
Ultra-Fast Charge Transfer in P3HT Composites Using the Core Hole Clock Technique

 , , , and
, , , and
Abstract
:1. Introduction
1.1. Time-Resolved Photoemission Spectroscopy (TRPES)
1.2. Transient Absorption Spectroscopy (TAS)
1.3. Time-Resolved Photoluminescence (TRPL)
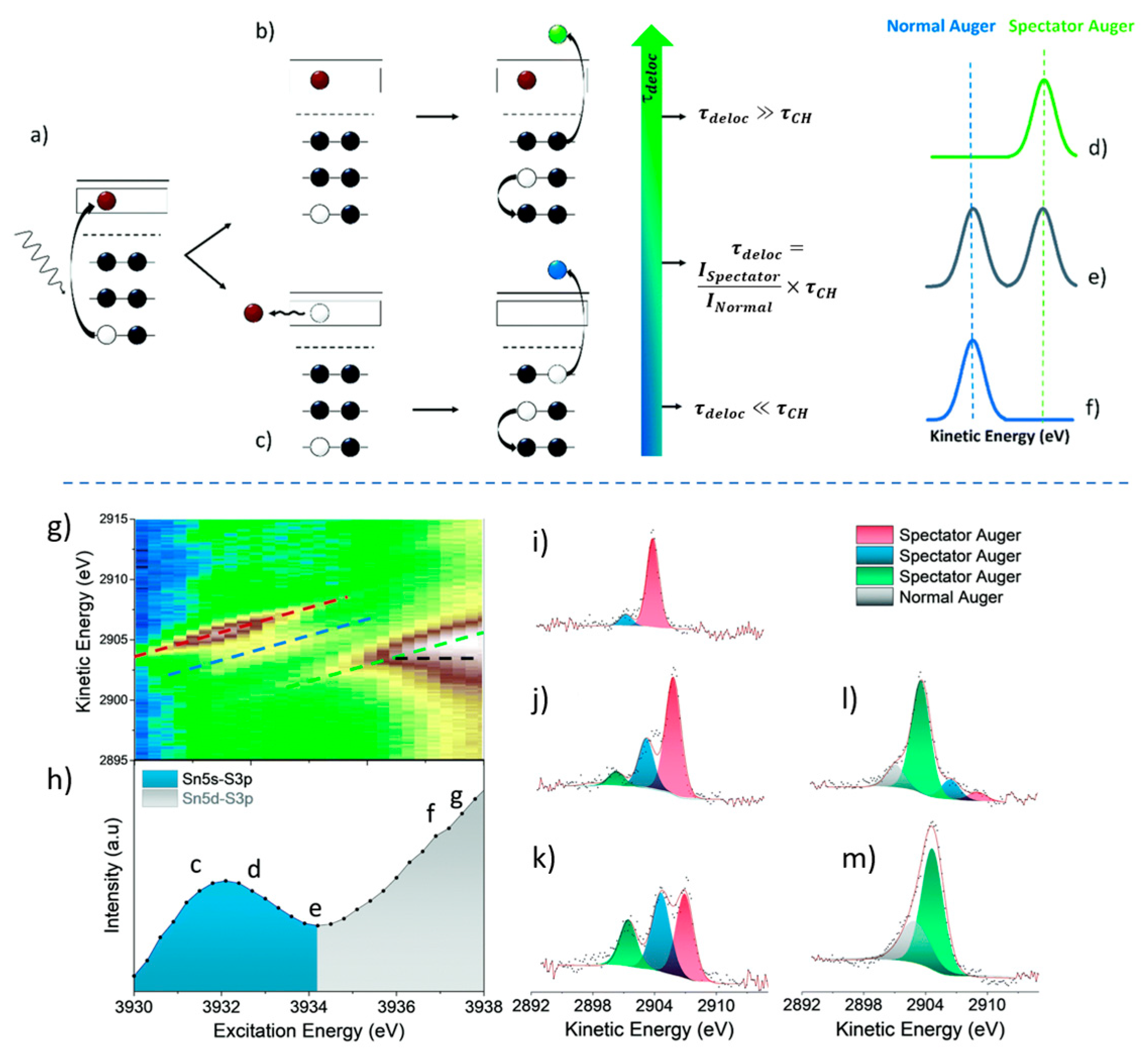
2. The Core Hole Clock Technique
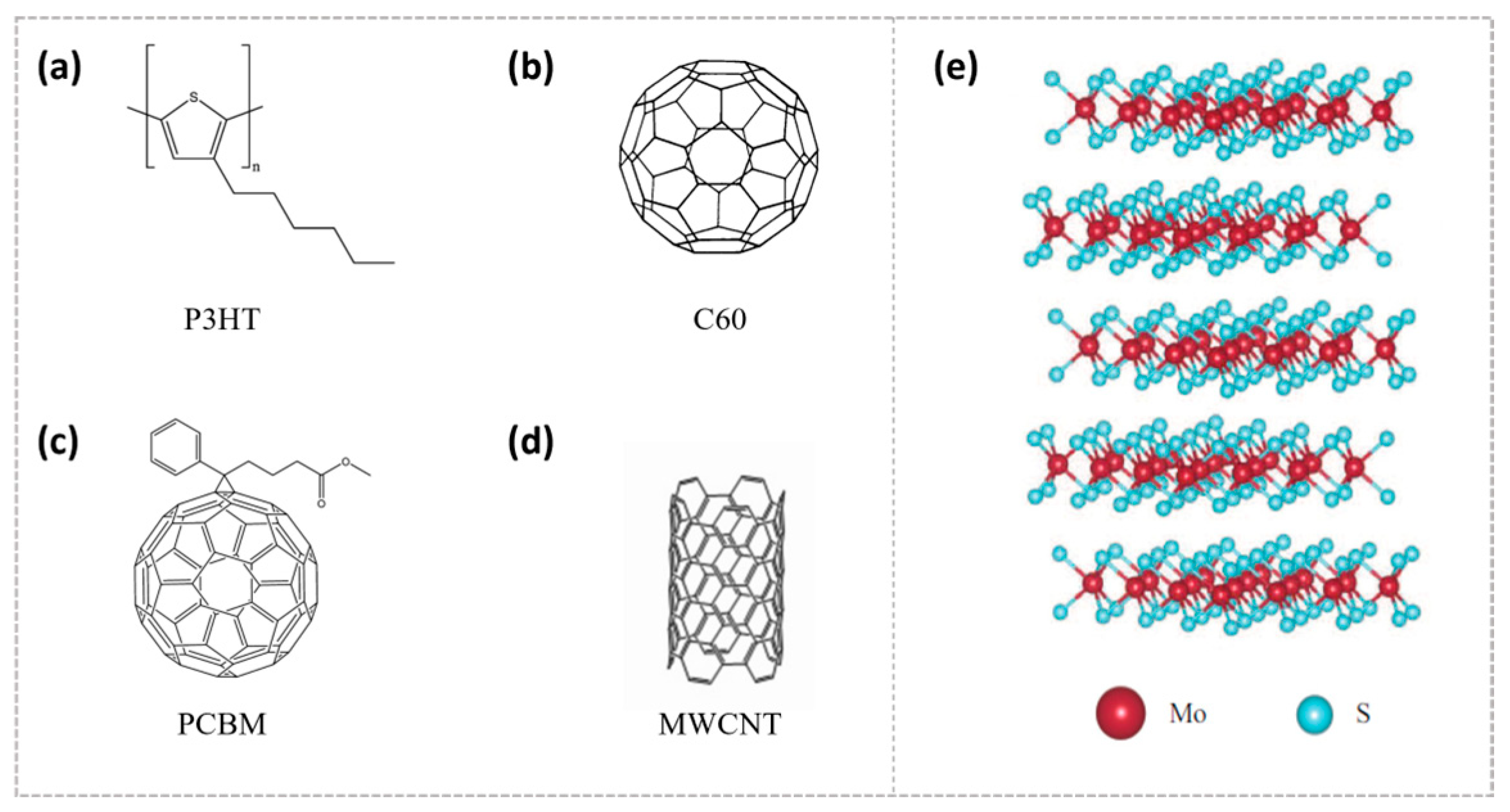
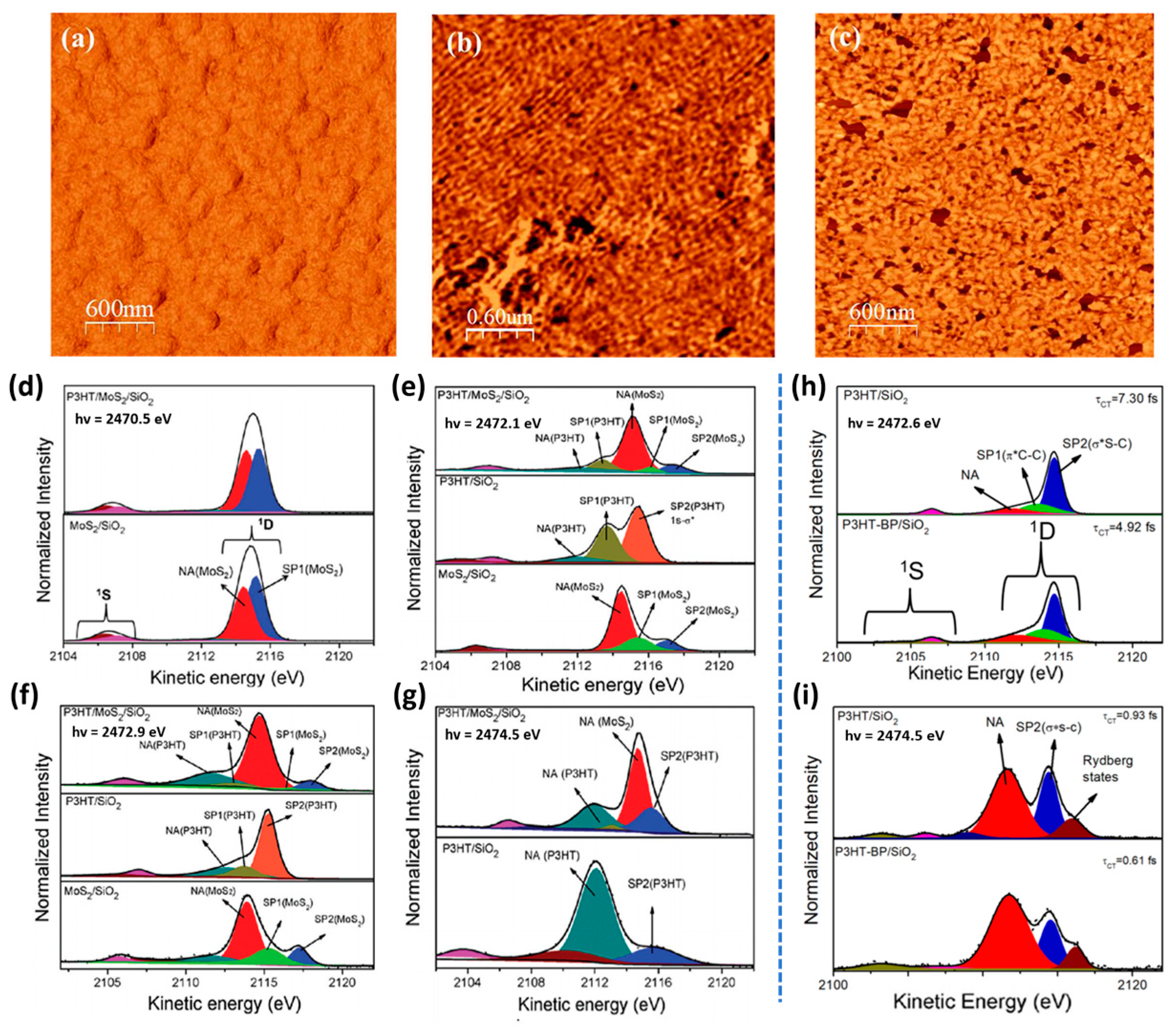
3. P3HT-Based Heterojunctions: Ultrafast Charge Transfer and Interfacial Interactions
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Xu, J.Y.; Tong, X.; Yu, P.; Wenya, G.E.; McGrath, T.; Fong, M.J.; Wu, J.; Wang, Z.M. Ultrafast dynamics of charge transfer and photochemical reactions in solar energy conversion. Adv. Sci. 2018, 5, 1800221. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Tan, J.; Zhang, L.; Xu, D.; Dong, J.; Ouyang, G. Ultrafast interfacial charge transfer and superior photoelectric conversion properties in one-dimensional Janus-MoSSe/WSe 2 van der Waals heterostructures. Phys. Rev. B 2023, 108, 045416. [Google Scholar] [CrossRef]
- Pan, Q.; Abdellah, M.; Cao, Y.; Lin, W.; Liu, Y.; Meng, J.; Zhou, Q.; Zhao, Q.; Yan, X.; Li, Z. Ultrafast charge transfer dynamics in 2D covalent organic frameworks/Re-complex hybrid photocatalyst. Nat. Commun. 2022, 13, 845. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Ma, Y.; Sun, W. Exploring the Dynamics of Charge Transfer in Photocatalysis: Applications of Femtosecond Transient Absorption Spectroscopy. Molecules 2024, 29, 3995. [Google Scholar] [CrossRef]
- Zhong, C.; Choi, H.; Kim, J.Y.; Woo, H.Y.; Nguyen, T.L.; Huang, F.; Cao, Y.; Heeger, A.J. Ultrafast charge transfer in operating bulk heterojunction solar cells. Adv. Mater. 2015, 27, 2036–2041. [Google Scholar] [CrossRef] [PubMed]
- Zharnikov, M. Femtosecond charge transfer dynamics in monomolecular films in the context of molecular electronics. Acc. Chem. Res. 2020, 53, 2975–2984. [Google Scholar] [CrossRef]
- Chu, W.; Tan, S.; Zheng, Q.; Fang, W.; Feng, Y.; Prezhdo, O.V.; Wang, B.; Li, X.-Z.; Zhao, J. Ultrafast charge transfer coupled to quantum proton motion at molecule/metal oxide interface. Sci. Adv. 2022, 8, eabo2675. [Google Scholar] [CrossRef]
- Hao, X.; Li, Y.; Ji, H.; Wang, T.; Fan, H.; Zhang, Q.; Yang, H.; Liu, L.; Zhang, T.; Wang, Y. Direct Visualization of Organometallic Intermediates on Cu (111) with Bond-Resolving Non-Contact Atomic Force Microscopy. Surfaces 2024, 7, 529–536. [Google Scholar] [CrossRef]
- Hao, X.; Li, Y.; Zhang, T.; Niu, M.; Yang, H.; Qiao, J.; Grazioli, C.; Guarnaccio, A.; Liu, L.; Zhang, Q. Exploring the characteristic “plug-in” configuration of an adsorbed starburst molecule. Phys. Chem. Chem. Phys. 2024, 26, 24151–24156. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Y.; Wang, T.; Zhao, Z.; Zhou, L.; Hou, B.; Ji, H.; Yang, H.; Zhang, T.; Sun, J.-T. Temperature-Driven Rotation Symmetry-Breaking States in an Atomic Kagome Metal KV3Sb5. Nano Lett. 2024, 24, 6560–6567. [Google Scholar] [CrossRef]
- Menzel, D. Ultrafast charge transfer at surfaces accessed by core electron spectroscopies. Chem. Soc. Rev. 2008, 37, 2212–2223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Svensson, P.H.; Brumboiu, I.E.; Lanzilotto, V.; Grazioli, C.; Guarnaccio, A.; Johansson, F.O.; Beranová, K.; Coreno, M.; de Simone, M. Clarifying the adsorption of triphenylamine on Au (111): Filling the HOMO–LUMO gap. J. Phys. Chem. C 2022, 126, 1635–1643. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Brumboiu, I.E.; Grazioli, C.; Guarnaccio, A.; Coreno, M.; de Simone, M.; Santagata, A.; Rensmo, H.; Brena, B.; Lanzilotto, V. Lone-pair delocalization effects within electron donor molecules: The case of triphenylamine and its thiophene-analog. J. Phys. Chem. C 2018, 122, 17706–17717. [Google Scholar] [CrossRef]
