Electronic and Molecular Adsorption Properties of Pt-Doped BC6N: An Ab-Initio Investigation


Abstract
1. Introduction
2. Computational Methods
3. Results
3.1. Pt-Doping
3.2. Molecules Adsorption on Pristine and Pt-Doped BC6N Systems
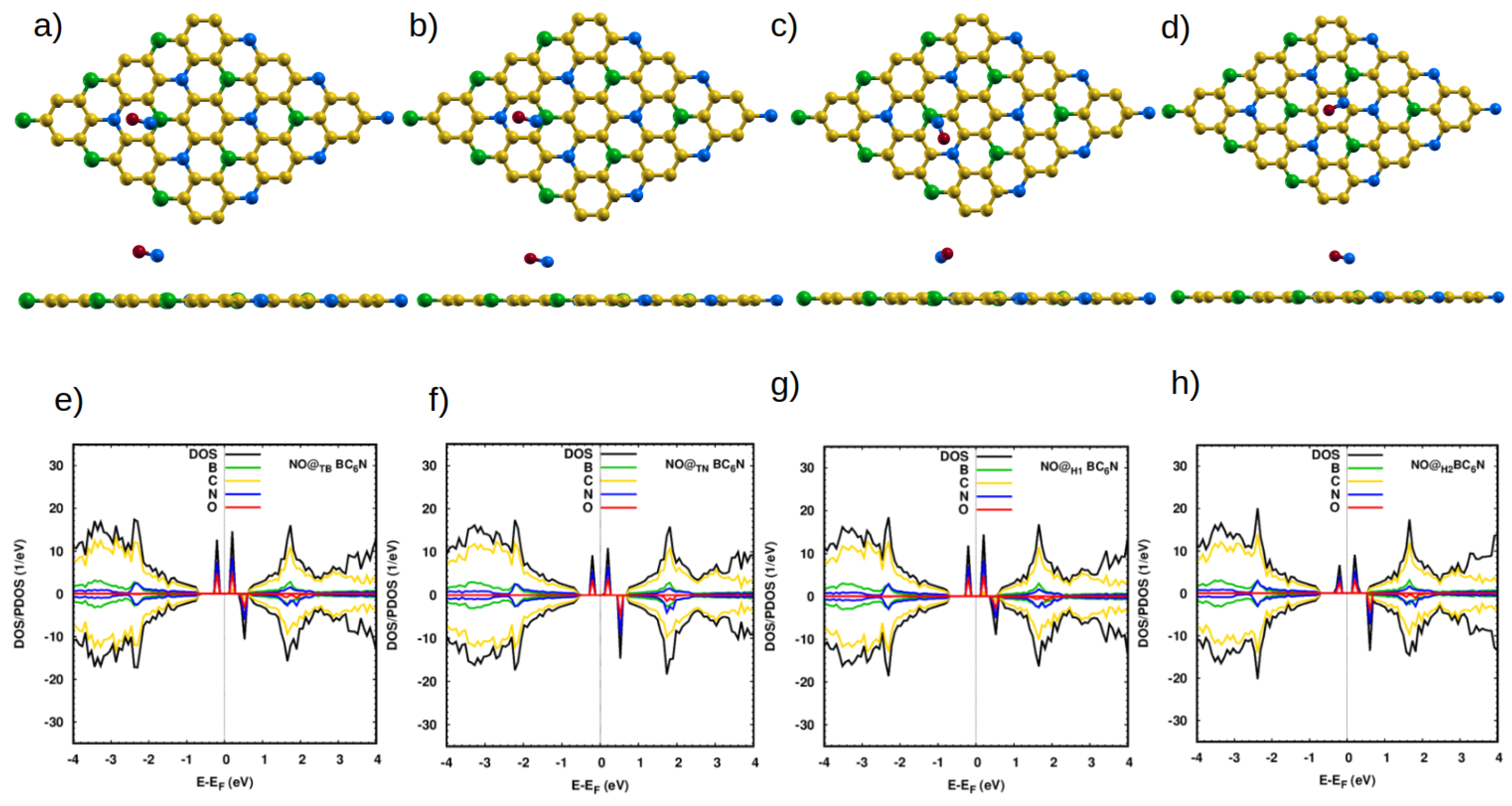
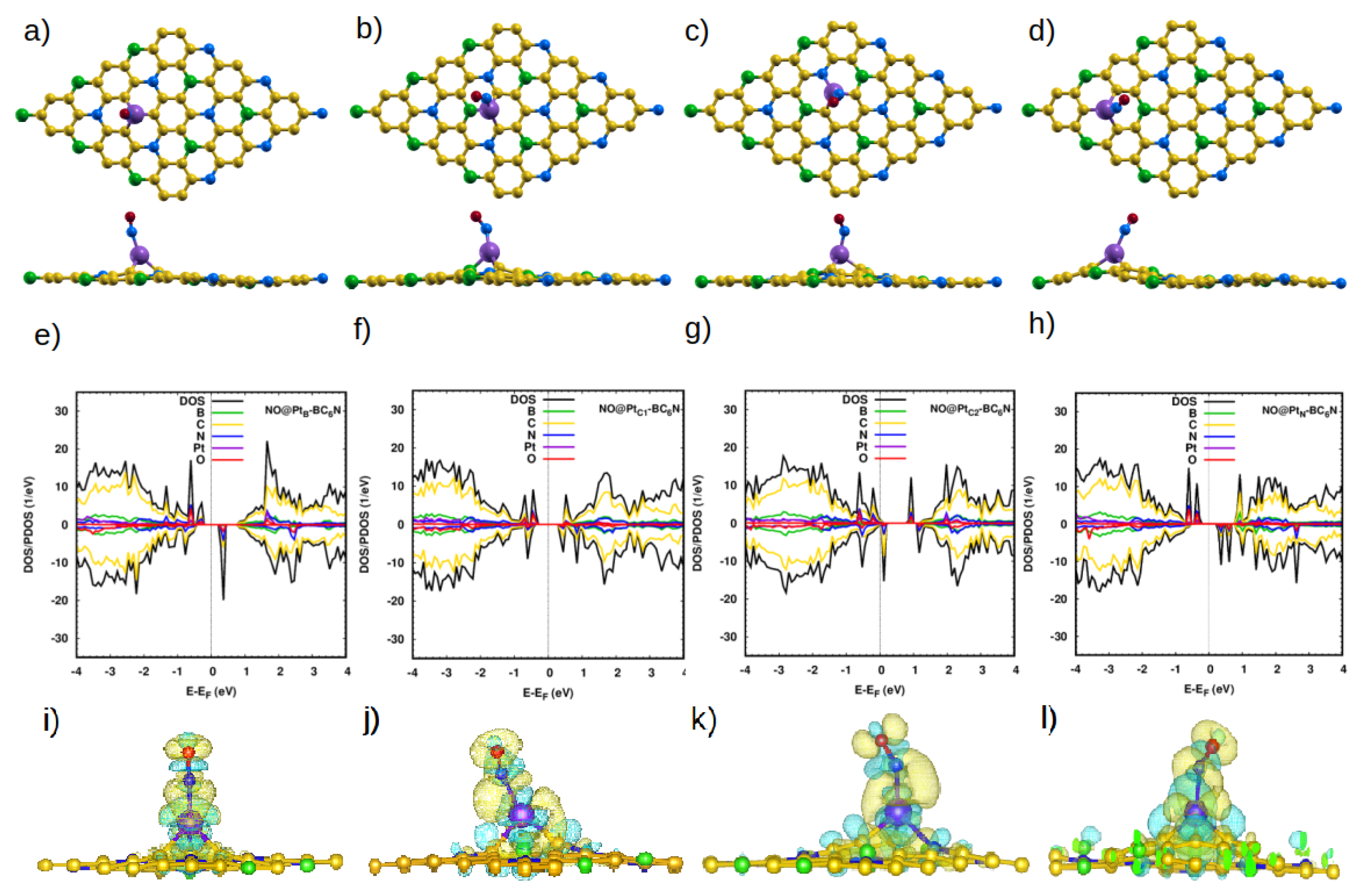
3.2.1. Adsorption of NO
3.2.2. Adsorption of NO2
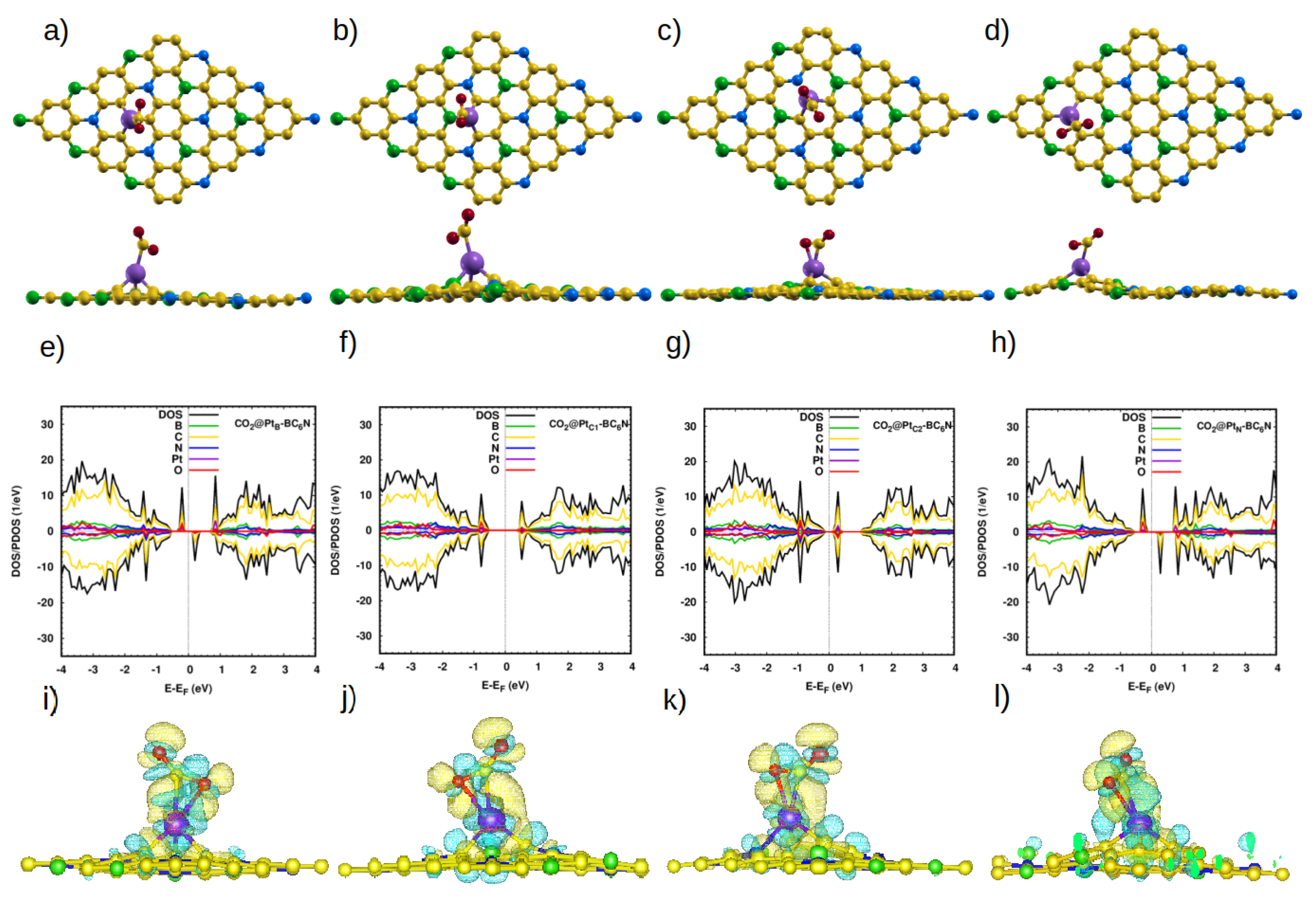
3.2.3. Adsorption of CO2
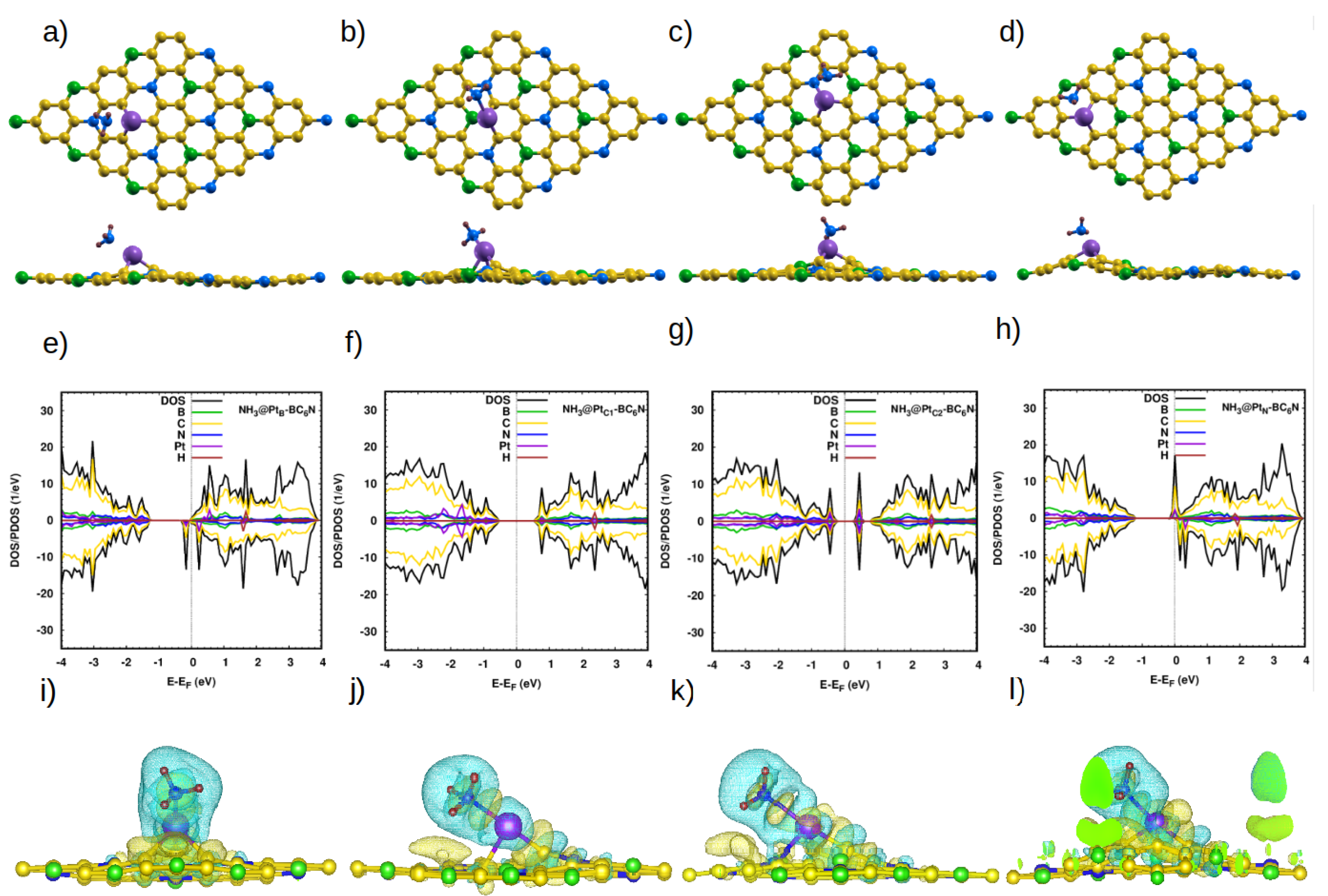
3.2.4. Adsorption of NH3
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhimanapati, G.R.; Lin, Z.; Meunier, V.; Jung, Y.; Cha, J.; Das, S.; Xiao, D.; Son, Y.; Strano, M.S.; Cooper, V.R.; et al. Recent advances in two-dimensional materials beyond graphene. ACS Nano 2015, 9, 11509–11539. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Lahiri, I.; Seelaboyina, R.; Kang, Y.S. Synthesis of graphene and its applications: A review. Crit. Rev. Solid State Mater. Sci. 2010, 35, 52–71. [Google Scholar] [CrossRef]
- Li, X.; Zhu, H. Two-dimensional MoS2: Properties, preparation, and applications. J. Mater. 2015, 1, 33–44. [Google Scholar] [CrossRef]
- Manzeli, S.; Ovchinnikov, D.; Pasquier, D.; Yazyev, O.V.; Kis, A. 2D transition metal dichalcogenides. Nat. Rev. Mater. 2017, 2, 17033. [Google Scholar] [CrossRef]
- Feng, J.W.; Liu, Y.J.; Wang, H.X.; Zhao, J.X.; Cai, Q.H.; Wang, X.Z. Gas adsorption on silicene: A theoretical study. Comput. Mater. Sci. 2014, 87, 218–226. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, W.; Hou, J.; Dai, Y.; Yang, J. Coronoid nanographene C216 as hydrogen purification membrane: A density functional theory study. Carbon 2018, 135, 112–117. [Google Scholar] [CrossRef]
