interaction in the z direction was truncated for the structures’ periodic in the x–y plane [37] in all simulations to ensure constant vacuum energy and avoid heavy simulations caused by large vacuum layer lengths.
The charge transfer Q between the substrate and adsorbate was obtained using the Bader algorithm [38]. The sign of the charge transfer Q is defined as positive when the charges transfer from the substrate to the VOC molecules, and vice versa. The work function is defined as follows:
where and are the vacuum energy and Fermi level, respectively.
Furthermore, a surface dipole moment analysis was carried out to study the behavior of the . Leung et al. [21] derived the relation between and the total surface dipole moment change as follows:
where e and are the unit charge and vacuum permittivity, respectively. Leung et al. [21] and Khazaei et al. [23] further divided into three components: (a) the surface dipole moment change induced by spatial charge redistribution , (b) the surface dipole moment induced by the pure adsorbate , and (c) the surface dipole moment induced by surface deformation after and before adsorption (). Then, Equation (2) turns into the following equation:
Each dipole moment component as a function of the z direction was calculated using the x–y planar average of the corresponding charge densities via the integral , where is set at the bottom of the cell. For the electron charge transfer, we have the following relationship [21]:
where is the charge density redistribution, is the total charge density of the adsorbate–substrate complex system, and / are the charge density for the isolated substrate/adsorbate computed by removing one component from the complex system without further optimization. Therefore, the component for the charge redistribution term is then calculated using . The total surface dipole moment change is defined as the total surface dipole moment difference between the adsorbate–substrate system and the isolated substrate (before adsorption), which has a corresponding total charge density of . Hence and in Equation (4) would be utilized for the calculation.
The resulting values of − , , and were normal to the substrate’s x–y plane, where a positive value indicates a dipole pointing out to the vacuum, and vice versa. Their corresponding values were obtained at , where these surface dipole moment components remained constant with increasing z length.
3. Results and Discussion
3.1. Physisorption Interaction Type
In order to find the most favorable configurations, we considered 7 to 12 different initial configurations for each adsorption case, including vertical, horizontal, and intermediate orientations of molecules and distinct adsorption sites. The configurations with the lowest adsorption energy were selected for further electronic property calculations. The is defined as , where is the total energy of the optimized adsorbate–substrate complex system, while and are the total energies of the isolated substrate and gas molecule, respectively. The adsorption energies for all initial configurations are depicted in Table S1.
The vertical distance between the lowest atom of the VOC molecules (always the H atom in our case) and the substrate plane (since there is no dramatic deformation of the substrate upon adsorption, as shown in Figures S1–S4) for these low-energy configurations are listed in Table 1. As shown in Table 1, all of the vertical distances are longer than typical N-H and C-H covalent bonds, such as ∼1.1 Å in methylamine [39,40,41], indicating the physisorption interaction type for our systems.

Table 1.
Vertical distance between the lowest atom on the VOC molecule along the slab direction and the substrate plane. Distances are in Å.
3.2. Work Function Change
The change in work function is calculated and used as a indicator for the substrate’s electronic response towards the VOC molecules. In principle, a large WFC upon adsorption implies a higher sensitivity of the substrate to adsorbate. Differences in the WFC between different molecules adsorbing on the same substrate provide information into the selectivity performance of this substrate towards these adsorbates.
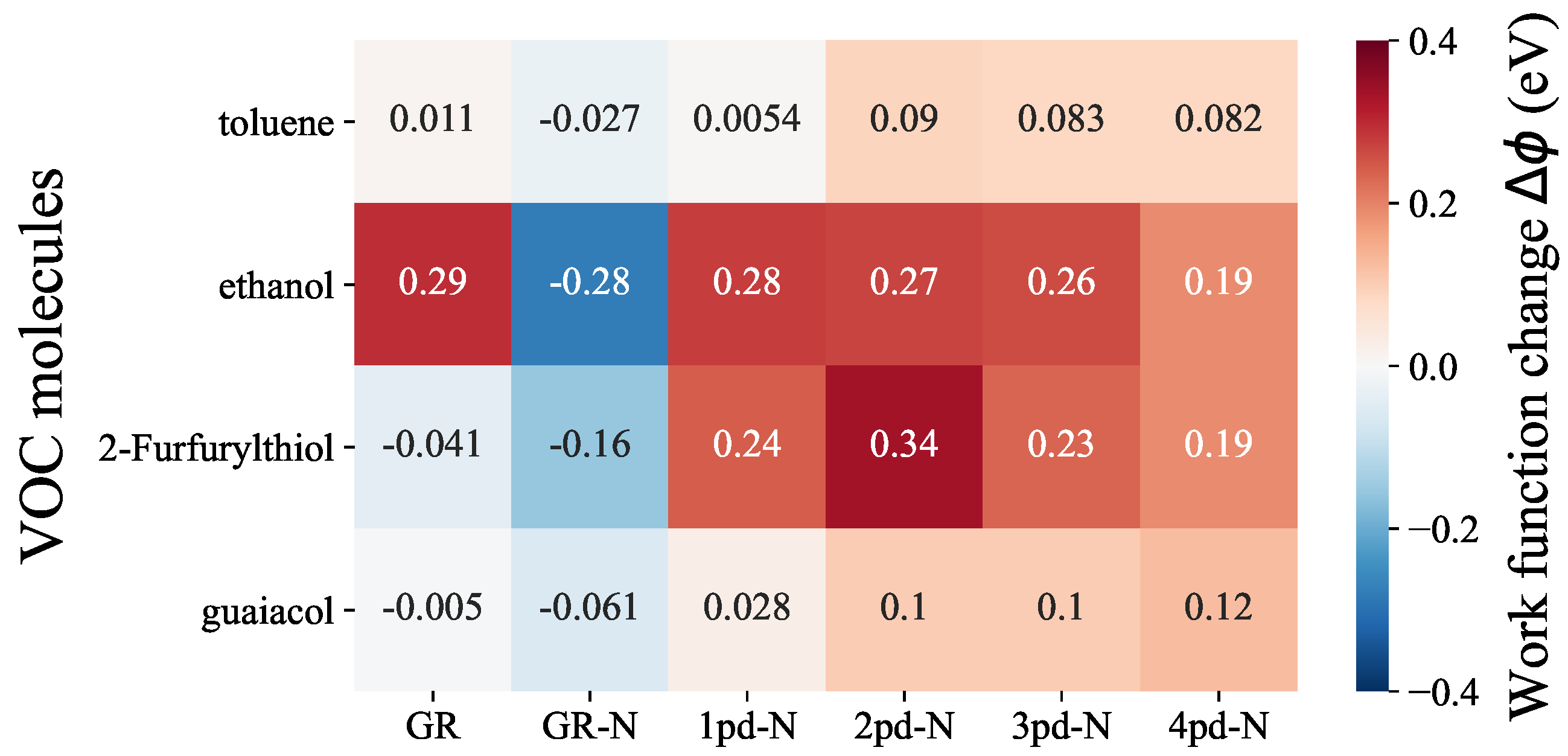
As depicted in Figure 2, within each molecule row, we can make comparisons regarding the sensitivity of each substrate to a specific VOC molecule, while within each column, the selectivity of a particular substrate towards various VOC molcules can be assessed. Upon examining the molecule rows, it becomes evident that toluene and guaiacol evoke rather smaller responses in all substrates, with values consistently falling below 0.1 eV compared to the other two, even though, there is still a subtle increase in values when transitioning from pristine graphene (GR) to N-doped graphene (GR-N) and multiple pd-N substrates. In contrast, ethanol and 2-Furfurylthiol exhibit overall higher responses compared to the other two odorants. In the ethanol row, the relatively large absolute values of can be attributed to the high electronegativity of the oxygen atom of ethanol, resulting in substantial charge transfer. Importantly, the values for ethanol are consistently large and nearly identical for all substrates, indicating that both graphene and N-doped graphene hold significant promise for the detection of ethanol. Regarding 2-Furfurylthiol adsorption, the richer variety of work function changes in this row suggests that the substrates demonstrate distinguishable responses. Notably, 2pd-N displays the most substantial of 0.34 eV, whereas GR exhibits almost no sensitivity to 2-Furfurylthiol, yielding a value of −0.041 eV. From a selectivity perspective (column-wise), GR-N and 2pd-N exhibit relatively superior discrimination capabilities towards ethanol and 2-Furfurylthiol. This is evident from the more pronounced differences in values in these columns compared to the toluene and guaiacol. Although the introduction of nitrogen into the substrates generally raises the values when compared to pure graphene, this does not necessarily enhance selectivity towards ethanol, 2-Furfurylthiol, toluene, or guaiacol. In fact, the stronger interaction between substrate and adsorbate, as reflected in the adsorption energy trend presented in Figure S5, was expected to lead to a stronger response, e.g., . However, either sensitivity or selectivity appears to deteriorate, particularly for 3pd-N and 4pd-N substrates, which does not satisfy our expectation regarding the enhancement by adding multiple pyrodinic-nitrogen atoms and enlarging the point defect concentration, as mentioned in the introduction.