- Hao, X.; Zhang, T.; Niu, M.; Han, X.; Yang, H.; Zhang, Q.; Hou, Y.; Grazioli, C.; Liu, L.; Qiao, J. Selective Formation of Homochiral Dimers by Intermolecular Charge Transfer on a hBN Nanomesh. ACS Nano 2024, 18, 11933–11940. [Google Scholar] [CrossRef]
- Zhang, T.; Grazioli, C.; Guarnaccio, A.; Brumboiu, I.E.; Lanzilotto, V.; Johansson, F.O.; Beranová, K.; Coreno, M.; de Simone, M.; Brena, B. m-MTDATA on Au (111): Spectroscopic Evidence of Molecule–Substrate Interactions. J. Phys. Chem. C 2022, 126, 3202–3210. [Google Scholar] [CrossRef]
- Wang, L.; Chen, W.; Wee, A.T.S. Charge transfer across the molecule/metal interface using the core hole clock technique. Surf. Sci. Rep. 2008, 63, 465–486. [Google Scholar] [CrossRef]
- Cao, L.; Gao, X.Y.; Wee, A.T.; Qi, D.C. Quantitative femtosecond charge transfer dynamics at organic/electrode interfaces studied by core-hole clock spectroscopy. Adv. Mater. 2014, 26, 7880–7888. [Google Scholar] [CrossRef]
- Colombo, D.P.; Bowman, R.M. Does interfacial charge transfer compete with charge carrier recombination? A femtosecond diffuse reflectance investigation of TiO2 nanoparticles. J. Phys. Chem. 1996, 100, 18445–18449. [Google Scholar] [CrossRef]
- Costantini, R.; Morgante, A.; Dell’Angela, M. Excitation density in time-resolved water window soft X-ray spectroscopies: Experimental constraints in the detection of excited states. J. Electron Spectrosc. Relat. Phenom. 2022, 254, 147141. [Google Scholar] [CrossRef]
- Miura, Y.; Yamamoto, Y.-I.; Karashima, S.; Orimo, N.; Hara, A.; Fukuoka, K.; Ishiyama, T.; Suzuki, T. Formation of long-lived dark states during electronic relaxation of pyrimidine nucleobases studied using extreme ultraviolet time-resolved photoelectron spectroscopy. J. Am. Chem. Soc. 2023, 145, 3369–3381. [Google Scholar] [CrossRef]
- Brauer, J.C.; Marchioro, A.; Paraecattil, A.A.; Oskouei, A.A.; Moser, J.-E. Dynamics of interfacial charge transfer states and carriers separation in dye-sensitized solar cells: A time-resolved terahertz spectroscopy study. J. Phys. Chem. C 2015, 119, 26266–26274. [Google Scholar] [CrossRef]
- Tajima, N.; Kanemoto, K. Time-Resolved Operando Spectroscopy for Dye-Sensitized Solar Cells from Multiple Perspectives. J. Phys. Chem. C 2022, 126, 7535–7541. [Google Scholar] [CrossRef]
- Khalili, K.; Inhester, L.; Arnold, C.; Welsch, R.; Andreasen, J.W.; Santra, R. Hole dynamics in a photovoltaic donor-acceptor couple revealed by simulated time-resolved X-ray absorption spectroscopy. Struct. Dyn. 2019, 6, 044102. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Kafle, T.R.; Kattel, B.; Chan, W.-L. A multidimensional view of charge transfer excitons at organic donor–acceptor interfaces. J. Am. Chem. Soc. 2017, 139, 4098–4106. [Google Scholar] [CrossRef]
- Pettine, J.; Maioli, P.; Vallée, F.; Del Fatti, N.; Nesbitt, D.J. Energy-resolved femtosecond hot electron dynamics in single plasmonic nanoparticles. ACS Nano 2023, 17, 10721–10732. [Google Scholar] [CrossRef]
- Manuel, A.P.; Shankar, K. Hot electrons in TiO2–noble metal nano-heterojunctions: Fundamental science and applications in photocatalysis. Nanomaterials 2021, 11, 1249. [Google Scholar] [CrossRef]
- Guo, Z.; Driver, T.; Beauvarlet, S.; Cesar, D.; Duris, J.; Franz, P.L.; Alexander, O.; Bohler, D.; Bostedt, C.; Averbukh, V. Experimental demonstration of attosecond pump–probe spectroscopy with an X-ray free-electron laser. Nat. Photonics 2024, 18, 691–697. [Google Scholar] [CrossRef]
