

Insoluble Network Skeleton and Soluble Components of Nylon 6,6-Sputtered Nanoparticles: Insights from Liquid-State and Solid-State NMR Analysis

 ,
,
Abstract

1. Introduction
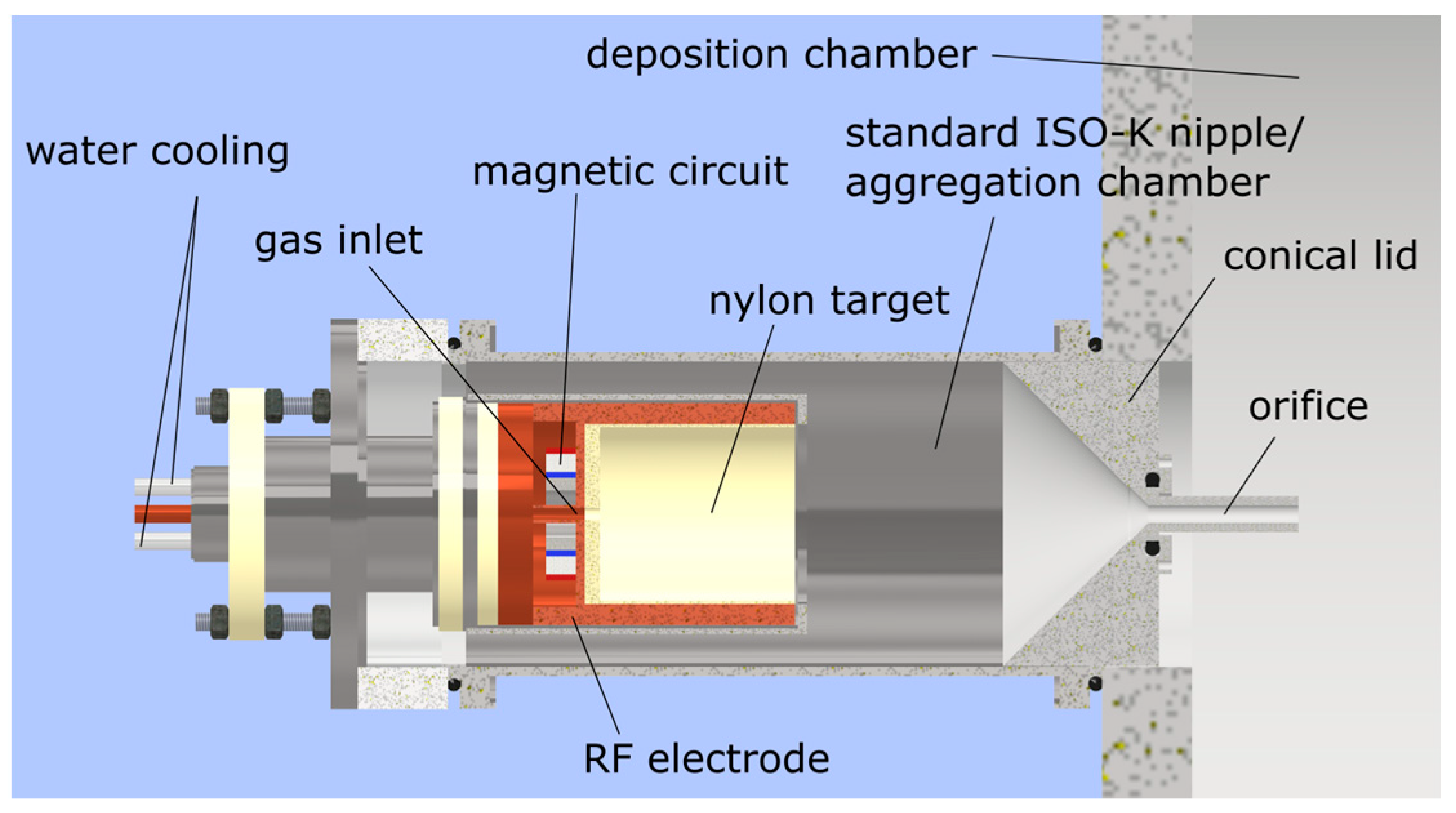
2. Materials and Methods
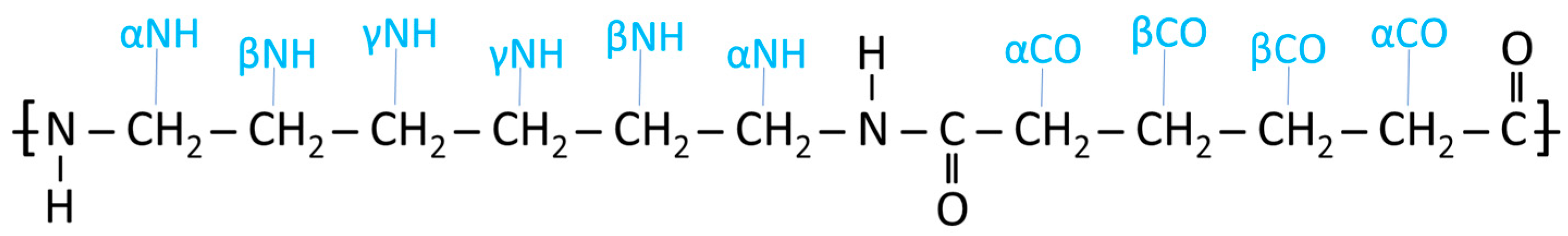
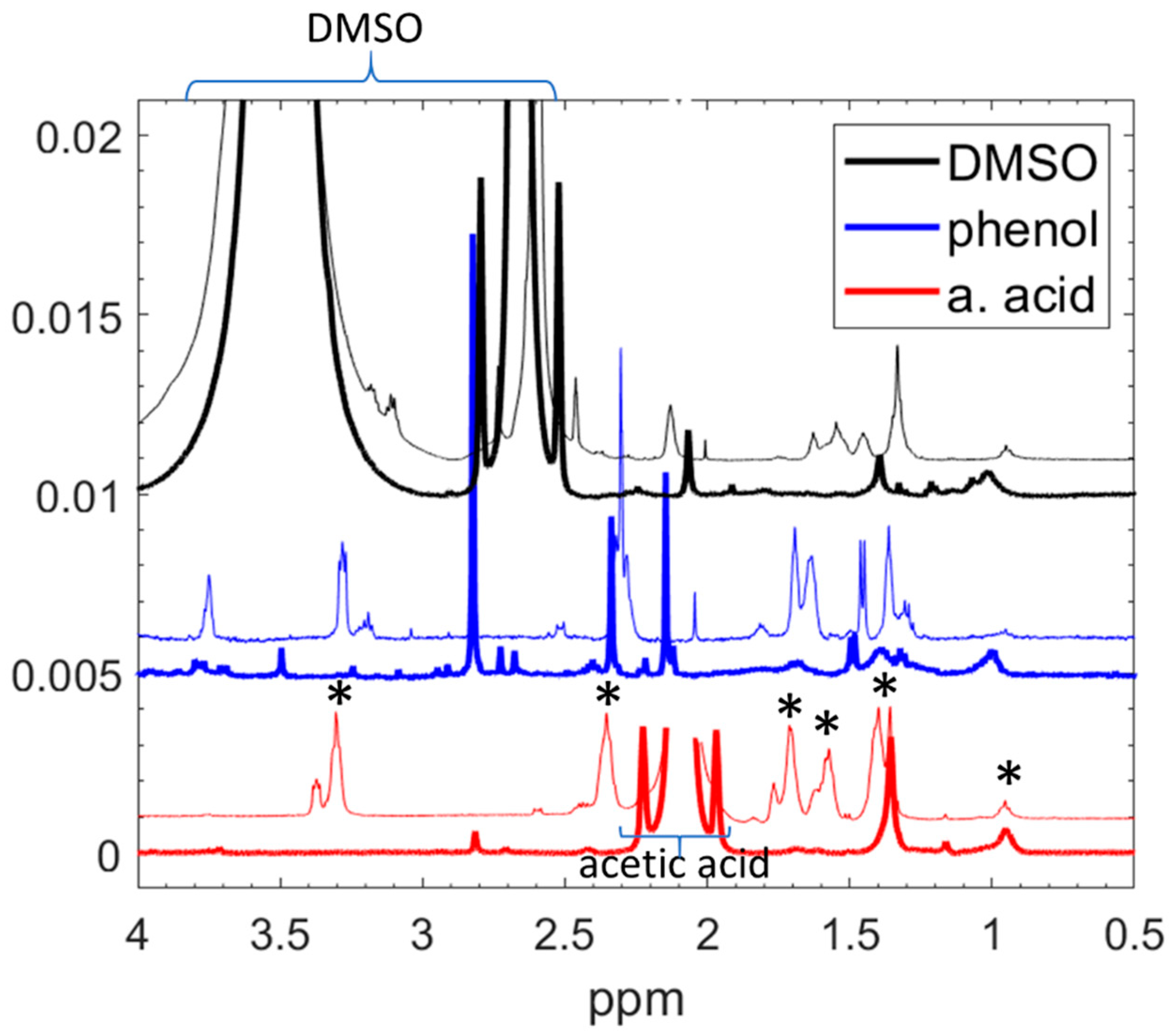
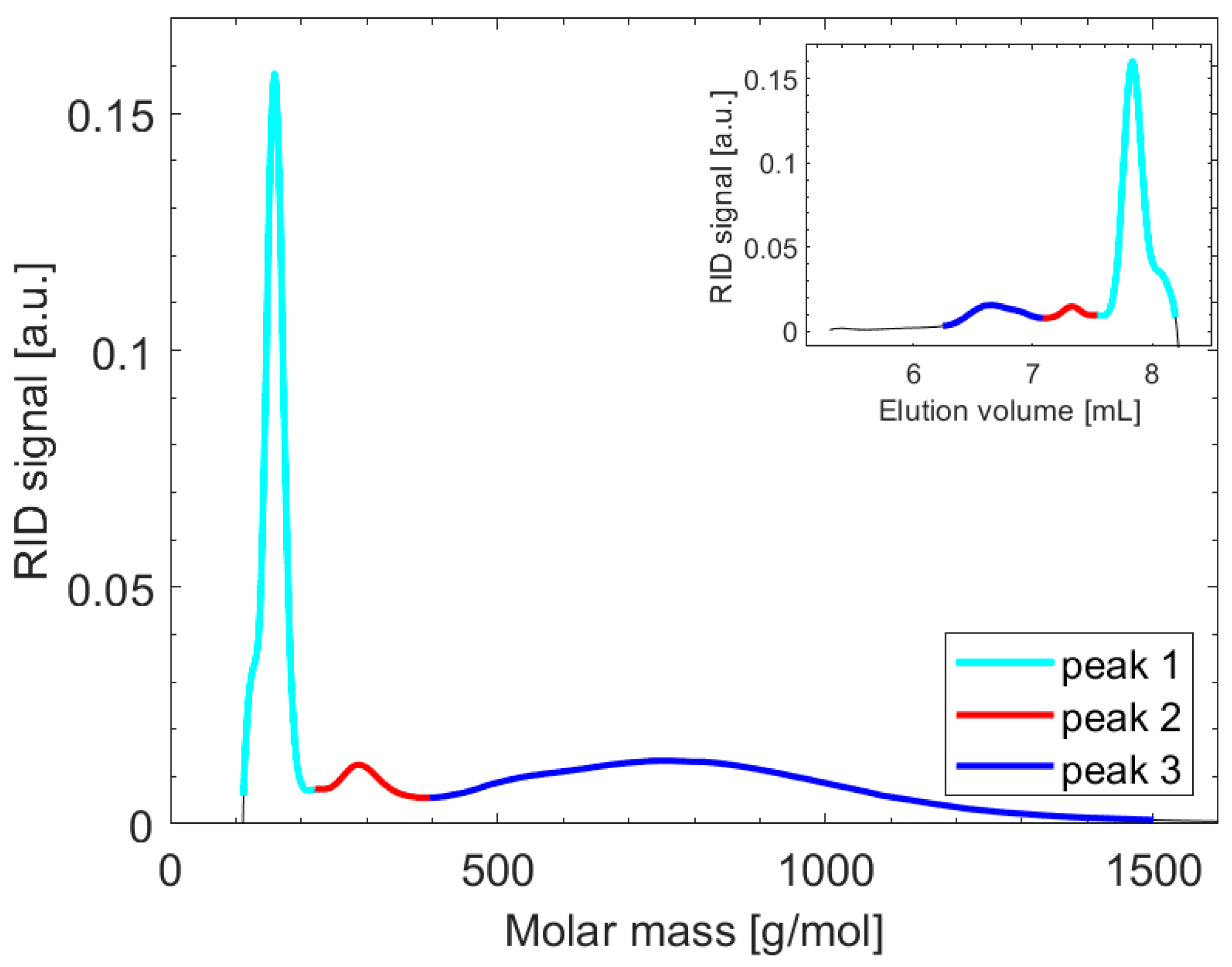