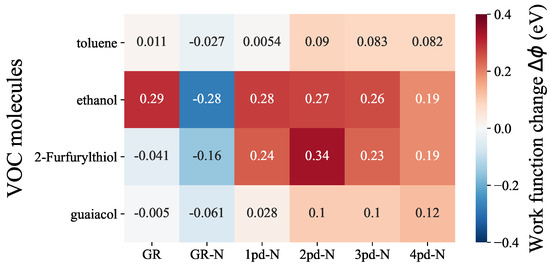
Figure 2.
Heatmap of work function change comparisons among different odorant–substrate combinations. The labeled number indicates the corresponding work function change value.
3.3. Charge Transfer vs. Work Function Change
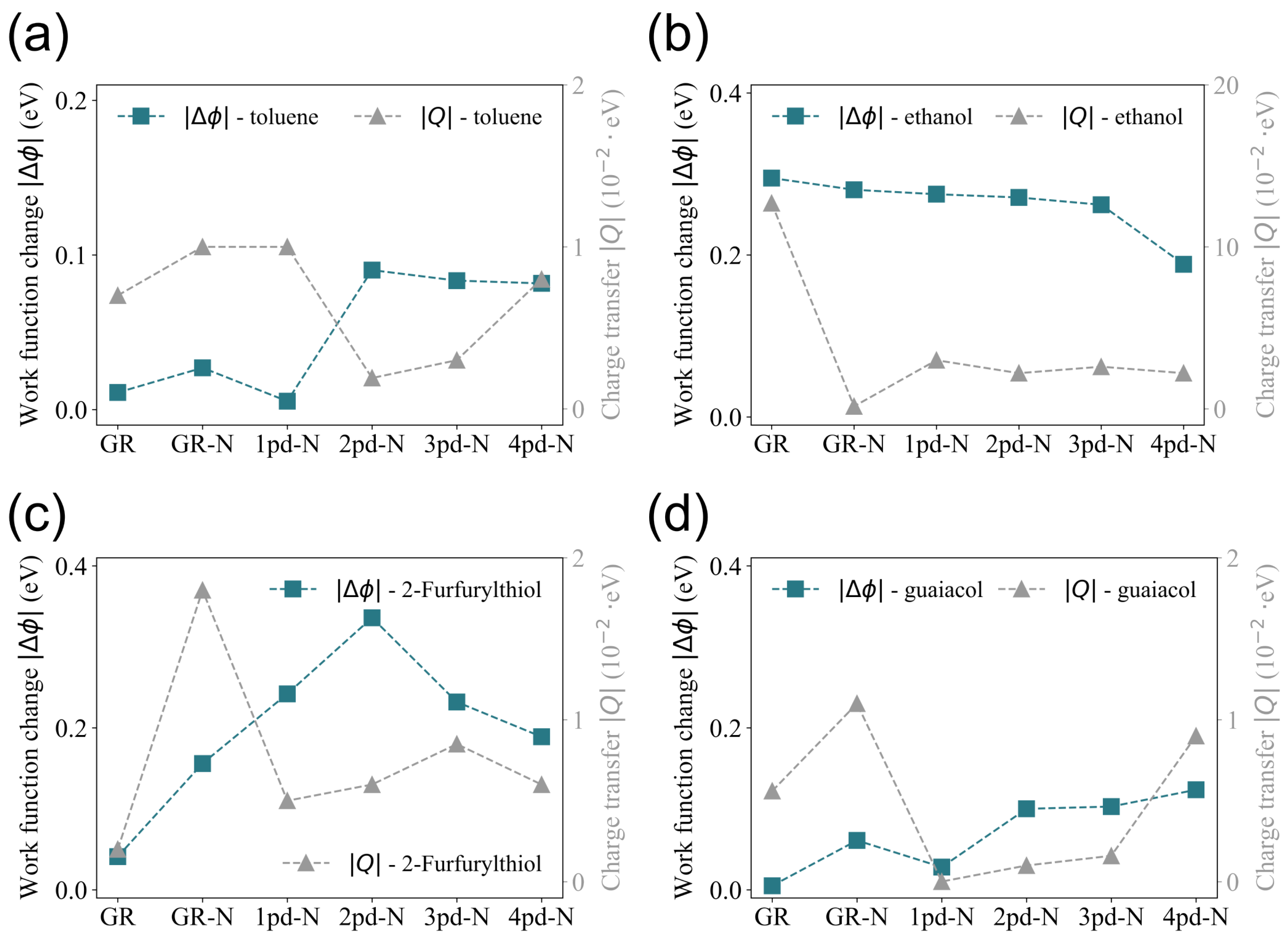
In order to obtain insights into the behavior of work function changes observed in Figure 2, the total net charge transfer Q is compared with the change in work function, since Q on the substrate is tightly correlated to . It is generally anticipated that the adsorption of molecules induces charge transfer as a result of the electronegativity difference between the adsorbate and adsorbent. Once the adsorbate donates electrons, the substrate likely needs less energy to spill out the electrons again; hence, the work function will decrease, leading to a negative , and vice versa. As a consequence, the charge transfer Q is anticipated to correlate directly with the magnitude of the change in the work function , implying that variations in should be mirrored by corresponding variations in Q.
