Mesoporous Silica Nanoparticles as Drug Delivery Vehicles in Cancer


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. MSN Synthesis and Characteristics
2.1. MSN Synthesis
2.2. Influence of MSN Characteristics on Biological Systems
3. Modifications to Control Cellular Uptake, Drug Release and Endosomal Escape
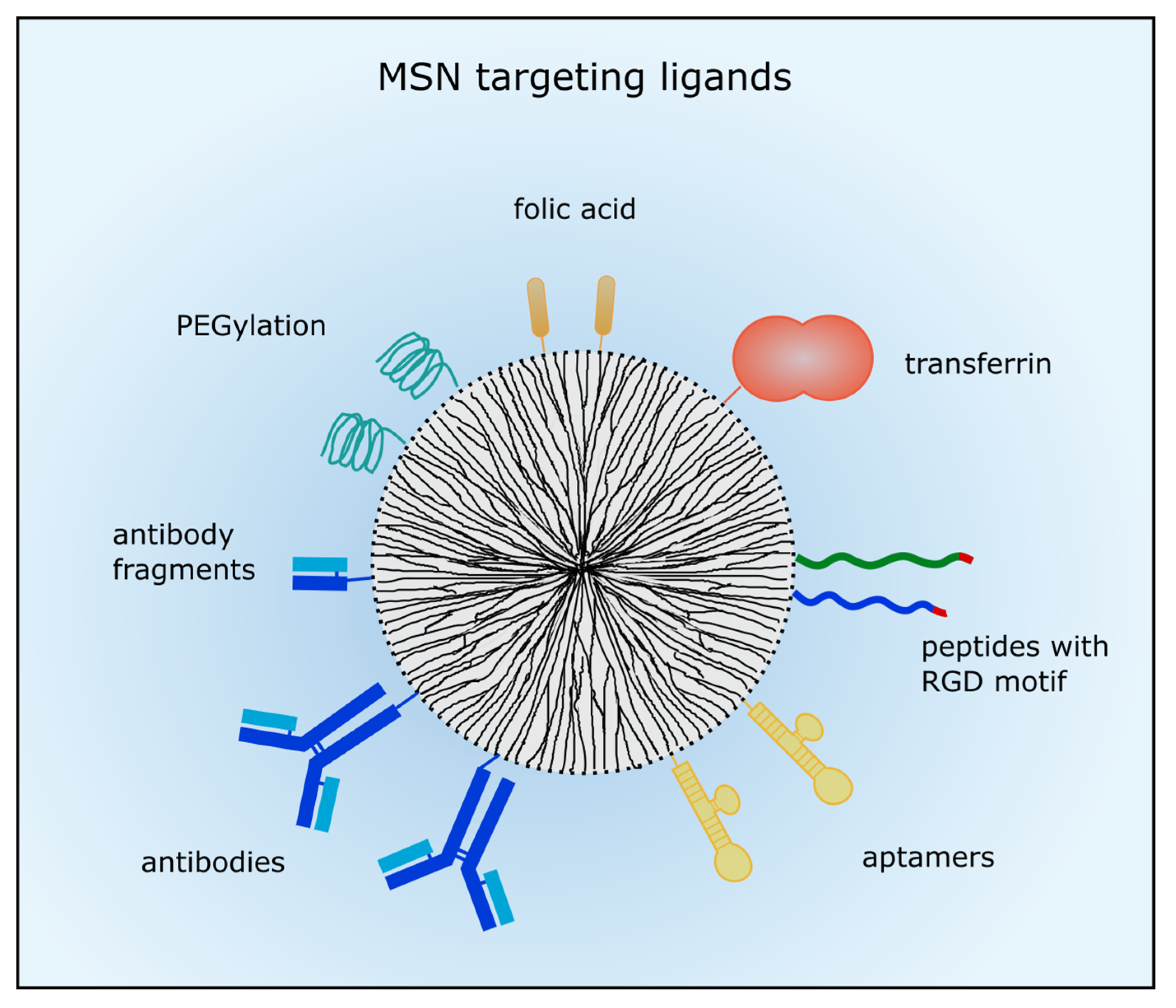
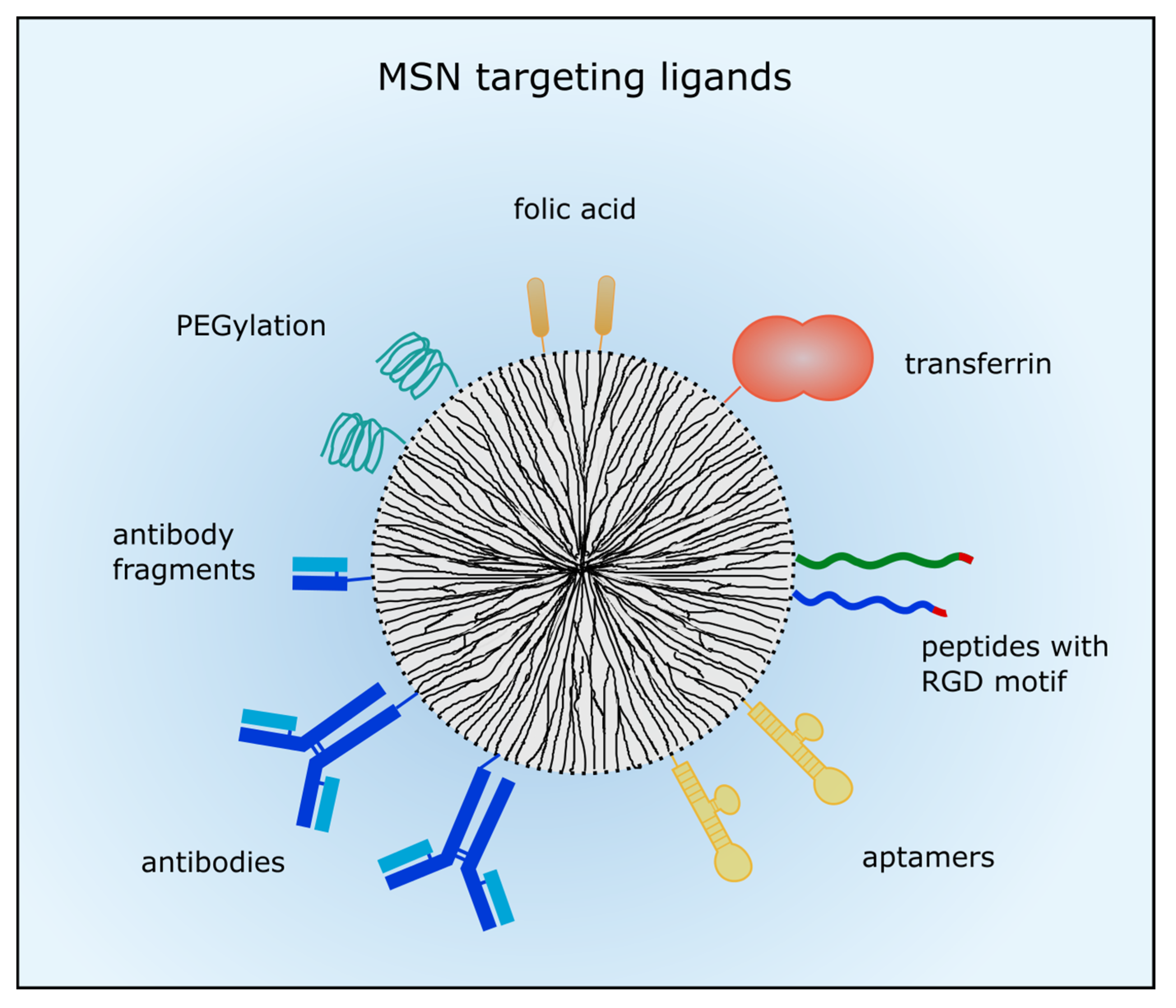
3.1. Passive and Active Targeting of MSNs
3.1.1. Passive Targeting
3.1.2. Active Targeting
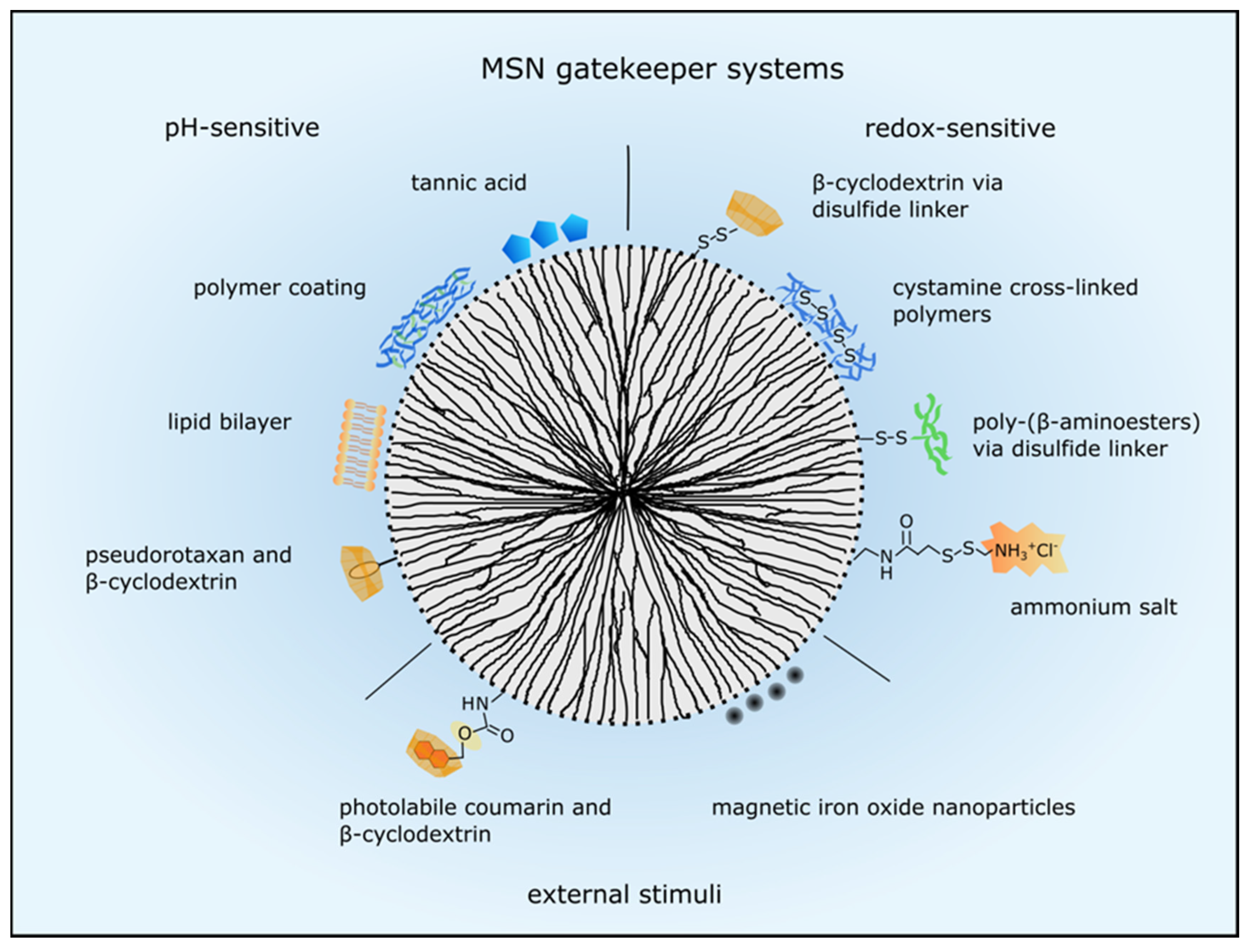
3.2. Systems for Controlled Drug Release
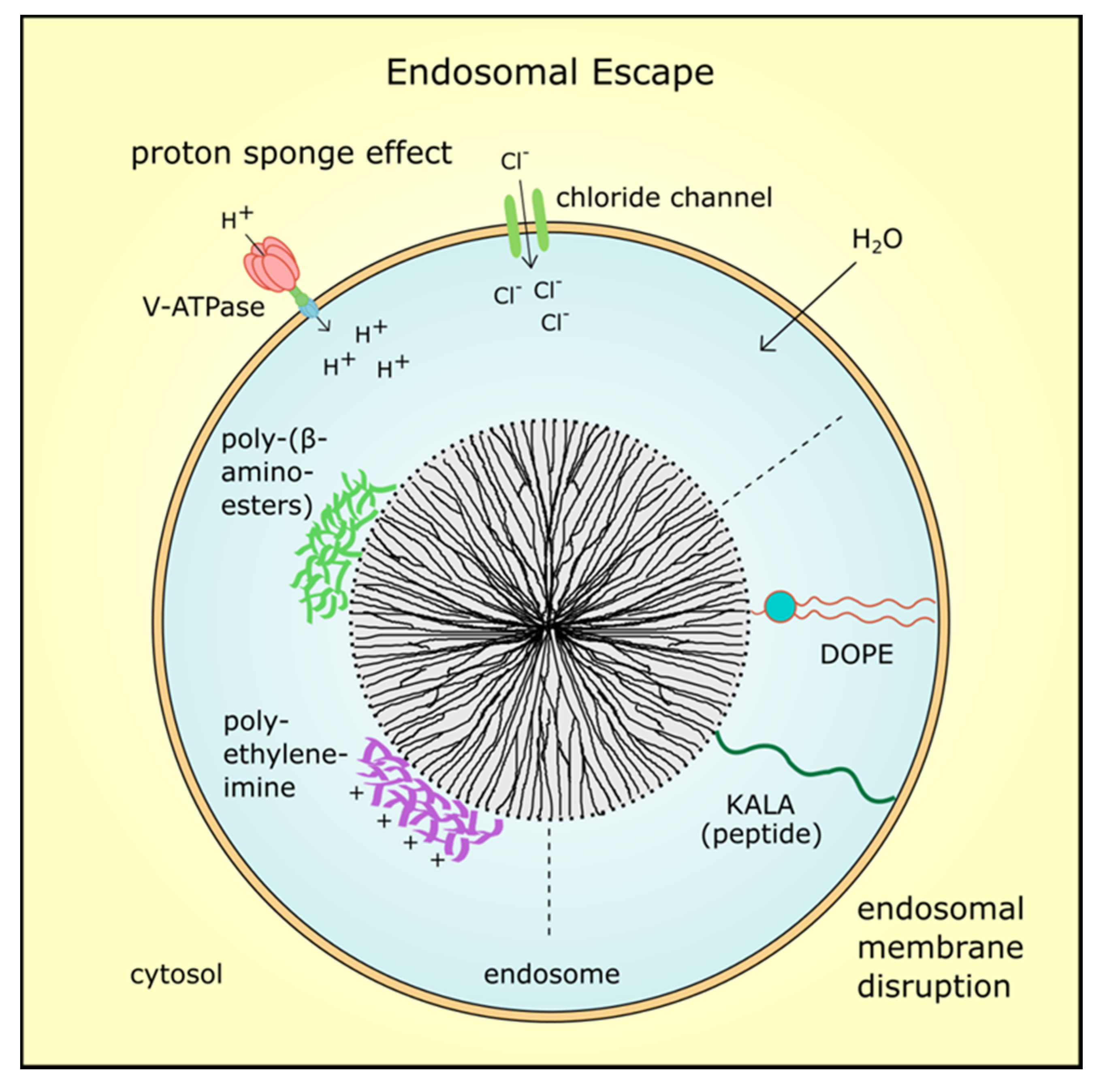
3.3. Endosomal Escape of MSN and Their Cargo
4. Biocompatibility of MSN