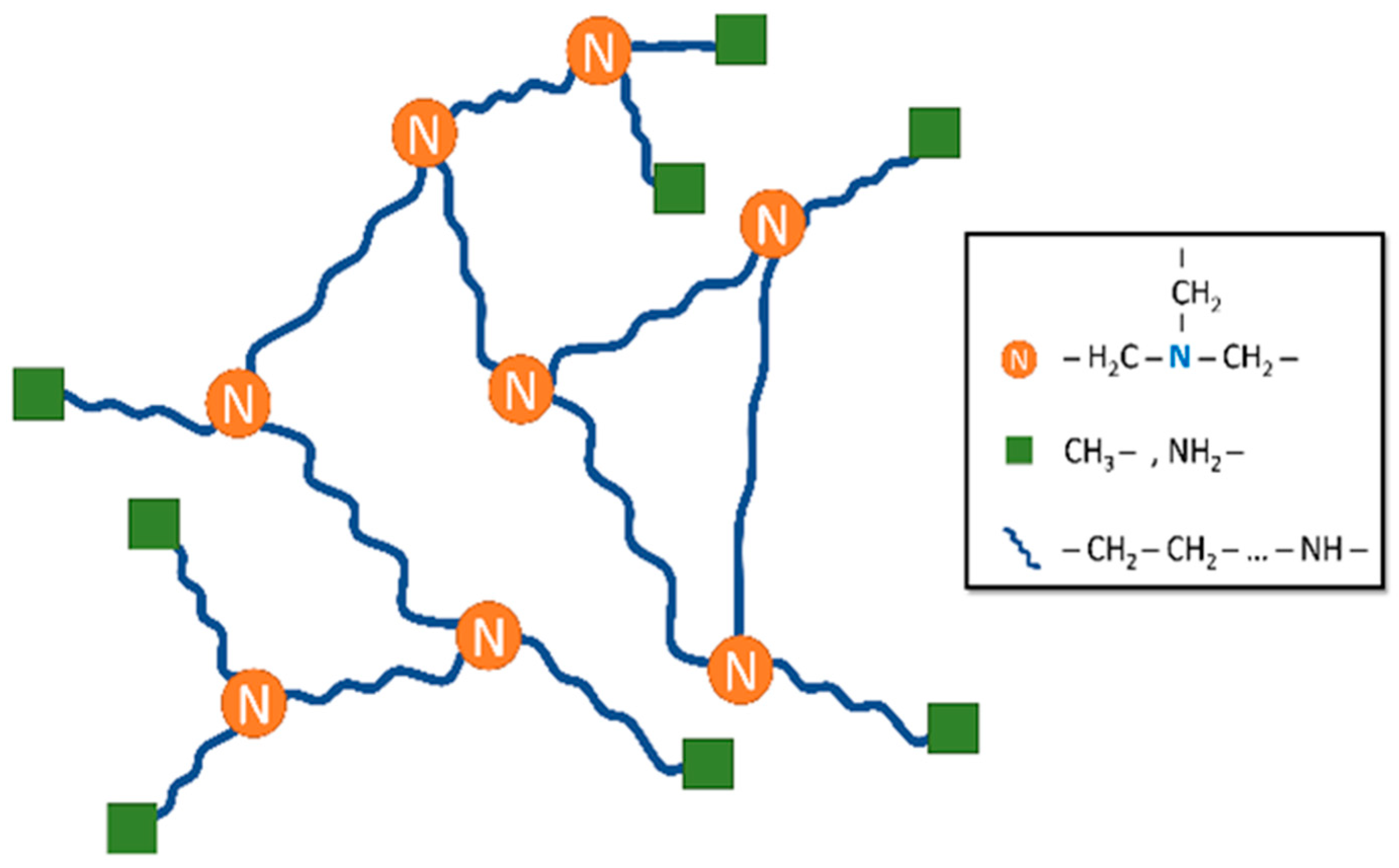
3. Results and Discussion
- The elemental composition of the source nylon 6,6, composed of linear chains, obtained using XPS is 75.0% carbon, 12.5% oxygen, and 12.5% nitrogen. The NPs exhibited a comparable composition, although they underwent a slight denitrogenation. They comprise 81% carbon, 12% oxygen, and 7% nitrogen.