However, this comparison is presented in Figure 3, which depicts an inconsistent trend between the variations in and Q across the different substrates (GR was taken as the reference point). This inconsistent correlation has also been observed in other studies [14]. For instance, while the change in work function and charge transfer for 2-Furfurylthiol on the GR, 1pd-N, 2pd-N, and 4pd-N substrates aligns with the above-mentioned expectations, discrepancies are observed for the GR-N and 3pd-N substrates. These anomalies suggest that the relationship between and Q is not universally correlated and might be influenced by additional factors.
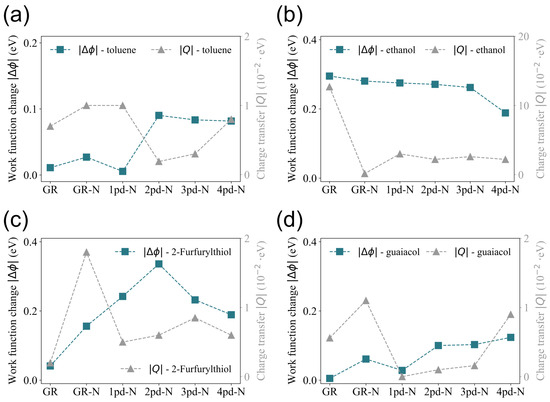
Figure 3.
Comparison of trends in the absolute value of work function change and charge transfer Q of the substrates towards the adsorption of (a) toluene, (b) ethanol, (c) 2-Furfurylthiol, and (d) guaiacol.
3.4. Surface Dipole Moment Change Validation
To further investigate the mechanisms underlying the observed behavior for N-doped graphene substrates, we carried out an analysis of surface dipole moment changes.
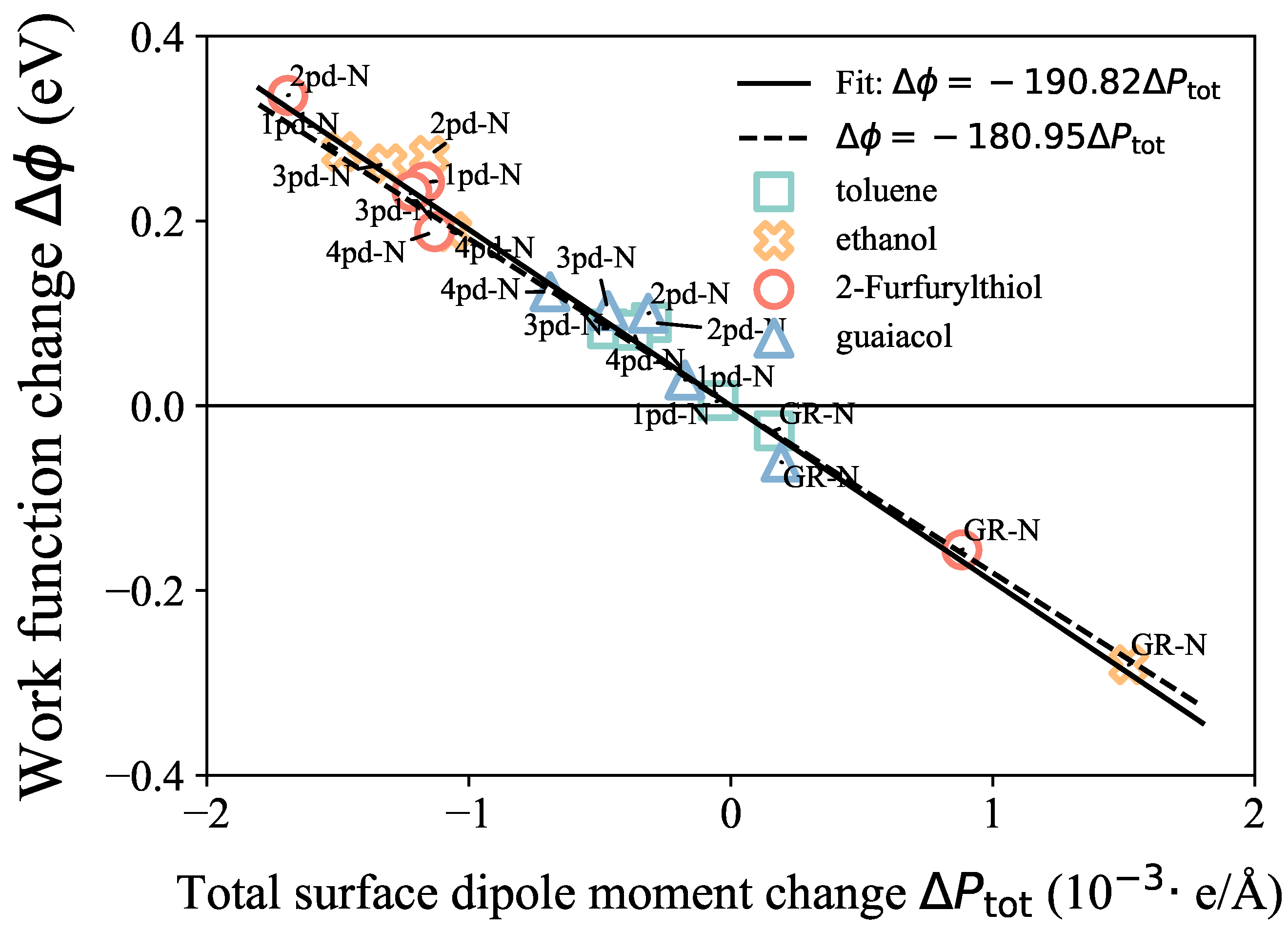
First, to validate the model on our systems, the linear correlation between the work function change and the surface dipole moment change is presented in Figure 4. The values were calculated by considering the difference in the total dipole moment between the adsorbate–substrate system and the isolated substrate, specifically . The proportionality coefficient obtained from our fitting result is −190.82 , as shown in Equation (2), which is in good agreement with the constant value of . These results indicate that this surface potential model is not only valid for chemisorbed systems of bulk, as Leung et al. [21] and Khazaei [23] reported, but is also applicable to the physisorbed 2D systems in this work due to the negligible shifts in the Fermi level for both kinds of systems. Based on this linear relationship, the trend of could approximate the trend of the work function change , and we could also obtain insights into the possible reasons for the trend through the components’ competition.
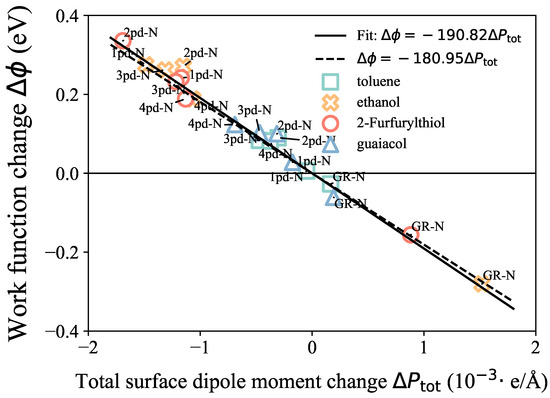
Figure 4.