5. Possible Challenges of MSN Application in Cancer Therapy
6. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA. Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [CrossRef] [PubMed]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Martin, F.J. Advantages of liposomal delivery systems for anthracyclines. Semin. Oncol. 2004, 31, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Susewind, M.; Schilmann, A.-M.; Heim, J.; Henkel, A.; Link, T.; Fischer, K.; Strand, D.; Kolb, U.; Tahir, M.N.; Brieger, J.; et al. Silica-coated Au@ZnO Janus particles and their stability in epithelial cells. J. Mater. Chem. B 2015, 3, 1813–1822. [Google Scholar] [CrossRef]
- Wang, Y.X.J.; Hussain, S.M.; Krestin, G.P. Superparamagnetic iron oxide contrast agents: Physicochemical characteristics and applications in MR imaging. Eur. Radiol. 2001, 11, 2319–2331. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.; Kostarelos, K.; Prato, M. Applications of carbon nanotubes in drug delivery. Curr. Opin. Chem. Biol. 2005, 9, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Bruchez, M., Jr.; Moronne, M.; Gin, P.; Weiss, S.; Alivisatos, A.P. Semiconductor Nanocrystals as Fluorescent Biological Labels. Science 1998, 281, 2013–2016. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Liong, M.; Zink, J.I.; Tamanoi, F. Mesoporous Silica Nanoparticles as a Delivery System for Hydrophobic Anticancer Drugs. Small 2007, 3, 1341–1346. [Google Scholar] [CrossRef] [PubMed]
- Targeted Silica Nanoparticles for Image-Guided Intraoperative Sentinel Lymph Node Mapping in Head and Neck Melanoma, Breast and Gynecologic Malignancies. Available online: https://clinicaltrials.gov/ct2/show/NCT02106598?term=silica+nanoparticle&rank=1 (accessed on 6 April 2017).
- US Food and Drug Administration GRAS Substances (SCOGS) Database—Select Committee on GRAS Substances (SCOGS) Opinion: Silicates. Available online: https://www.fda.gov/food/ingredientspackaginglabeling/gras/scogs/ucm260849.htm (accessed on 7 April 2017).