- Lara-Astiaso, M.; Galli, M.; Trabattoni, A.; Palacios, A.; Ayuso, D.; Frassetto, F.; Poletto, L.; De Camillis, S.; Greenwood, J.; Decleva, P. Attosecond pump–probe spectroscopy of charge dynamics in tryptophan. J. Phys. Chem. Lett. 2018, 9, 4570–4577. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, B.; Zhang, L.; Yu, J. Femtosecond transient absorption spectroscopy investigation into the electron transfer mechanism in photocatalysis. Chem. Commun. 2023, 59, 688–699. [Google Scholar] [CrossRef]
- Cheng, C.; Zhang, J.; Zhu, B.; Liang, G.; Zhang, L.; Yu, J. Verifying the charge-transfer mechanism in S-Scheme heterojunctions using femtosecond transient absorption spectroscopy. Angew. Chem. Int. Ed. 2023, 62, e202218688. [Google Scholar] [CrossRef]
- Huang, B.; Fu, X.; Wang, K.; Wang, L.; Zhang, H.; Liu, Z.; Liu, B.; Li, J. Chemically bonded BiVO4/Bi19Cl3S27 heterojunction with fast hole extraction dynamics for continuous CO2 photoreduction. Adv. Powder Mater. 2024, 3, 100140. [Google Scholar] [CrossRef]
- Xu, H.-Q.; Hu, J.; Wang, D.; Li, Z.; Zhang, Q.; Luo, Y.; Yu, S.-H.; Jiang, H.-L. Visible-light photoreduction of CO2 in a metal–organic framework: Boosting electron–hole separation via electron trap states. J. Am. Chem. Soc. 2015, 137, 13440–13443. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, W.; Liu, J.; Fujimori, Y.; Higashino, T.; Imahori, H.; Jiang, X.; Zhao, J.; Sakurai, T.; Hattori, Y. A new class of epitaxial porphyrin metal–organic framework thin films with extremely high photocarrier generation efficiency: Promising materials for all-solid-state solar cells. J. Mater. Chem. A 2016, 4, 12739–12747. [Google Scholar] [CrossRef]
- Moore, G.J.; Günther, F.; Yallum, K.M.; Causa’, M.; Jungbluth, A.; Réhault, J.; Riede, M.; Ortmann, F.; Banerji, N. Direct visualization of the charge transfer state dynamics in dilute-donor organic photovoltaic blends. Nat. Commun. 2024, 15, 9851. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kamat, P.V.; Janáky, C.; Samu, G.F. Charge transfer kinetics in halide perovskites: On the constraints of time-resolved spectroscopy measurements. ACS Energy Lett. 2024, 9, 3187–3203. [Google Scholar] [CrossRef]
- Björneholm, O.; Nilsson, A.; Sandell, A.; Hernnäs, B.; Mrtensson, N. Determination of time scales for charge-transfer screening in physisorbed molecules. Phys. Rev. Lett. 1992, 68, 1892. [Google Scholar] [CrossRef]
- Brühwiler, P.; Karis, O.; Mårtensson, N. Charge-transfer dynamics studied using resonant core spectroscopies. Rev. Mod. Phys. 2002, 74, 703. [Google Scholar] [CrossRef]
- Ohno, M. Deexcitation processes in adsorbates. Phys. Rev. B 1994, 50, 2566. [Google Scholar] [CrossRef]
- Karis, O.; Nilsson, A.; Weinelt, M.; Wiell, T.; Puglia, C.; Wassdahl, N.; Mårtensson, N.; Samant, M.; Stöhr, J. One-step and two-step description of deexcitation processes in weakly interacting systems. Phys. Rev. Lett. 1996, 76, 1380. [Google Scholar] [CrossRef]
- Muchova, E.; Gopakumar, G.; Unger, I.; Öhrwall, G.; Céolin, D.; Trinter, F.; Wilkinson, I.; Chatzigeorgiou, E.; Slavíček, P.; Hergenhahn, U. Attosecond formation of charge-transfer-to-solvent states of aqueous ions probed using the core-hole-clock technique. Nat. Commun. 2024, 15, 8903. [Google Scholar] [CrossRef]
- Tendo, S.; Niozu, A.; Yoshioka, K.; Tabuse, M.; Adachi, J.-I.; Tanaka, H.; Wada, S.-I. Comparative study of electron transport through aromatic molecules on gold nanoparticles: Insights from soft X-ray spectroscopy of condensed nanoparticle films versus flat monolayer films. Phys. Chem. Chem. Phys. 2025, 27, 388–396. [Google Scholar] [CrossRef]