- Kumar, R.; Sahoo, S.; Joanni, E.; Singh, R.K.; Yadav, R.M.; Verma, R.K.; Singh, D.P.; Tan, W.K.; Perez del Pino, A.; Moshkalev, S.A.; et al. A review on synthesis of graphene, h-BN and MoS 2 for energy storage applications: Recent progress and perspectives. Nano Res. 2019, 12, 2655–2694. [Google Scholar] [CrossRef]
- Susarla, S.; Kutana, A.; Hachtel, J.A.; Kochat, V.; Apte, A.; Vajtai, R.; Idrobo, J.C.; Yakobson, B.I.; Tiwary, C.S.; Ajayan, P.M. Quaternary 2D transition metal dichalcogenides (TMDs) with tunable bandgap. Adv. Mater. 2017, 29, 1702457. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Wang, Y.; Li, X.; Li, H.; Wang, Z.; Tang, Z.; Ma, L.; Mo, F.; Zhi, C. Recent Progress of MX ene-Based Nanomaterials in Flexible Energy Storage and Electronic Devices. Energy Environ. Mater. 2018, 1, 183–195. [Google Scholar] [CrossRef]
- Salim, O.; Mahmoud, K.; Pant, K.; Joshi, R. Introduction to MXenes: Synthesis and characteristics. Mater. Today Chem. 2019, 14, 100191. [Google Scholar] [CrossRef]
- Hong, Y.L.; Liu, Z.; Wang, L.; Zhou, T.; Ma, W.; Xu, C.; Feng, S.; Chen, L.; Chen, M.L.; Sun, D.M.; et al. Chemical vapor deposition of layered two-dimensional MoSi2N4 materials. Science 2020, 369, 670. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, A.; Sardroodi, J.J. The adsorption of sulfur trioxide and ozone molecules on stanene nanosheets investigated by DFT: Applications to gas sensor devices. Phys. E Low-Dimens. Syst. Nanostruct. 2019, 108, 382–390. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, H.; Tong, Y.; Zhao, L.; Zhang, Y.; Qiu, Y.; Lin, X. First-principles investigations of metal (V, Nb, Ta)-doped monolayer MoS2: Structural stability, electronic properties and adsorption of gas molecules. Appl. Surf. Sci. 2017, 419, 522–530. [Google Scholar] [CrossRef]