Linear correlation between work function change (y-axis) and surface dipole moment change (x-axis). The straight solid and dashed lines present the fitted and referred linear correlation, respectively.
3.5. Surface Dipole Moment Change Decomposition
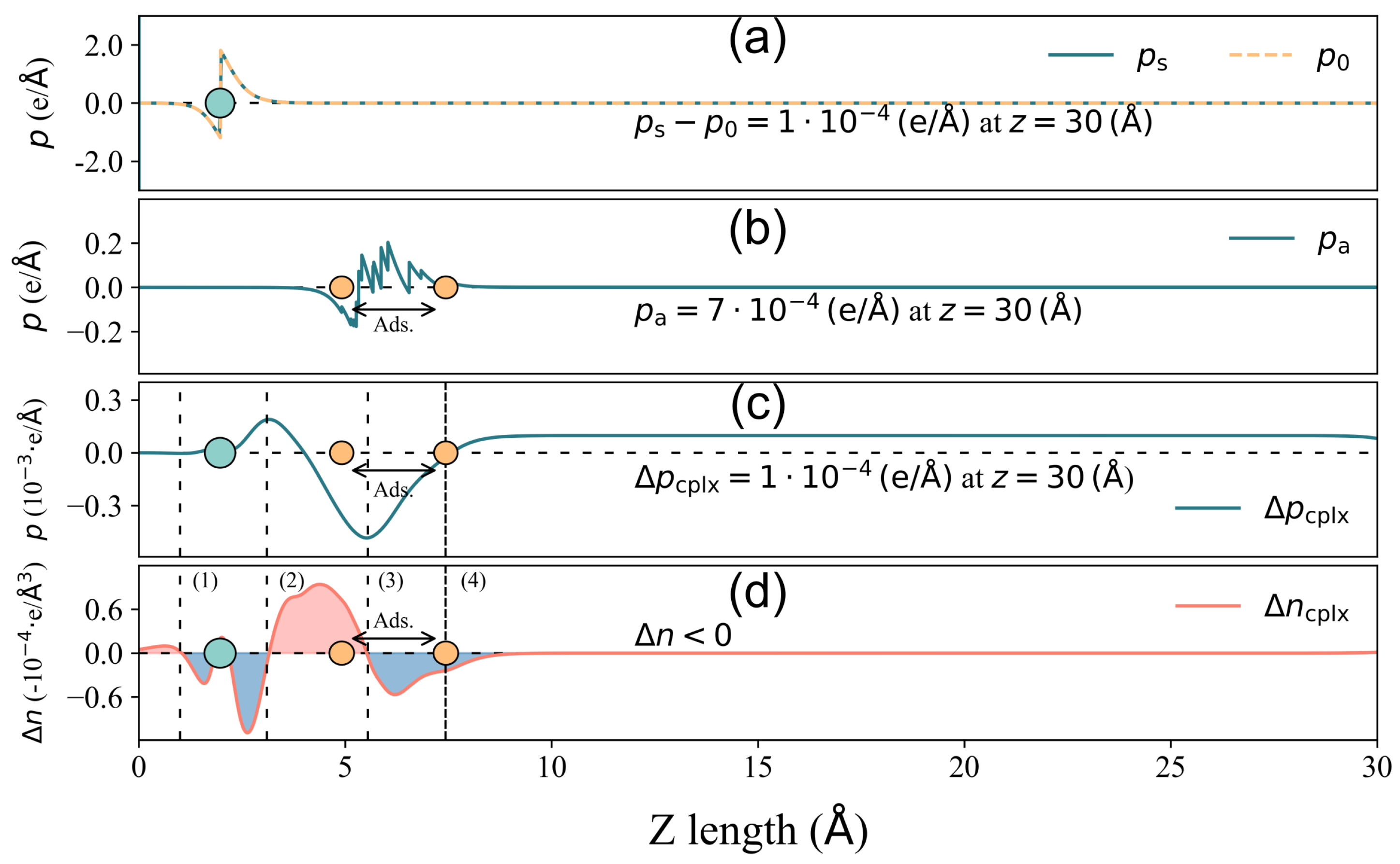
Now, to further investigate the correlation behavior between Q and in Figure 3, we take 2-Furfurylthiol adsorbed on GR-N and 4pd-N as typical examples, which have and Q pairs of −0.156 eV, 0.018 e and 0.189 eV, −0.006 e, respectively. Their corresponding descriptions of components , , and for total surface dipole moment change decomposition are displayed in Figure 5 and Figure 6.
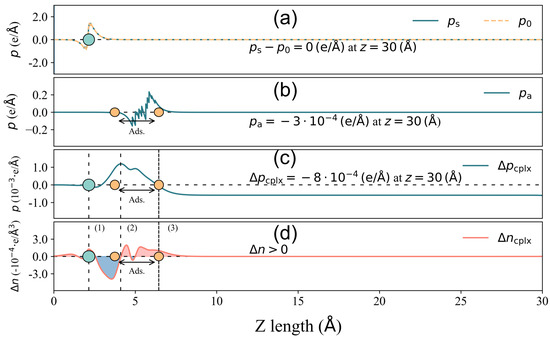
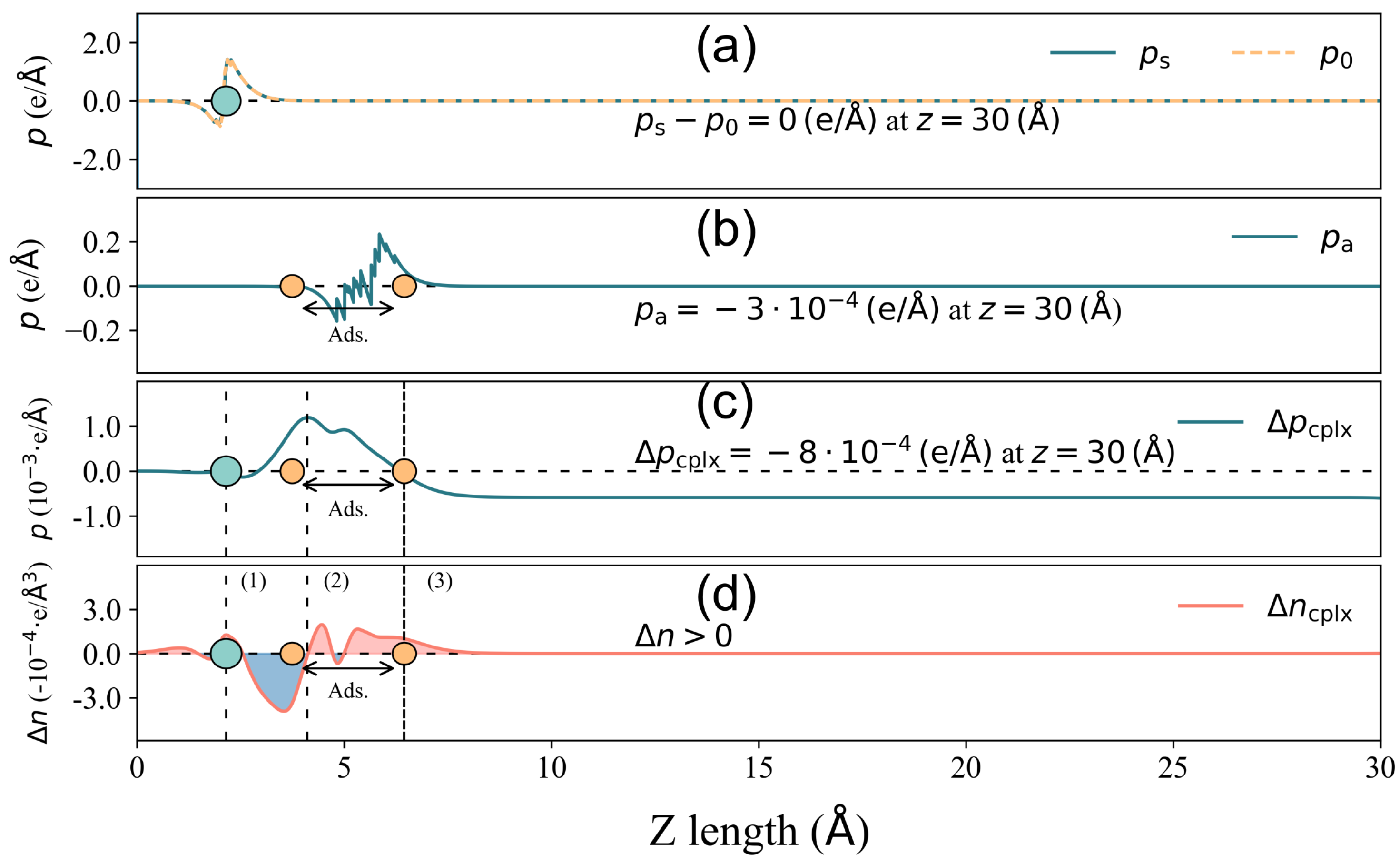
Figure 5.
Total surface dipole moment change decomposition for 2-Furfurylthiol adsorbed on 4pd-N substrate. (a) Components of surface deformation − . (b) Surface dipole moment resulting from the deformation of the VOC molecule upon adsorption . (c) owing to the spatial charge redistribution in the complex adsorbate–adsorbent system. (d) Charge density difference distribution of the x–y planar average along the z direction. The grey circle is the location of the substrate, and the area between the two orange circles is the adsorbate location, where the lowest and highest atoms of the VOC molecule are denoted by the two atoms. In (c,d), the curve is divided into several regions: (1), (3) blue areas indicating electron depletion and (2) red area indicating electron accumulation. The dense dashed line denotes the highest position for , and the sign of is labeled in (d).
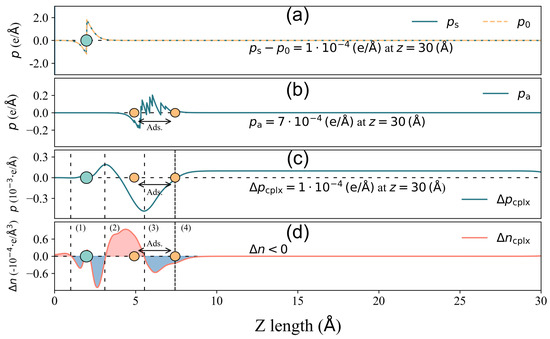
Figure 6.
The same as Figure 5, but for GR-N.
In the case of 4pd-N, as depicted in Figure 5, the electron accumulation/depletion in regions (1) and (3)/(2) results in a tiny total net charge transfer Q of e from the substrate to the odorant. The charge density changes in regions (1) and (2) effectively compensate for each other, leading to a zero value of until the onset of region (3) in panel (c). The charge redistribution in the outer layer of region (3) contributes significantly to the final value of , amounting to , which is considerably larger than the other two components ( and ). Hence, in this case, it is the charge redistribution mechanism that primarily drives the surface dipole moment change under a small Q. In the GR-N case, as illustrated in Figure 6, electrons accumulate/deplete in regions (2)/(1), (3), and (4). Similar to the 4pd-N case, charge redistribution (depletion) within the outer layer of the adsorbate in region (4) leads to , pointing out to the vacuum. However, the spatial charge redistribution results in a greater net charge transfer Q of from the adsorbate to the adsorbent, which is three times higher than the Q observed in the 4pd-N case. Nonetheless, the pure adsorbate-induced dipole moment, , dominates the change in comparison to .