- Cao, L.; Zhang, Z.; Thompson, D.; Qi, D.-C.; Nijhui, C.A. Resolving charge transfer mechanisms in molecular tunnel junctions using dynamic charge transfer and static current–voltage measurements. J. Mater. Chem. C 2024, 12, 1701–1709. [Google Scholar] [CrossRef]
- Oropeza, F.E.; Barawi, M.; Alfonso-González, E.; Trigo, J.F.; Guillén, C.; Saiz, F.; Villar-Garcia, I.J. Understanding ultrafast charge transfer processes in SnS and SnS2: Using the core hole clock method to measure attosecond orbital-dependent electron delocalisation in semiconducting layered materials. J. Mater. Chem. C 2021, 9, 11859–11872. [Google Scholar] [CrossRef]
- Velasquez, N.; Nunes, F.B.; Travnikova, O.; Ismail, I.; Guillemin, R.; Martins, J.B.; Céolin, D.; Journel, L.; Fillaud, L.; Koulentianos, D. X-ray induced ultrafast charge transfer in thiophene-based conjugated polymers controlled by core-hole clock spectroscopy. Phys. Chem. Chem. Phys. 2024, 26, 1234–1244. [Google Scholar] [CrossRef]
- Berggren, E.; Weng, Y.-C.; Li, Q.; Yang, C.-Y.; Johansson, F.O.; Cappel, U.B.; Berggren, M.; Fabiano, S.; Lindblad, A. Charge Transfer in the P (g42T-T): BBL Organic Polymer Heterojunction Measured with Core-Hole Clock Spectroscopy. J. Phys. Chem. C 2023, 127, 23733–23742. [Google Scholar] [CrossRef]
- Xue, J.; Bao, J. Interfacial charge transfer of heterojunction photocatalysts: Characterization and calculation. Surf. Interfaces 2021, 25, 101265. [Google Scholar] [CrossRef]
- Schnadt, J.; Brühwiler, P.A.; Patthey, L.; O’Shea, J.N.; Södergren, S.; Odelius, M.; Ahuja, R.; Karis, O.; Bässler, M.; Persson, P. Experimental evidence for sub-3-fs charge transfer from an aromatic adsorbate to a semiconductor. Nature 2002, 418, 620–623. [Google Scholar] [CrossRef]
- Borges, B.; Veiga, A.G.; Tzounis, L.; Laskarakis, A.; Logothetidis, S.; Rocco, M. Molecular orientation and ultrafast charge transfer dynamics studies on the P3HT: PCBM blend. J. Phys. Chem. C 2016, 120, 25078–25082. [Google Scholar] [CrossRef]
- Shrotriya, V.; Ouyang, J.; Tseng, R.J.; Li, G.; Yang, Y. Absorption spectra modification in poly (3-hexylthiophene): Methanofullerene blend thin films. Chem. Phys. Lett. 2005, 411, 138–143. [Google Scholar] [CrossRef]
- Tremel, K.; Ludwigs, S. Morphology of P3HT in thin films in relation to optical and electrical properties. P3HT Revisit.–Mol. Scale Sol. Cell Devices 2014, 265, 39–82. [Google Scholar]
- Garcia-Basabe, Y.; Parra, G.G.; Barioni, M.B.; Mendoza, C.D.; Vicentin, F.C.; Larrudé, D.G. Species selective charge transfer dynamics in a P3HT/MoS 2 van der Waals heterojunction: Fluorescence lifetime microscopy and core hole clock spectroscopy approaches. Phys. Chem. Chem. Phys. 2019, 21, 23521–23532. [Google Scholar] [CrossRef] [PubMed]
- Eknapakul, T.; King, P.; Asakawa, M.; Buaphet, P.; He, R.-H.; Mo, S.-K.; Takagi, H.; Shen, K.; Baumberger, F.; Sasagawa, T. Electronic structure of a quasi-freestanding MoS2 monolayer. Nano Lett. 2014, 14, 1312–1316. [Google Scholar] [CrossRef]
- Ren, S.; Bernardi, M.; Lunt, R.R.; Bulovic, V.; Grossman, J.C.; Gradecak, S. Toward efficient carbon nanotube/P3HT solar cells: Active layer morphology, electrical, and optical properties. Nano Lett. 2011, 11, 5316–5321. [Google Scholar] [CrossRef] [PubMed]
- Berger, P.; Kim, M. Polymer solar cells: P3HT: PCBM and beyond. J. Renew. Sustain. Energy 2018, 10, 013508. [Google Scholar] [CrossRef]