- Yadav, S.; Tam, J.; Singh, C.V. A first principles study of hydrogen storage on lithium decorated two dimensional carbon allotropes. Int. J. Hydrogen Energy 2015, 40, 6128–6136. [Google Scholar] [CrossRef]
- Fadlallah, M.M.; Abdelrahman, A.G.; Schwingenschlögl, U.; Maarouf, A.A. Graphene and graphene nanomesh supported nickel clusters: Electronic, magnetic, and hydrogen storage properties. Nanotechnology 2019, 30, 085709. [Google Scholar] [CrossRef]
- Donarelli, M.; Ottaviano, L. 2D materials for gas sensing applications: A review on graphene oxide, MoS2, WS2 and phosphorene. Sensors 2018, 18, 3638. [Google Scholar] [CrossRef]
- Wang, B.; Gu, Y.; Chen, L.; Ji, L.; Zhu, H.; Sun, Q. Gas sensing devices based on two-dimensional materials: A review. Nanotechnology 2022, 33, 252001. [Google Scholar] [CrossRef]
- Zhang, L.; Khan, K.; Zou, J.; Zhang, H.; Li, Y. Recent advances in emerging 2D material-based gas sensors: Potential in disease diagnosis. Adv. Mater. Interfaces 2019, 6, 1901329. [Google Scholar] [CrossRef]
- Rahimi, R.; Solimannejad, M. Hydrogen storage on pristine and Li-decorated BC6N monolayer from first-principles insights. Mol. Phys. 2021, 119, e1827177. [Google Scholar] [CrossRef]
- Bafekry, A.; Naseri, M.; Fadlallah, M.M.; Abdolhosseini Sarsari, I.; Faraji, M.; Bagheri Khatibani, A.; Ghergherehchi, M.; Gogova, D. A novel two-dimensional boron–carbon–nitride (BCN) monolayer: A first-principles insight. J. Appl. Phys. 2021, 130, 114301. [Google Scholar] [CrossRef]
- Lu, Y.; Yu, Y.; Zhu, X.; Wang, M. Two predicted two-dimensional BCN structures: A first-principles study. Phys. E Low-Dimens. Syst. Nanostruct. 2021, 125, 114413. [Google Scholar] [CrossRef]