From both cases, it is shown that the total net charge transfer Q does not necessarily correlate with the total surface dipole moment change (and hence the work function change ).
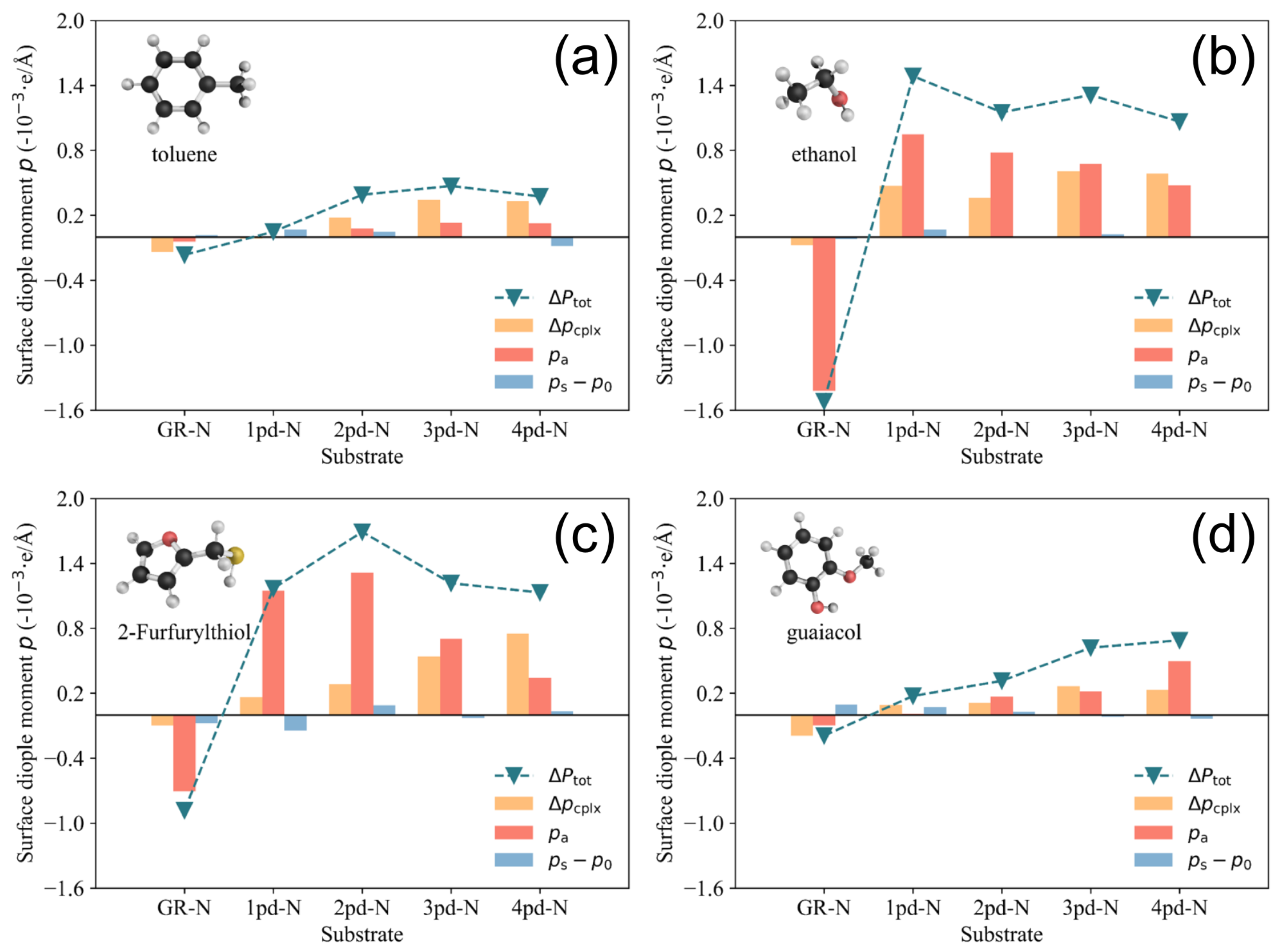
Furthermore, there is almost no contribution from , indicating that the adsorption of 2-Furfurylthiol induces negligible substrate deformation. As for other cases, the mechanism of charge redistribution could be more complicated, but the competition between and remains, which results in the inconsistent scaling trend between charge transfer and total surface dipole moment change (work function change ). In order to make an overall comparison of the surface dipole moment change between various cases, the value of and its corresponding components are presented in Figure 7. To make a direct comparison between and , the minus sign is assigned to the unit in Figure 7.
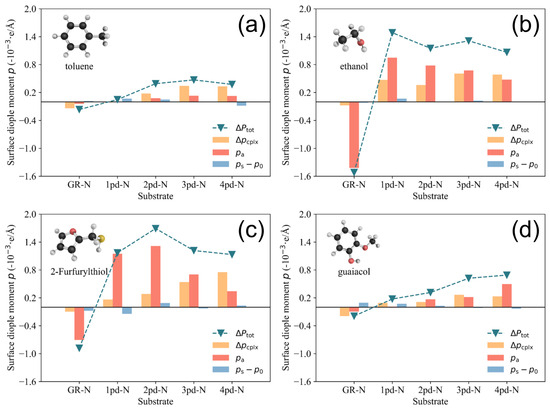
Figure 7.
Histograms of total surface dipole moment change decomposition for (a) toluene, (b) ethanol, (c) 2-Furfurylthiol, and (d) guaiacol. The variation in the negative total surface dipole moment change and the distribution of the components , , and are presented.
As illustrated in Figure 7, the component (represented by the blue bar), which characterizes the surface dipole moment change induced by surface deformation, is not as sensitive as the other two components, which could typically yield large-value increments by changing the substrates. It is worth noting that the positive and components observed in the GR-N columns in all four subfigures lead to a positive (and hence negative in Figure 2) upon the adsorption of the odorants. This is in contrast to other pyridinic-N-doped substrates, as discussed in Figure 5, which yield negative values for these surface dipole moment components and finally result in a positive . A notable general trend is the continuous increase in (depicted by the orange bar) as a function of the number of pyridinic-N atoms for the four odorants. This phenomenon can be attributed to the growing number of pyridinic-N atoms, which introduces additional dangling bonds due to the presence of an unpaired electron localized at these pyridinic-N sites [42]. These dangling bonds facilitate substantial charge rearrangement, resulting in the amplification of . However, it is important to note that the increasing value does not always lead to a monotonic increase in the change of the total surface dipole moment, . In the cases of 2-Furfurylthiol and ethanol adsorption on 3pd-N and 4pd-N substrates, as shown in (c) and (d) of Figure 7, the components decrease, while increases. This phenomenon might explain why the work function change for the adsorption of 2-Furfurylthiol and ethanol on the 3pd-N and 4pd-N substrates does not exhibit a continuous increase as more pyridinic-N atoms are added.