- Top of Form
- The high-resolution XPS of the original nylon 6,6 showed the following bonds: primarily C–C, C–H with some C–N, and N–C = O. The C1s spectrum of the nylon-sputtered NPs was similar but slightly wider and showed the contribution of unsaturated carbon (C = C, C ≡ C) and C = N, C ≡ N, and C–O species, which are not present in the original polymer. The FTIR spectrum supported these findings, showing O-H and CH3 groups. The broadening of the peaks suggested a random structure neighbouring the groups corresponding to the observed peaks.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Haberland, H.; Karrais, M.; Mall, M. A new type of cluster and cluster ion source. Z. Für Phys. D At. Mol. Clust. 1991, 20, 413–415. [Google Scholar] [CrossRef]
- Haberland, H.; Karrais, M.; Mall, M.; Thurner, Y. Thin films from energetic cluster impact: A feasibility study. J. Vac. Sci. Technol. A Vac. Surf. Film. 1992, 10, 3266–3271. [Google Scholar] [CrossRef]
- Haberland, H.; Mall, M.; Moseler, M.; Qiang, Y.; Reiners, T.; Thurner, Y. Filling of micron-sized contact holes with copper by energetic cluster impact. J. Vac. Sci. Technol. A Vac. Surf. Film. 1994, 12, 2925–2930. [Google Scholar] [CrossRef]
- Binns, C. Nanoclusters deposited on surfaces. Surf. Sci. Rep. 2001, 44, 1–49. [Google Scholar] [CrossRef]
- Drache, S.; Stranak, V.; Berg, F.; Hubicka, Z.; Tichy, M.; Helm, C.A.; Hippler, R. Pulsed gas aggregation for improved nanocluster growth and flux. Phys. Status Solidi 2014, 211, 1189–1193. [Google Scholar] [CrossRef]
- Ganeva, M.; Pipa, A.V.; Hippler, R. The influence of target erosion on the mass spectra of clusters formed in the planar DC magnetron sputtering source. Surf. Coatings Technol. 2012, 213, 41–47. [Google Scholar] [CrossRef]
- Huttel, Y. (Ed.) Gas-Phase Synthesis of Nanoparticles, 1st ed.; Wiley-VCH: Leipzig, Germany, 2017. [Google Scholar]
- Acsente, T.; Negrea, R.F.; Nistor, L.C.; Logofatu, C.; Matei, E.; Birjega, R.; Grisolia, C.; Dinescu, G. Synthesis of flower-like tungsten nanoparticles by magnetron sputtering combined with gas aggregation. Eur. Phys. J. D 2015, 69, 161. [Google Scholar] [CrossRef]
- Peter, T.; Polonskyi, O.; Gojdka, B.; Ahadi, A.M.; Strunskus, T.; Zaporojtchenko, V.; Biederman, H.; Faupel, F. Influence of reactive gas admixture on transition metal cluster nucleation in a gas aggregation cluster source. J. Appl. Phys. 2012, 112, 114321. [Google Scholar] [CrossRef]
- Polonskyi, O.; Peter, T.; Ahadi, A.M.; Hinz, A.; Strunskus, T.; Zaporojtchenko, V.; Biederman, H.; Faupel, F. Huge increase in gas phase nanoparticle generation by pulsed direct current sputtering in a reactive gas admixture. Appl. Phys. Lett. 2013, 103, 033118. [Google Scholar] [CrossRef]
- Ahadi, A.M.; Zaporojtchenko, V.; Peter, T.; Polonskyi, O.; Strunskus, T.; Faupel, F. Role of oxygen admixture in stabilizing TiOx nanoparticle deposition from a gas aggregation source. J. Nanoparticle Res. 2013, 15, 2125. [Google Scholar] [CrossRef]