- Bafekry, A.; Stampfl, C. Band-gap control of graphenelike borocarbonitride g-BC6N bilayers by electrical gating. Phys. Rev. B 2020, 102, 195411. [Google Scholar] [CrossRef]
- Rani, P.; Jindal, V. Designing band gap of graphene by B and N dopant atoms. RSC Adv. 2013, 3, 802–812. [Google Scholar] [CrossRef]
- Deng, X.; Wu, Y.; Dai, J.; Kang, D.; Zhang, D. Electronic structure tuning and band gap opening of graphene by hole/electron codoping. Phys. Lett. A 2011, 375, 3890–3894. [Google Scholar] [CrossRef]
- Kouvetakis, J.; Sasaki, T.; Shen, C.; Hagiwara, R.; Lerner, M.; Krishnan, K.; Bartlett, N. Novel aspects of graphite intercalation by fluorine and fluorides and new B/C, C/N and B/C/N materials based on the graphite network. Synth. Met. 1989, 34, 1–7. [Google Scholar] [CrossRef]
- Sadeghi, S.N.; Allaei, S.M.V.; Zebarjadi, M.; Esfarjani, K. Ultra-high lattice thermal conductivity and the effect of pressure in superhard hexagonal BC 2 N. J. Mater. Chem. C 2020, 8, 15705–15716. [Google Scholar] [CrossRef]
- Nehate, S.; Saikumar, A.; Prakash, A.; Sundaram, K. A review of boron carbon nitride thin films and progress in nanomaterials. Mater. Today Adv. 2020, 8, 100106. [Google Scholar] [CrossRef]
- Wang, J.; Chen, C.; Yang, C.; Fan, Y.; Liu, D.; Lei, W. Boron carbon nitride (BCN) nanomaterials: Structures, synthesis and energy applications. Curr. Graphene Sci. 2018, 2, 3–14. [Google Scholar] [CrossRef]
- Matsui, K.; Oda, S.; Yoshiura, K.; Nakajima, K.; Yasuda, N.; Hatakeyama, T. One-shot multiple borylation toward BN-doped nanographenes. J. Am. Chem. Soc. 2018, 140, 1195–1198. [Google Scholar] [CrossRef]