- Mir, A.M.; Bashir, F.; Khanday, F.A.; Zahoor, F.; Hanif, M.; May, Z. Design and optimization of high performance P3HT: PCBM Polymer Solar Cell using P3HT buffer layer. IEEE Access 2024, 12, 10961–10969. [Google Scholar] [CrossRef]
- Garcia-Basabe, Y.; Ceolin, D.; Zarbin, A.J.; Roman, L.S.; Rocco, M.L.M. Ultrafast interface charge transfer dynamics on P3HT/MWCNT nanocomposites probed by resonant Auger spectroscopy. RSC Adv. 2018, 8, 26416–26422. [Google Scholar] [CrossRef]
- Singh, V.; Arora, S.; Arora, M.; Sharma, V.; Tandon, R. Optimizing P3HT/PCBM/MWCNT films for increased stability in polymer bulk heterojunction solar cells. Phys. Lett. A 2014, 378, 3046–3054. [Google Scholar] [CrossRef]
- Tiwari, S.P.; Verma, R.; Alam, M.B.; Kumari, R.; Sinha, O.; Srivastava, R. Charge transport study of P3HT blended MoS2. Vacuum 2017, 146, 474–477. [Google Scholar] [CrossRef]
- Chaudhary, V.; Pandey, R.K.; Sahu, P.K.; Prakash, R.; Kumar, N.; Kumar Singh, A. MoS2 assisted self-assembled poly (3-hexylthiophene) thin films at an air/liquid interface for high-performance field-effect transistors under ambient conditions. J. Phys. Chem. C 2020, 124, 8101–8109. [Google Scholar] [CrossRef]
- Tachikawa, H.; Kawabata, H.; Abe, S.; Watanabe, I. Mechanism of Carrier Formation in P3HT-C60-PCBM Solar Cells. Nanomaterials 2024, 14, 1400. [Google Scholar] [CrossRef]
- Kroto, H.W.; Allaf, A.; Balm, S. C60: Buckminsterfullerene. Chem. Rev. 1991, 91, 1213–1235. [Google Scholar] [CrossRef]
- Çakır, Ü.; Kestel, F.; Kızılduman, B.K.; Bicil, Z.; Doğan, M. Multi walled carbon nanotubes functionalized by hydroxyl and Schiff base and their hydrogen storage properties. Diam. Relat. Mater. 2021, 120, 108604. [Google Scholar] [CrossRef]
- Borges, B.; Roman, L.; Rocco, M. Femtosecond and attosecond electron transfer dynamics of semiconductors probed by the core-hole clock spectroscopy. Top. Catal. 2019, 62, 1004–1010. [Google Scholar] [CrossRef]
- Stranks, S.D.; Weisspfennig, C.; Parkinson, P.; Johnston, M.B.; Herz, L.M.; Nicholas, R.J. Ultrafast charge separation at a polymer− single-walled carbon nanotube molecular junction. Nano Lett. 2011, 11, 66–72. [Google Scholar] [CrossRef]
- Kafle, T.R.; Kattel, B.; Lane, S.D.; Wang, T.; Zhao, H.; Chan, W.-L. Charge transfer exciton and spin flipping at organic–transition-metal dichalcogenide interfaces. ACS Nano 2017, 11, 10184–10192. [Google Scholar] [CrossRef]
- Cho, M. Coherent two-dimensional optical spectroscopy. Chem. Rev. 2008, 108, 1331–1418. [Google Scholar] [CrossRef]
- Geneaux, R.; Marroux, H.J.; Guggenmos, A.; Neumark, D.M.; Leone, S.R. Transient absorption spectroscopy using high harmonic generation: A review of ultrafast X-ray dynamics in molecules and solids. Philos. Trans. R. Soc. A 2019, 377, 20170463. [Google Scholar] [CrossRef]
- Föhlisch, A.; Vijayalakshmi, S.; Hennies, F.; Wurth, W.; Medicherla, V.; Drube, W. Verification of the core-hole-clock method using two different time references: Attosecond charge transfer in c (4 × 2) S/Ru (0 0 0 1). Chem. Phys. Lett. 2007, 434, 214–217. [Google Scholar] [CrossRef]
- Lindblad, R. Electronic Structures and Energy Level Alignment in Mesoscopic Solar Cells: A Hard and Soft X-ray Photoelectron Spectroscopy Study. Ph.D. Thesis, Uppsala University, Uppsala, Sweden, 2014. [Google Scholar]
- Elder, F.; Gurewitsch, A.; Langmuir, R.; Pollock, H. Radiation from electrons in a synchrotron. Phys. Rev. 1947, 71, 829. [Google Scholar] [CrossRef]