- Ahadi, A.M.; Polonskyi, O.; Schürmann, U.; Strunskus, T.; Faupel, F. Stable production of TiOx nanoparticles with narrow size distribution by reactive pulsed DC magnetron sputtering. J. Phys. D. Appl. Phys. 2015, 48, 035501. [Google Scholar] [CrossRef]
- Wan, J.; Ma, Z.; Han, M.; Hong, J.; Wang, G. Poly (N-vinyl-2-pyrrolidone) cluster-assembled nanostructured films prepared by low energy cluster beam deposition. Solid State Commun. 2002, 121, 251–256. [Google Scholar] [CrossRef]
- Polonskyi, O.; Kylián, O.; Solař, P.; Artemenko, A.; Kousal, J.; Slavínská, D.; Choukourov, A.; Biederman, H. Nylon-sputtered nanoparticles: Fabrication and basic properties. J. Phys. D. Appl. Phys. 2012, 45, 495301. [Google Scholar] [CrossRef]
- Solař, P.; Melnichuk, I.; Artemenko, A.; Polonskyi, O.; Kylián, O.; Choukourov, A.; Slavínská, D.; Biederman, H. Nylon-sputtered plasma polymer particles produced by a semi-hollow cathode gas aggregation source. Vacuum 2015, 111, 124–130. [Google Scholar] [CrossRef]
- Vahl, A.; Strobel, J.; Reichstein, W.; Polonskyi, O.; Strunskus, T.; Kienle, L.; Faupel, F. Single target sputter deposition of alloy nanoparticles with adjustable composition via a gas aggregation cluster source. Nanotechnology 2017, 28, 175703. [Google Scholar] [CrossRef]
- Llamosa, D.; Ruano, M.; Martínez, L.; Mayoral, A.; Roman, E.; García-Hernández, M.; Huttel, Y. The ultimate step towards a tailored engineering of core@shell and core@shell@shell nanoparticles. Nanoscale 2014, 6, 13483–13486. [Google Scholar] [CrossRef]
- Bai, J.; Wang, J.-P. High-magnetic-moment core-shell-type FeCo–Au/Ag nanoparticles. Appl. Phys. Lett. 2005, 87, 152502. [Google Scholar] [CrossRef]
- Balasubramanian, B.; Kraemer, K.L.; Reding, N.A.; Skomski, R.; Ducharme, S.; Sellmyer, D.J. Synthesis of monodisperse TiO2-paraffin core-shell nanoparticles for improved dielectric properties. ACS Nano 2010, 4, 1893–1900. [Google Scholar] [CrossRef]
- Mehdi, M.A.S.; Still, R.H. Thermal degradation of polymers: Part XXVI—Vacuum pyrolysis of poly(m-acetoxystyrene): Poroducts volatile at ambient temperature. Polym. Degrad. Stab. 1985, 10, 369–380. [Google Scholar] [CrossRef]
- Biederman, H.; Osada, Y. Plasma Polymerization Processes, 1st ed.; Elsevier Science: Amsterdam, The Netherlands, 1992. [Google Scholar]
- Hiratsuka, H.; Akovali, G.; Shen, M.; Bell, A.T. Plasma polymerization of some simple saturated hydrocarbons. J. Appl. Polym. Sci. 1978, 22, 917–925. [Google Scholar] [CrossRef]
- Siow, K.S.; Britcher, L.; Kumar, S.; Griesser, H.J. Plasma Methods for the Generation of Chemically Reactive Surfaces for Biomolecule Immobilization and Cell Colonization—A Review. Plasma Process. Polym. 2006, 3, 392–418. [Google Scholar] [CrossRef]
- Meyer-Plath, A.; Schröder, K.; Finke, B.; Ohl, A. Current trends in biomaterial surface functionalization—Nitrogen-containing plasma assisted processes with enhanced selectivity. Vacuum 2003, 71, 391–406. [Google Scholar] [CrossRef]
- Artemenko, A.; Kylián, O.; Kousal, J.; Choukourov, A.; Polonskyi, O.; Slavinska, D.; Biederman, H. Deposition of amino-rich coatings by RF magnetron sputtering of Nylon: Investigation of their properties related to biomedical applications. Surf. Coatings Technol. 2011, 205, S529–S533. [Google Scholar] [CrossRef]