- Aghaei, S.; Aasi, A.; Farhangdoust, S.; Panchapakesan, B. Graphene-like BC6N nanosheets are potential candidates for detection of volatile organic compounds (VOCs) in human breath: A DFT study. Appl. Surf. Sci. 2021, 536, 147756. [Google Scholar] [CrossRef]
- Bafekry, A. Graphene-like BC6N single-layer: Tunable electronic and magnetic properties via thickness, gating, topological defects, and adatom/molecule. Phys. E Low-Dimens. Syst. Nanostruct. 2020, 118, 113850. [Google Scholar] [CrossRef]
- Li, Y.; Gu, Q.; Johannessen, B.; Zheng, Z.; Li, C.; Luo, Y.; Zhang, Z.; Zhang, Q.; Fan, H.; Luo, W.; et al. Synergistic Pt doping and phase conversion engineering in two-dimensional MoS2 for efficient hydrogen evolution. Nano Energy 2021, 84, 105898. [Google Scholar] [CrossRef]
- Deng, J.; Li, H.; Xiao, J.; Tu, Y.; Deng, D.; Yang, H.; Tian, H.; Li, J.; Ren, P.; Bao, X. Triggering the electrocatalytic hydrogen evolution activity of the inert two-dimensional MoS2 surface via single-atom metal doping. Energy Environ. Sci. 2015, 8, 1594–1601. [Google Scholar] [CrossRef]
- Wang, X.; Zeng, F.; Qiu, H.; Guo, X.; Yao, Q.; Li, L.; Tang, J. Adsorption study of SF6 molecules on Pt-doped two-dimensional material Ti3C2Tx Mxene. AIP Adv. 2023, 13, 085217. [Google Scholar] [CrossRef]
- Wu, P.; Huang, M. Investigation of adsorption behaviors, and electronic and magnetic properties for small gas molecules adsorbed on Pt-doped arsenene by density functional calculations. RSC Adv. 2023, 13, 3807–3817. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Dion, M.; Rydberg, H.; Schröder, E.; Langreth, D.C.; Lundqvist, B.I. Van der Waals density functional for general geometries. Phys. Rev. Lett. 2004, 92, 246401. [Google Scholar] [CrossRef]