- Toffoli, D.; Guarnaccio, A.; Grazioli, C.; Zhang, T.; Johansson, F.; De Simone, M.; Coreno, M.; Santagata, A.; D’Auria, M.; Puglia, C. Electronic structure characterization of a thiophene benzo-annulated series of common building blocks for donor and acceptor compounds studied by gas phase photoelectron and photoabsorption synchrotron spectroscopies. J. Phys. Chem. A 2018, 122, 8745–8761. [Google Scholar] [CrossRef]
- Cappel, U.B.; Svanström, S.; Lanzilotto, V.; Johansson, F.O.; Aitola, K.; Philippe, B.; Giangrisostomi, E.; Ovsyannikov, R.; Leitner, T.; Föhlisch, A. Partially reversible photoinduced chemical changes in a mixed-ion perovskite material for solar cells. ACS Appl. Mater. Interfaces 2017, 9, 34970–34978. [Google Scholar] [CrossRef]
- Vilmercati, P.; Cvetko, D.; Cossaro, A.; Morgante, A. Heterostructured organic interfaces probed by resonant photoemission. Surf. Sci. 2009, 603, 1542–1556. [Google Scholar] [CrossRef]
- Fadley, C.S. X-ray photoelectron spectroscopy: Progress and perspectives. J. Electron Spectrosc. Relat. Phenom. 2010, 178, 2–32. [Google Scholar] [CrossRef]
- Doniach, S.; Sunjic, M. Many-electron singularity in X-ray photoemission and X-ray line spectra from metals. J. Phys. C Solid State Phys. 1970, 3, 285. [Google Scholar] [CrossRef]
- Campbell, J.; Papp, T. Widths of the atomic K–N7 levels. At. Data Nucl. Data Tables 2001, 77, 1–56. [Google Scholar] [CrossRef]
- Zhang, Z.; Cao, L.; Chen, X.; Thompson, D.; Qi, D.; Nijhuis, C.A. Energy-level alignment and orbital-selective femtosecond charge transfer dynamics of redox-active molecules on Au, Ag, and Pt metal surfaces. J. Phys. Chem. C 2021, 125, 18474–18482. [Google Scholar] [CrossRef]
- Wächter, T.; Tröster, A.; Hock, S.; Terfort, A.; Zharnikov, M. Dynamics of electron transfer in self-assembled monolayers with acene backbone. J. Phys. Chem. C 2018, 122, 4105–4115. [Google Scholar] [CrossRef]
- Mekuye, B.; Abera, B. Nanomaterials: An overview of synthesis, classification, characterization, and applications. Nano Sel. 2023, 4, 486–501. [Google Scholar] [CrossRef]
- Ajayi, T.M.; Shirato, N.; Rojas, T.; Wieghold, S.; Cheng, X.; Latt, K.Z.; Trainer, D.J.; Dandu, N.K.; Li, Y.; Premarathna, S. Characterization of just one atom using synchrotron X-rays. Nature 2023, 618, 69–73. [Google Scholar] [CrossRef]
- Sloboda, T.; Johansson, F.O.; Kammlander, B.; Berggren, E.; Svanström, S.; Fernández, A.G.; Lindblad, A.; Cappel, U.B. Unravelling the ultrafast charge dynamics in PbS quantum dots through resonant Auger mapping of the sulfur K-edge. RSC Adv. 2022, 12, 31671–31679. [Google Scholar] [CrossRef]
- Cao, L.; Yang, M.; Yuan, L.; Nerngchamnong, N.; Feng, Y.-P.; Wee, A.T.; Qi, D.-C.; Nijhuis, C.A. Orbital dependent ultrafast charge transfer dynamics of ferrocenyl-functionalized SAMs on gold studied by core-hole clock spectroscopy. J. Phys. Condens. Matter 2016, 28, 094006. [Google Scholar] [CrossRef]
- Wang, F.; Wang, Z.; Jiang, C.; Yin, L.; Cheng, R.; Zhan, X.; Xu, K.; Wang, F.; Zhang, Y.; He, J. Progress on Electronic and Optoelectronic Devices of 2D Layered Semiconducting Materials. Small 2017, 13, 1604298. [Google Scholar] [CrossRef] [PubMed]
- Ota, H.; Salehi, E.; Fujimoto, M.; Hayashi, K.; Horigome, T.; Iwayama, H.; Katoh, M.; Kondo, N.; Makita, S.; Matsui, F. UVSOR synchrotron facility update. J. Phys. Conf. Ser. 2022, 2380, 012003. [Google Scholar] [CrossRef]
- Ternov, I.M. Synchrotron radiation. Phys. Uspekhi 1995, 38, 409. [Google Scholar] [CrossRef]
- Zaumseil, J. P3HT and other polythiophene field-effect transistors. In P3HT Revisited—From Molecular Scale to Solar Cell Devices; Springer: Berlin/Heidelberg, Germany, 2014; pp. 107–137. [Google Scholar]