In Figure 2, we have already seen the difference for toluene/guaiacol and 2-Furfuryl-thiol/ethanol. As for toluene, it is a weakly polarized molecule with the smallest intrinsic dipole moment; therefore, the charge rearrangements resulted in varying only within a limited range despite the increasing enhancement of nitrogen doping. Similarly, even though guaiacol has the largest in-plane intrinsic dipole moment among the four molecules, it contributes only to a limited extent to the spatial charge redistribution, as the phenyl ring of guaiacol, as well as toluene, is positioned horizontally on the substrates to maximize the van der Waals interaction and minimize the adsorption energy .
Additionally, the high electronegative oxygen atoms of guaiacol do not contribute to the charge redistribution. The reason for this could be that the phenol hydrogen atom and oxygen atom adjacent to the methyl group already construct a robust intramolecular hydrogen bond [43], which leads to a weaker interaction with the substrate. Therefore, though the sensitivity of guaiacol is slightly greater than that of toluene due to the increase in the component, their tiny and values limit the corresponding (hence ) within a small range. In contrast, there is a significantly larger increase in and for the larger polarized 2-Furfurylthiol and ethanol compared to toluene and guaiacol, which could be attributed to the presence of strong polar functional groups on the odorants such as S and −OH. However, for these VOCs molcules, as shown in (b) and (c) in Figure 7, there is a drop in the component as the pyridinic-N atoms number increases, especially on 3pd-N and 4pd-N substrates. Even though the remains increasing, the (hence the ) becomes unchanged due to the decline in . This might explain why the selectivity ultimately deteriorates while continuing to induce the pyrodinic-N atoms into substrates, as reflected in the work function change shown in Figure 2.
3.6. Design Principle of for VOC Sensing
By validating the linear relationship between and and understanding the underlying mechanism, we propose possible design principles of sensing materials and VOC pairs with their expected values.
- : One could induce multiple highly electronegative atoms into the adsorption site on the substrate, since it will bring more dangling bonds and yield charge redistribution, and vice versa, as reflected in the values presented in Figure 7.
- : While the out-of-plane dipole moment of VOC molecules can be tuned, adjusting the sign of by substituting different types of dopant atoms (e.g., pyridinic-N and pyrrolic-N) could further enhance the directional contrast of the difference.
- : Increasing the value is not recommended, even though this term also has a large influence on the value, as large deformation of the substrate is detrimental to the stability and durability of sensing materials.