- Aasi, A.; Mehdi Aghaei, S.; Panchapakesan, B. Outstanding performance of transition-metal-decorated single-layer graphene-like BC6N nanosheets for disease biomarker detection in human breath. ACS Omega 2021, 6, 4696–4707. [Google Scholar] [CrossRef] [PubMed]
- Babar, V.; Sharma, S.; Schwingenschlögl, U. Gas sensing performance of pristine and monovacant C6BN monolayers evaluated by density functional theory and the nonequilibrium green’s function formalism. J. Phys. Chem. C 2020, 124, 5853–5860. [Google Scholar] [CrossRef]
- Degler, D.; Pereira de Carvalho, H.W.; Kvashnina, K.; Grunwaldt, J.D.; Weimar, U.; Barsan, N. Structure and chemistry of surface-doped Pt:SnO2 gas sensing materials. RSC Adv. 2016, 6, 28149–28155. [Google Scholar] [CrossRef]
- Ma, D.; Ju, W.; Li, T.; Zhang, X.; He, C.; Ma, B.; Lu, Z.; Yang, Z. The adsorption of CO and NO on the MoS2 monolayer doped with Au, Pt, Pd, or Ni: A first-principles study. Appl. Surf. Sci. 2016, 383, 98–105. [Google Scholar] [CrossRef]
- Ni, J.; Wang, W.; Quintana, M.; Jia, F.; Song, S. Adsorption of small gas molecules on strained monolayer WSe2 doped with Pd, Ag, Au, and Pt: A computational investigation. Appl. Surf. Sci. 2020, 514, 145911. [Google Scholar] [CrossRef]
- Cui, H.; Zhu, H.; Jia, P. Adsorption and sensing of SO2 and SOF2 molecule by Pt-doped HfSe2 monolayer: A first-principles study. Appl. Surf. Sci. 2020, 530, 147242. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Systems | Mag | (up) | (dn) | ||
|---|---|---|---|---|---|
| BC6N | - | - | 0.0 | 1.3 | 1.3 |
| PtB-BC6N | 0.08 | 87 | 0.9 | 0.2 | 1.3 |
| PtC1-BC6N | 0.10 | 88 | 0.0 | 1.1 | 1.1 |
| PtC2-BC6N | 0.43 | 94 | 0.0 | 0.5 | 0.5 |