- Kylián, O.; Kousal, J.; Artemenko, A.; Choukourov, A.; Petr, M.; Polonskyi, O.; Slavinska, D.; Biederman, H. Deposition of amino-rich coatings by RF magnetron sputtering of Nylon: In-situ characterization of the deposition process. Surf. Coat. Technol. 2011, 205, S558–S561. [Google Scholar] [CrossRef]
- Choukourov, A.; Biederman, H.; Kholodkov, I.; Slavinska, D.; Trchova, M.; Hollander, A. Properties of amine-containing coatings prepared by plasma polymerization. J. Appl. Polym. Sci. 2004, 92, 979–990. [Google Scholar] [CrossRef]
- Girard-Lauriault, P.; Mwale, F.; Iordanova, M.; Demers, C.; Desjardins, P.; Wertheimer, M.R. Atmospheric Pressure Deposition of Micropatterned Nitrogen-Rich Plasma-Polymer Films for Tissue Engineering. Plasma Process. Polym. 2005, 2, 263–270. [Google Scholar] [CrossRef]
- Truica-Marasescu, F.; Wertheimer, M.R. Nitrogen-Rich Plasma-Polymer Films for Biomedical Applications. Plasma Process. Polym. 2008, 5, 44–57. [Google Scholar] [CrossRef]
- Denis, L.; Marsal, P.; Olivier, Y.; Godfroid, T.; Lazzaroni, R.; Hecq, M.; Cornil, J.; Snyders, R. Deposition of Functional Organic Thin Films by Pulsed Plasma Polymerization: A Joint Theoretical and Experimental Study. Plasma Process. Polym. 2010, 7, 172–181. [Google Scholar] [CrossRef]
- Král’, M.; Ogino, A.; Nagatsu, M. Effect of hydrogen on amino group introduction onto the polyethylene surface by surface-wave plasma chemical modification. J. Phys. D Appl. Phys. 2008, 41, 105213. [Google Scholar] [CrossRef]
- Chen, Q.; Förch, R.; Knoll, W. Characterization of Pulsed Plasma Polymerization Allylamine as an Adhesion Layer for DNA Adsorption/Hybridization. Chem. Mater. 2004, 16, 614–620. [Google Scholar] [CrossRef]
- Choukourov, A.; Biederman, H.; Slavinska, D.; Hanley, L.; Grinevich, A.; Boldyryeva, H.; Mackova, A. Mechanistic Studies of Plasma Polymerization of Allylamine. J. Phys. Chem. B 2005, 109, 23086–23095. [Google Scholar] [CrossRef] [PubMed]
- Coad, B.R.; Jasieniak, M.; Griesser, S.S.; Griesser, H.J. Controlled covalent surface immobilisation of proteins and peptides using plasma methods. Surf. Coat. Technol. 2013, 233, 169–177. [Google Scholar] [CrossRef]
- Muguruma, H. Plasma-Polymerized Films for Biochip Design. Plasma Process. Polym. 2010, 7, 151–162. [Google Scholar] [CrossRef]
- Remes, Z.; Choukourov, A.; Stuchlik, J.; Potmesil, J.; Vanecek, M. Nanocrystalline diamond surface functionalization in radio frequency plasma. Diam. Relat. Mater. 2006, 15, 745–748. [Google Scholar] [CrossRef]
- Solař, P.; Škorvánková, K.; Kuzminova, A.; Kylián, O. Challenges in the deposition of plasma polymer nanoparticles using gas aggregation source: Rebounding upon impact and how to land them on a substrate. Plasma Process. Polym. 2023, 20, e2300070. [Google Scholar] [CrossRef]
- Zekonyte, J. Sputtering and Surface Modification of Thermoplastic Polymers with Low Energy Ion Beams. Ph.D. Thesis, Christian-Albrechts-Universität zu Kiel, Kiel, Germany, 2005. Available online: https://d-nb.info/977267776/34 (accessed on 31 January 2024).