4. Conclusions
In this work, the work function change (WFC) is calculated as a electronic response for four VOC molcules—toluene, ethanol, 2-Furfurylthiol, and guaiacol—physisorbed onto pristine and N-doped graphene substrates using DFT. 2-Furfurylthiol and ethanol induced a significantly larger WFC than toluene and guaiacol. Among the N-doped substrates, GR-N and 2pd-N exhibited good discrimination toward sensing 2-Furfurylthiol and ethanol. However, the sensitivity was not further enhanced by introducing more pyridinic-N atoms into the substrate, especially for 3pd-N and 4pd-N. To explain this, the surface dipole moment analysis for the 2-Furfurylthiol and ethanol cases showed that the charge redistribution scale (characterized by the ) increases, while the induced adsorbate dipole () drops with respect to the increasing number of pyridinic-N atoms. This gives rise to the non-increasing sensitivity of 2-Furfurylthiol and ethanol on the 3pd-N and 4pd-N substrates. This shows that the competition between the surface dipole moment components leads to the work function change behavior on our N-doped graphene samples. Finally, based on this knowledge, we proposed possible design principles of WFC for VOCs interacting with N-doped graphene by tuning the surface dipole moment components’ values. These computational findings suggest a new way to investigate the work function change behavior for 2D materials and hence offer principles for the rational design of materials and their responses to VOCs.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/nano14221778/s1, Table S1: Adsorption energy for all adsorption cases. The lowest is highlighted in bold. The adsorption on graphene serves as a reference case. Therefore, only four initial configurations were taken into account. Figure S1: The most energy-favorable adsorption configurations for toluene adsorption. Figure S2: The most energy-favorable adsorption configurations for ethanol adsorption. Figure S3: The most energy-favorable adsorption configurations for 2-Furfurylthiol adsorption. Figure S4: The most energy-favorable adsorption configurations for guaiacol adsorption. Table S2: Adsorption energy and Charge transfer Q comparison for the odor molecules with the most stable configurations over pristine graphene, N-gra, 1pd-N, 2pd-N, 3pd-N, and 4pd-N substrates. Positive values of charge transfer indicate electron transfer from the molecule to the substrate, while negative values indicate electron transfer from the substrate to the molecule. Figure S5: Adsorption energy comparison for the odorants adsorption on different substrates is shown.
Author Contributions
L.C.: Methodology, investigation, visualization, data curation, writing—original draft. D.B.: Investigation, methodology, validation, software, Writing—review and editing. A.R.: Conceptualization, methodology, validation, supervision. A.D.: Conceptualization, investigation, methodology, supervision, writing—review and editing. R.B.: Conceptualization, software, supervision. R.G.: Project administration, supervision, writing—review and editing. M.K.: Conceptualization, methodology. G.C.: Project administration, funding acquisition, supervision, writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the European Union Horizon Europe EIC Pathfinder Open project, “Smart Electronic Olfaction for Body Odor Diagnostics” (SMELLODI, grant agreement ID: 101046369); the Volkswagen Foundation for the Qualification Concept “Olfactorial Perceptronics” (Project ID 9B396); and the DFG Collaborative Research Centers project, “Understanding and Predicting the Formation of Synthetic 2D Materials: From Molecules to Material” (CRC 1415 no. 417590517).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgments
We acknowledge the Center for Information Services and High Performance Computing (ZIH) at TU Dresden for providing the computational resources.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Liu, X.; Cheng, S.; Liu, H.; Hu, S.; Zhang, D.; Ning, H. A survey on gas sensing technology. Sensors 2012, 12, 9635–9665. [Google Scholar] [CrossRef] [PubMed]
- Pöschl, U.; Shiraiwa, M. Multiphase chemistry at the atmosphere–biosphere interface influencing climate and public health in the anthropocene. Chem. Rev. 2015, 115, 4440–4475. [Google Scholar]
- He, C.; Cheng, J.; Zhang, X.; Douthwaite, M.; Pattisson, S.; Hao, Z. Recent advances in the catalytic oxidation of volatile organic compounds: A review based on pollutant sorts and sources. Chem. Rev. 2019, 119, 4471–4568. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Jiang, C.; Wei, S.h. Gas sensing in 2D materials. Appl. Phys. Rev. 2017, 4, 021304. [Google Scholar] [CrossRef]
- Donarelli, M.; Ottaviano, L. 2D materials for gas sensing applications: A review on graphene oxide, MoS2, WS2 and phosphorene. Sensors 2018, 18, 3638. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Jacobs, R.; Ma, T.; Chen, D.; Booske, J.; Morgan, D. Work Function: Fundamentals, Measurement, Calculation, Engineering, and Applications. Phys. Rev. Appl. 2023, 19, 037001. [Google Scholar] [CrossRef]
- Gurlo, A.; Sahm, M.; Oprea, A.; Barsan, N.; Weimar, U. A p- to n-transition on α-Fe2O3-based thick film sensors studied by conductance and work function change measurements. Sens. Actuators B 2004, 102, 291–298. [Google Scholar] [CrossRef]
- Chou, P.C.; Chen, H.I.; Liu, I.P.; Chen, W.C.; Chen, C.C.; Liou, J.K.; Lai, C.J.; Liu, W.C. On a Schottky diode-type hydrogen sensor with pyramid-like Pd nanostructures. Int. J. Hydrogen Energy 2015, 40, 9006–9012. [Google Scholar] [CrossRef]
- Kumar, A. Palladium-based trench gate MOSFET for highly sensitive hydrogen gas sensor. Mater. Sci. Semicond. Process. 2020, 120, 105274. [Google Scholar] [CrossRef]
- Pour, G.B.; Aval, L.F.; Eslami, S. Sensitive capacitive-type hydrogen sensor based on Ni thin film in different hydrogen concentrations. Curr. Nanosci. 2018, 14, 136–142. [Google Scholar] [CrossRef]
- Sahm, T.; Gurlo, A.; Bârsan, N.; Weimar, U. Basics of oxygen and SnO2 interaction; work function change and conductivity measurements. Sens. Actuators B 2006, 118, 78–83. [Google Scholar] [CrossRef]
- Meng, J.; Li, Z. Schottky-Contacted Nanowire Sensors. Adv. Mater. 2020, 32, 2000130. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Wu, J.; Yu, Y.X. DFT exploration of sensor performances of two-dimensional WO3 to ten small gases in terms of work function and band gap changes and I-V responses. Appl. Surf. Sci. 2021, 546, 149104. [Google Scholar] [CrossRef]