| PtN-BC6N | 0.39 | 51 | 1.0 | 0.2 | 1.4 |
| Systems | d | X | Mag | (up) | (dn) | ||
|---|---|---|---|---|---|---|---|
| @TBBC6N | 2.8 | N | −0.04 | 0.2 | 1.0 | 0.4 | 1.1 |
| @TNBC6N | 3.1 | N | 0.00 | 0.1 | 1.0 | 1.1 | 0.4 |
| @H1BC6N | 3.1 | N | −0.04 | 0.1 | 1.0 | 0.3 | 1.2 |
| @H2BC6N | 2.9 | N | −0.05 | 0.2 | 1.0 | 0.5 | 1.3 |
| @PtB-BC6N | 1.9 | N | −0.20 | 2.7 | 2.1 | 1.1 | 0.8 |
| @PtC1-BC6N | 1.9 | N | −0.21 | 2.3 | 1.0 | 0.9 | 0.9 |
| @PtC2-BC6N | 1.9 | N | −0.15 | 2.3 | 0.7 | 0.2 | - |
| @PtN-BC6N | 2.0 | N | −0.20 | 2.1 | 2.1 | 1.2 | 0.9 |
| Systems | d | X | Mag | (up) | (dn) | |||
|---|---|---|---|---|---|---|---|---|
| @TBBC6N | 2.9 | 126.7 | O | −0.07 | 0.1 | 0.9 | 1.4 | - |
| @TNBC6N | 3.0 | 126.9 | O | −0.16 | 0.1 | 0.9 | 1.4 | - |
| @H1BC6N | 3.0 | 126.9 | O | −0.08 | 0.1 | −0.9 | - | 1.3 |
| @H2BC6N | 2.8 | 128.6 | N | −0.11 | 0.1 | 0.9 | 0.3 | 1.3 |
| @PtB-BC6N | 2.0 | 112.3 | O | −0.43 | 3.0 | 0.0 | 1.3 | 1.3 |
| @PtC1-BC6N | 2.1 | 123.3 | N | −0.34 | 2.5 | 0.2 | - | - |
| @PtC2-BC6N | 2.0 | 124.8 | N | −0.30 | 2.5 | 0.0 | - | - |
| @PtN-BC6N | 2.3 | 111.8 | O | −0.41 | 3.3 | 0.0 | 1.2 | 1.2 |
| Systems | d | X | Mag | (up) | (dn) | |||
|---|---|---|---|---|---|---|---|---|
| @TBBC6N | 3.3 | 179.3 | C | −0.02 | 0.2 | 0.0 | 1.3 | 1.3 |
| @TNBC6N | 3.2 | 179.8 | C | −0.01 | 0.2 | 0.0 | 1.3 | 1.3 |
| @H1BC6N | 3.2 | 179.3 | C | −0.02 | 0.2 | 0.0 | 1.3 | 1.3 |
| @H2BC6N | 3.2 | 179.3 | C | −0.02 | 0.2 | 0.0 | 1.3 | 1.3 |
| @PtB-BC6N | 2.2 | 144.0 | C | −0.38 | 0.7 | 1.0 | 1.0 | 0.8 |
| @PtC1-BC6N | 2.1 | 141.5 | C | −0.42 | 0.8 | 0.0 | 1.1 | 1.1 |
| @PtC2-BC6N | 2.1 | 141.0 | C | −0.42 | 0.6 | 0.0 | 0.4 | 0.4 |