- Artemenko, A. XPS Analysis of Plasma Polymers and Nanocomposite Films without Breaking Vacuum; Charles University: Prague, Czech Republic, 2013. [Google Scholar]
- Hanuš, J.; Hanyková, L.; Choukourov, A.; Kousal, J.; Polonskyi, O.; Slavínská, D.; Biederman, H. NMR Study of Polyethylene-Like Plasma Polymer Films. Plasma Process. Polym. 2009, 6, S362–S365. [Google Scholar] [CrossRef]
- Roualdes, S.; Berjoan, R.; Durand, J. 29Si NMR and Si2p XPS correlation in polysiloxane membranes prepared by plasma enhanced chemical vapor deposition. Sep. Purif. Technol. 2001, 25, 391–397. [Google Scholar] [CrossRef]
- Duan, P.; Cao, X.; Pham, H.; Datye, A.; Schmidt-Rohr, K. Protective Carbon Overlayers from 2,3-Naphthalenediol Pyrolysis on Mesoporous SiO2 and Al2O3 Analyzed by Solid-State NMR. Materials 2018, 11, 980. [Google Scholar] [CrossRef]
- Widdifield, C.M.; Kaur, N. NMR of nanoparticles. In Comprehensive Inorganic Chemistry III; Elsevier: Amsterdam, The Netherlands, 2023; pp. 398–449. [Google Scholar] [CrossRef]
- Marchetti, A.; Chen, J.; Pang, Z.; Li, S.; Ling, D.; Deng, F.; Kong, X. Understanding Surface and Interfacial Chemistry in Functional Nanomaterials via Solid-State NMR. Adv. Mater. 2017, 29, 1605895. [Google Scholar] [CrossRef]
- Mirau, P.A. A Practical Guide to Understanding the NMR of Polymers, 1st ed.; Wiley-Interscience: Hoboken, NJ, USA, 2004. [Google Scholar]
- Davis, R.D.; Jarrett, W.L.; Mathias, L.J. Solution 13 C NMR spectroscopy of polyamide homopolymers (nylons 6, 11, 12, 66, 69, 610 and 612) and several commercial copolymers. Polymer 2001, 42, 2621–2626. [Google Scholar] [CrossRef]
- Krejsa, M.R.; Udipi, K.; Middleton, J.C. NMR Analysis of UV- and Heat-Aged Nylon-6,6. Macromolecules 1997, 30, 4695–4703. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, J.; Tang, L.; Huang, J.; Fang, Y.; Ji, P.; Wang, C.; Wang, H. A Novel Synthetic Strategy for Preparing Polyamide 6 (PA6)-Based Polymer with Transesterification. Polymers 2019, 11, 978. [Google Scholar] [CrossRef] [PubMed]
- Serov, A.; Choukourov, A.; Melnichuk, I.; Shelemin, A.; Kuzminova, A.; Kylián, O.; Hanuš, J.; Kousal, J.; Drábik, M.; Slavínská, D.; et al. Poly(tetrafluoroethylene) sputtering in a gas aggregation source for fabrication of nano-structured deposits. Surf. Coat. Technol. 2014, 254, 319–326. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assignment in Figure 6 | Position in the Spectra (ppm) | Chemical Group | Relative Molar Abundance of the Chemical Groups |
|---|---|---|---|
| A | 0.96 | CH3–C– | 5 |
| B | 1.2 | NH2–C–, –C–NH–C– | >1–2 |
| C | 1.3 | –C–CH2–C– | 20 |
| acetic acid | 2.1 | CH3–N–, (acetic acid) | approx. 2 |
| D | 2.8 | –N–CH2–C–, –C–CH–NH2 | CH3 | 2.5–5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šťastná, J.; Škorvánková, K.; Kuzminova, A.; Hanuš, J.; Hanyková, L.; Krakovský, I.; Solař, P. Insoluble Network Skeleton and Soluble Components of Nylon 6,6-Sputtered Nanoparticles: Insights from Liquid-State and Solid-State NMR Analysis. Nanomaterials 2024, 14, 497. https://doi.org/10.3390/nano14060497
Šťastná J, Škorvánková K, Kuzminova A, Hanuš J, Hanyková L, Krakovský I, Solař P. Insoluble Network Skeleton and Soluble Components of Nylon 6,6-Sputtered Nanoparticles: Insights from Liquid-State and Solid-State NMR Analysis. Nanomaterials. 2024; 14(6):497. https://doi.org/10.3390/nano14060497
Chicago/Turabian StyleŠťastná, Julie, Kateřina Škorvánková, Anna Kuzminova, Jan Hanuš, Lenka Hanyková, Ivan Krakovský, and Pavel Solař. 2024. "Insoluble Network Skeleton and Soluble Components of Nylon 6,6-Sputtered Nanoparticles: Insights from Liquid-State and Solid-State NMR Analysis" Nanomaterials 14, no. 6: 497. https://doi.org/10.3390/nano14060497
APA StyleŠťastná, J., Škorvánková, K., Kuzminova, A., Hanuš, J., Hanyková, L., Krakovský, I., & Solař, P. (2024). Insoluble Network Skeleton and Soluble Components of Nylon 6,6-Sputtered Nanoparticles: Insights from Liquid-State and Solid-State NMR Analysis. Nanomaterials, 14(6), 497. https://doi.org/10.3390/nano14060497