- Nath, U.; Sarma, M. Pyridinic Dominance N-Doped Graphene: A Potential Material for SO2 Gas Detection. J. Phys. Chem. A 2023, 127, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Cid, B.J.; Santana, J.E.; Arellano, L.G.; Miranda, Á.; Pérez-Figueroa, S.E.; Iturrios, M.I.; Pérez, L.A.; Cruz-Irisson, M. Metal-decorated siligene as work function type sensor for NH3 detection: A DFT approach. Appl. Surf. Sci. 2023, 610, 155541. [Google Scholar] [CrossRef]
- Kalwar, B.A.; Fangzong, W.; Soomro, A.M.; Naich, M.R.; Saeed, M.H.; Ahmed, I. Highly sensitive work function type room temperature gas sensor based on Ti doped hBN monolayer for sensing CO2, CO, H2S, HF and NO. A DFT study. RSC Adv. 2022, 12, 34185–34199. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Jia, P.; Peng, X.; Li, P. Adsorption and sensing of CO and C2H2 by S-defected SnS2 monolayer for DGA in transformer oil: A DFT study. Mater. Chem. Phys. 2020, 249, 123006. [Google Scholar] [CrossRef]
- Ni, J.; Wang, W.; Quintana, M.; Jia, F.; Song, S. Adsorption of small gas molecules on strained monolayer WSe2 doped with Pd, Ag, Au, and Pt: A computational investigation. Appl. Surf. Sci. 2020, 514, 145911. [Google Scholar] [CrossRef]
- Reji, R.P.; Balaji, S.K.C.; Sivalingam, Y.; Kawazoe, Y.; Velappa Jayaraman, S. First-principles density functional theory calculations on the potential of Sc2CO2 MXene nanosheets as a dual-mode sensor for detection of volatile organic compounds in exhaled human breath. ACS Appl. Nano Mater. 2023, 6, 5345–5356. [Google Scholar] [CrossRef]
- Lin, L.; Feng, Z.; Dong, Z.; Hu, C.; Han, L.; Tao, H. DFT study on the adsorption of CO, NO2, SO2 and NH3 by Te vacancy and metal atom doped MoTe2 monolayers. Phys. E 2023, 145, 115489. [Google Scholar] [CrossRef]
- Leung, C.; Kao, L.; Su, S.; Feng, J.; Chan, T. Relationship between surface dipole, work function and charge transfer: Some exceptions to an established rule. Phys. Rev. B Condens. Matter Mater. Phys. 2003, 68, 195408. [Google Scholar] [CrossRef]
- Li, H.; Zhang, X.; Xu, Y.; Lei, G.; Tsumori, K.; Isobe, M.; Shimizu, A.; Cui, Z.; Zhu, Y.; Hu, J.; et al. Theoretical calculation of cesium deposition and co-deposition with electronegative elements on the plasma grid in negative ion sources. Nucl. Mater. Energy 2023, 34, 101334. [Google Scholar] [CrossRef]
- Khazaei, M.; Arai, M.; Sasaki, T.; Ranjbar, A.; Liang, Y.; Yunoki, S. OH-terminated two-dimensional transition metal carbides and nitrides as ultralow work function materials. Phys. Rev. B Condens. Matter 2015, 92, 075411. [Google Scholar] [CrossRef]
- Gong, C.; Colombo, L.; Wallace, R.M.; Cho, K. The Unusual Mechanism of Partial Fermi Level Pinning at Metal–MoS2 Interfaces. Nano Lett. 2014, 14, 1714–1720. [Google Scholar] [CrossRef] [PubMed]
- Basera, P.; Traoré, B.; Even, J.; Katan, C. Interfacial engineering to modulate surface dipoles, work functions and dielectric confinement of halide perovskites. Nanoscale 2023, 15, 11884–11897. [Google Scholar] [CrossRef]
- Chen, W.Y.; Li, L.; Huang, T.; Yang, Z.X.; Zhang, T.; Huang, G.F.; Hu, W.; Huang, W.Q. Extending Schottky–Mott rule to van der Waals heterostructures of 2D Janus materials: Influence of intrinsic dipoles. Appl. Phys. Lett. 2023, 123, 171601. [Google Scholar] [CrossRef]
- Traoré, B.; Basera, P.; Ramadan, A.J.; Snaith, H.J.; Katan, C.; Even, J. A Theoretical Framework for Microscopic Surface and Interface Dipoles, Work Functions, and Valence Band Alignments in 2D and 3D Halide Perovskite Heterostructures. ACS Energy Lett. 2021, 7, 349–357. [Google Scholar] [CrossRef]
- Pappas, G.P.; Herbert, R.J.; Henderson, W.; Koenig, J.; Stover, B.; Barnhart, S. The respiratory effects of volatile organic compounds. Int. J. Occup. Environ. Health 2000, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dorfner, R.; Ferge, T.; Kettrup, A.; Zimmermann, R.; Yeretzian, C. Real-time monitoring of 4-vinylguaiacol, guaiacol, and phenol during coffee roasting by resonant laser ionization time-of-flight mass spectrometry. J. Agric. Food Chem. 2003, 51, 5768–5773. [Google Scholar] [CrossRef]
- Liu, X.Y.; Zhang, J.M.; Xu, K.W.; Ji, V. Improving SO2 gas sensing properties of graphene by introducing dopant and defect: A first-principles study. Appl. Surf. Sci. 2014, 313, 405–410. [Google Scholar] [CrossRef]
- Tit, N.; Said, K.; Mahmoud, N.M.; Kouser, S.; Yamani, Z.H. Ab-initio investigation of adsorption of CO and CO2 molecules on graphene: Role of intrinsic defects on gas sensing. Appl. Surf. Sci. 2017, 394, 219–230. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Condens. Matter Phys. 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Condens. Matter Phys. 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Baseggio, O.; Bonfà, P.; Brunato, D.; Car, R.; Carnimeo, I.; Cavazzoni, C.; Gironcoli, S.D.; Delugas, P.; Ruffino, F.F.; et al. Quantum ESPRESSO toward the exascale. J. Condens. Matter Phys. 2020, 152, 154105. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B Condens. Matter 1996, 54, 16533. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Sohier, T.; Calandra, M.; Mauri, F. Density functional perturbation theory for gated two-dimensional heterostructures: Theoretical developments and application to flexural phonons in graphene. Phys. Rev. B Condens. Matter 2017, 96, 075448. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Itoh, T. Molecular structure of methylamine. J. Phys. Soc. Jpn. 1956, 11, 264–271. [Google Scholar] [CrossRef]
- Dzade, N.Y.; Roldan, A.; De Leeuw, N.H. Adsorption of methylamine on mackinawite (FeS) surfaces: A density functional theory study. J. Chem. Phys. 2013, 139, 124708. [Google Scholar] [CrossRef]
- Iijima, T.; Jimbo, H.; Taguchi, M. The molecular structure of methylamine in the vapour phase. J. Mol. Struct. 1986, 144, 381–383. [Google Scholar] [CrossRef]
- Yutomo, E.B.; Noor, F.A.; Winata, T. Effect of the number of nitrogen dopants on the electronic and magnetic properties of graphitic and pyridinic N-doped graphene–A density-functional study. RSC Adv. 2021, 11, 18371–18380. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Wang, K.; Yang, Z.; Wu, S.; Han, D. Quantum chemical study on the ozonolysis mechanism of guaiacol and the structure-reactivity relationship of phenols with hydroxyl, methoxy, and methyl substituents. Chem. Eng. J. 2021, 420, 127629. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).