- Kumari, P.; Hajduk, B.; Jarka, P.; Bednarski, H.; Janeczek, H.; Łapkowski, M.; Waśkiewicz, S. A Supramolecular Approach to Enhance the Optoelectronic Properties of P3HT-b-PEG Block Copolymer for Organic Field-Effect Transistors. ACS Omega 2024, 9, 39023–39032. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Yang, Y.; Durstock, M.; Baek, J.-B.; Dai, L. Soluble P3HT-grafted graphene for efficient bilayer−heterojunction photovoltaic devices. ACS Nano 2010, 4, 5633–5640. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, Z.; Zhang, X.; Yang, L.; Zhang, N.; Pan, G.; Yin, S.; Chen, Y.; Wei, J. Polymer photovoltaic cells based on solution-processable graphene and P3HT. Adv. Funct. Mater. 2009, 19, 894–904. [Google Scholar] [CrossRef]
- El Gemayel, M.; Narita, A.; Dössel, L.F.; Sundaram, R.S.; Kiersnowski, A.; Pisula, W.; Hansen, M.R.; Ferrari, A.C.; Orgiu, E.; Feng, X. Graphene nanoribbon blends with P3HT for organic electronics. Nanoscale 2014, 6, 6301–6314. [Google Scholar] [CrossRef]
- Smith, M.K.; Singh, V.; Kalaitzidou, K.; Cola, B.A. High thermal and electrical conductivity of template fabricated P3HT/MWCNT composite nanofibers. ACS Appl. Mater. Interfaces 2016, 8, 14788–14794. [Google Scholar] [CrossRef]
- Nurazzi, N.; Abdullah, N.; Demon, S.; Halim, N.; Mohamad, I. The Influence of Reaction Time on Non-Covalent Functionalisation of P3HT/MWCNT Nanocomposites. Polymers 2021, 13, 1916. [Google Scholar] [CrossRef]
- Chan, S.-H.; Lai, C.-S.; Chen, H.-L.; Ting, C.; Chen, C.-P. Highly efficient P3HT: C60 solar cell free of annealing process. Macromolecules 2011, 44, 8886–8891. [Google Scholar] [CrossRef]
- Yang, Z.; Fu, K.; Yu, J.; Shi, X.; Zhou, P.; Cheng, Z. Facile preparation of nanoporous C60/P3HT thin films from PLA-b-C60-b-P3HT triblock copolymers. Appl. Surf. Sci. 2018, 458, 70–76. [Google Scholar] [CrossRef]
- Dang, M.T.; Hirsch, L.; Wantz, G. P3HT: PCBM, Best Seller in Polymer Photovoltaic Research; Wiley Online Library: Hoboken, NJ, USA, 2011. [Google Scholar]
- Chi, D.; Qu, S.; Wang, Z.; Wang, J. High efficiency P3HT: PCBM solar cells with an inserted PCBM layer. J. Mater. Chem. C 2014, 2, 4383–4387. [Google Scholar] [CrossRef]
- Mahakul, P.C.; Sa, K.; Das, B.; Mahanandia, P. Structural investigation of the enhanced electrical, optical and electrochemical properties of MWCNT incorporated Poly [3-hexylthiophene-2, 5-diyl] composites. Mater. Chem. Phys. 2017, 199, 477–484. [Google Scholar] [CrossRef]
- Lüer, L.; Hoseinkhani, S.; Meneghetti, M.; Lanzani, G. Dynamical screening of the exciton resonance in conjugated polymers/carbon nanotubes composites. Phys. Rev. B Condens. Matter Mater. Phys. 2010, 81, 155411. [Google Scholar] [CrossRef]
- Bindl, D.J.; Safron, N.S.; Arnold, M.S. Dissociating excitons photogenerated in semiconducting carbon nanotubes at polymeric photovoltaic heterojunction interfaces. ACS Nano 2010, 4, 5657–5664. [Google Scholar] [CrossRef]
- Liirò-Peluso, L.; Wrigley, J.; Amabilino, D.B.; Beton, P.H. Submolecular resolution imaging of P3HT: PCBM nanostructured films by atomic force microscopy: Implications for organic solar cells. ACS Appl. Nano Mater. 2022, 5, 13794–13804. [Google Scholar] [CrossRef]
- Garcia-Basabe, Y.; Gordo, V.O.; Daminelli, L.M.; Mendoza, C.D.; Vicentin, F.C.; Matusalem, F.; Rocha, A.R.; de Matos, C.J.; Larrudé, D.G. Interfacial electronic coupling and band alignment of P3HT and exfoliated black phosphorous van der Waals heterojunctions. Appl. Surf. Sci. 2021, 541, 148455. [Google Scholar] [CrossRef]
- Zeng, L.; Zhang, X.; Liu, Y.; Yang, X.; Wang, J.; Liu, Q.; Luo, Q.; Jing, C.; Yu, X.-F.; Qu, G. Surface and interface control of black phosphorus. Chem 2022, 8, 632–662. [Google Scholar] [CrossRef]