| @PtN-BC6N | 2.2 | 146.4 | C | −0.33 | 0.4 | 1.0 | 1.0 | 0.9 |
| Systems | d | X | Mag | (up) | (dn) | |||
|---|---|---|---|---|---|---|---|---|
| @TBBC6N | 2.7 | 102.2 | H | −0.01 | 0.2 | 0.0 | 1.3 | 1.3 |
| @TNBC6N | 2.7 | 106.4 | H | −0.01 | 0.2 | 0.0 | 1.3 | 1.3 |
| @H1BC6N | 2.7 | 106.2 | H | −0.01 | 0.2 | 0.0 | 1.3 | 1.3 |
| @H2BC6N | 2.8 | 106.2 | H | −0.01 | 0.2 | 0.0 | 1.3 | 1.3 |
| @PtB-BC6N | 2.2 | 107.3 | N | 0.22 | 1.5 | −0.9 | 1.3 | 0.2 |
| @PtC1-BC6N | 2.2 | 107.1 | N | 0.23 | 1.6 | 0.0 | 1.1 | 1.1 |
| @PtC2-BC6N | 2.3 | 107.3 | N | 0.21 | 1.3 | 0.0 | 0.7 | 0.7 |
| @PtN-BC6N | 2.3 | 106.9 | N | 0.22 | 1.2 | 0.9 | - | 1.4 |
| Gas | |
|---|---|
| NO2 | 2.8 |
| NO | 2.4 |
| NH3 | 1.4 |
| CO2 | 0.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alghamdi, N.M.; Fadlallah, M.M.; Al-qahtani, H.M.; Maarouf, A.A. Electronic and Molecular Adsorption Properties of Pt-Doped BC6N: An Ab-Initio Investigation. Nanomaterials 2024, 14, 762. https://doi.org/10.3390/nano14090762
Alghamdi NM, Fadlallah MM, Al-qahtani HM, Maarouf AA. Electronic and Molecular Adsorption Properties of Pt-Doped BC6N: An Ab-Initio Investigation. Nanomaterials. 2024; 14(9):762. https://doi.org/10.3390/nano14090762
Chicago/Turabian StyleAlghamdi, Nada M., Mohamed M. Fadlallah, Hind M. Al-qahtani, and Ahmed A. Maarouf. 2024. "Electronic and Molecular Adsorption Properties of Pt-Doped BC6N: An Ab-Initio Investigation" Nanomaterials 14, no. 9: 762. https://doi.org/10.3390/nano14090762
APA StyleAlghamdi, N. M., Fadlallah, M. M., Al-qahtani, H. M., & Maarouf, A. A. (2024). Electronic and Molecular Adsorption Properties of Pt-Doped BC6N: An Ab-Initio Investigation. Nanomaterials, 14(9), 762. https://doi.org/10.3390/nano14090762