- Zhou, S.; Bao, C.; Fan, B.; Zhou, H.; Gao, Q.; Zhong, H.; Lin, T.; Liu, H.; Yu, P.; Tang, P. Pseudospin-selective Floquet band engineering in black phosphorus. Nature 2023, 614, 75–80. [Google Scholar] [CrossRef]
- Chen, C.; Yin, Y.; Zhang, R.; Yuan, Q.; Xu, Y.; Zhang, Y.; Chen, J.; Zhang, Y.; Li, C.; Wang, J. Growth of single-crystal black phosphorus and its alloy films through sustained feedstock release. Nat. Mater. 2023, 22, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Scittarelli, D.; Coiai, S.; Cicogna, F.; Legnaioli, S.; Dell’Angela, M.; Verdini, A.; Costantini, R.; Serrano-Ruiz, M.; Passaglia, E.; Caporali, M. Nanohybrids of 2D Black Phosphorus with Phthalocyanines: Role of Interfacial Interactions in Heterostructure Development. Chem. A Eur. J. 2024, 31, e202403570. [Google Scholar] [CrossRef]
- Zhu, X.-Y.; Yang, Q.; Muntwiler, M. Charge-transfer excitons at organic semiconductor surfaces and interfaces. Acc. Chem. Res. 2009, 42, 1779–1787. [Google Scholar] [CrossRef]
- Oevering, H.; Verhoeven, J.; Paddon-Row, M.; Warman, J. Charge-transfer absorption and emission resulting from long-range through-bond interaction; exploring the relation between electronic coupling andelectron-transfer in bridged donor-acceptor systems. Tetrahedron 1989, 45, 4751–4766. [Google Scholar] [CrossRef]
- Difley, S.; Van Voorhis, T. Exciton/charge-transfer electronic couplings in organic semiconductors. J. Chem. Theory Comput. 2011, 7, 594–601. [Google Scholar] [CrossRef]
- Endicott, J.F.; Chen, Y.-J.; Xie, P. Electron-transfer spectroscopy: Donor–acceptor electronic coupling, reorganizational energies, reaction pathways and dynamics. Coord. Chem. Rev. 2005, 249, 343–373. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Photon Energy (eV) | (fs) | ||
|---|---|---|---|
| P3HT | P3HT/PCBM | P3HT/Fe-MWCNT-5% (fs) | |
| 2470.8 | - | 7.19 | - |
| 2471.2 | - | 8.49 | - |
| 2471.9 | 4.7 | - | 6.5 |
| 2472 | - | 5.76 | - |
| 2473.2 | 8.9 | 1.69 | 5.3 |
| 2474.4 | 5.5 | - | 7.6 |
| 2479.2 | - | 0.62 | - |
| 2490 | - | 0.22 | - |
| Photon Energy (eV) | (fs) | |||
|---|---|---|---|---|
| P3HT/MoS2/SiO2 | ||||
| MoS2/SiO2 | P3HT/SiO2 | MoS2 | P3HT | |
| 2470.5 | 1.32 | - | 1.25 | - |
| 2472.1 | 0.62 | 11.3 | 0.34 | 2.41 |
| 2472.9 | 0.5 | 4.13 | 0.2 | 0.45 |
| 2474.5 | - | 0.36 | - | 0.32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Hao, X.; Cao, X.; Wang, T.; Fan, H.; Zhan, L.; Zhou, Z.; Yang, H.; Zhang, Q.; Costantini, R.; et al. Ultra-Fast Charge Transfer in P3HT Composites Using the Core Hole Clock Technique. Nanomaterials 2025, 15, 433. https://doi.org/10.3390/nano15060433
Li Y, Hao X, Cao X, Wang T, Fan H, Zhan L, Zhou Z, Yang H, Zhang Q, Costantini R, et al. Ultra-Fast Charge Transfer in P3HT Composites Using the Core Hole Clock Technique. Nanomaterials. 2025; 15(6):433. https://doi.org/10.3390/nano15060433
Chicago/Turabian StyleLi, Yan, Xiaoyu Hao, Xiongbai Cao, Tingting Wang, Haolong Fan, Lingtao Zhan, Zhenru Zhou, Huixia Yang, Quanzhen Zhang, Roberto Costantini, and et al. 2025. "Ultra-Fast Charge Transfer in P3HT Composites Using the Core Hole Clock Technique" Nanomaterials 15, no. 6: 433. https://doi.org/10.3390/nano15060433
APA StyleLi, Y., Hao, X., Cao, X., Wang, T., Fan, H., Zhan, L., Zhou, Z., Yang, H., Zhang, Q., Costantini, R., Grazioli, C., Zhang, T., & Wang, Y. (2025). Ultra-Fast Charge Transfer in P3HT Composites Using the Core Hole Clock Technique. Nanomaterials, 15(6), 433. https://doi.org/10.3390/nano15060433