- Liberman, A.; Mendez, N.; Trogler, W.C.; Kummel, A.C. Synthesis and surface functionalization of silica nanoparticles for nanomedicine. Surf. Sci. Rep. 2014, 69, 132–158. [Google Scholar] [CrossRef] [PubMed]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 1992, 359, 710–712. [Google Scholar] [CrossRef]
- Tang, F.; Li, L.; Chen, D. Mesoporous silica nanoparticles: Synthesis, biocompatibility and drug delivery. Adv. Mater. 2012, 24, 1504–1534. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Feng, J.; Huo, Q.; Melosh, N.; Fredrickson, G.; Chmelka, B.; Stucky, G. Triblock copolymer syntheses of mesoporous silica with periodic 50 to 300 angstrom pores. Science 1998, 279, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Na, H.-K.; Kim, M.-H.; Park, K.; Ryoo, S.-R.; Lee, K.E.; Jeon, H.; Ryoo, R.; Hyeon, C.; Min, D.-H. Efficient functional delivery of siRNA using mesoporous silica nanoparticles with ultralarge pores. Small 2012, 8, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Meng, Q.; Chen, Y.; Zhang, L.; Li, M.; Cai, X.; Li, Y.; Yu, P.; Zhang, L.; Shi, J. Large Pore-Sized Hollow Mesoporous Organosilica for Redox-Responsive Gene Delivery and Synergistic Cancer Chemotherapy. Adv. Mater. 2016, 28, 1963–1969. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.M.; Shahin, S.A.; Wen, W.; Finlay, J.B.; Dong, J.; Wang, R.; Dellinger, T.H.; Zink, J.I.; Tamanoi, F.; Glackin, C.A. Nanoparticle delivery of siRNA against TWIST to reduce drug resistance and tumor growth in ovarian cancer models. Nanomed. Nanotechnol. Biol. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yu, D.; Jin, C.; Song, X.; Cheng, J.; Zhao, X.; Qi, X.; Zhang, G. A dual responsive targeted drug delivery system based on smart polymer coated mesoporous silica for laryngeal carcinoma treatment. New J. Chem. 2014, 38, 4830–4836. [Google Scholar] [CrossRef]
- Ebabe Elle, R.; Rahmani, S.; Lauret, C.; Morena, M.; Bidel, L.P.R.; Boulahtouf, A.; Balaguer, P.; Cristol, J.P.; Durand, J.O.; Charnay, C.; et al. Functionalized Mesoporous Silica Nanoparticle with Antioxidants as a New Carrier That Generates Lower Oxidative Stress Impact on Cells. Mol. Pharm. 2016, 13, 2647–2660. [Google Scholar] [CrossRef] [PubMed]
- Albanese, A.; Tang, P.S.; Chan, W.C.W. The Effect of Nanoparticle Size, Shape, and Surface Chemistry on Biological Systems. Annu. Rev. Biomed. Eng. 2012, 14, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Wang, T.; Cao, Y.; Wang, X.; Zhang, Y.; Liu, Y.; Huo, Q. Enzyme and voltage stimuli-responsive controlled release system based on β-cyclodextrin-capped mesoporous silica nanoparticles. Dalton Trans. 2015, 44, 4355–4361. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Cheng, Q.; Jiang, Q.; Huang, Y.; Yang, Z.; Han, S.; Zhao, Y.; Guo, S.; Liang, Z.; Dong, A. Intracellular cleavable poly(2-dimethylaminoethyl methacrylate) functionalized mesoporous silica nanoparticles for efficient siRNA delivery in vitro and in vivo. Nanoscale 2013, 5, 4291–4301. [Google Scholar] [CrossRef] [PubMed]
- Möller, K.; Müller, K.; Engelke, H.; Bräuchle, C.; Wagner, E.; Bein, T. Highly efficient siRNA delivery from core-shell mesoporous silica nanoparticles with multifunctional polymer caps. Nanoscale 2016, 8, 4007–4019. [Google Scholar] [CrossRef] [PubMed]
- Ashley, C.E.; Carnes, E.C.; Phillips, G.K.; Padilla, D.; Durfee, P.N.; Brown, P.A.; Hanna, T.N.; Liu, J.; Phillips, B.; Carter, M.B.; et al. The targeted delivery of multicomponent cargos to cancer cells by nanoporous particle-supported lipid bilayers. Nat. Mater. 2011, 10, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Kaszuba, M.; McKnight, D.; Connah, M.T.; McNeil-Watson, F.K.; Nobbmann, U. Measuring sub nanometre sizes using dynamic light scattering. J. Nanopart. Res. 2008, 10, 823–829. [Google Scholar] [CrossRef]
- Fröhlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [PubMed]
- Slowing, I.; Trewyn, B.G.; Lin, V.S.-Y. Effect of surface functionalization of MCM-41-type mesoporous silica nanoparticles on the endocytosis by human cancer cells. J. Am. Chem. Soc. 2006, 128, 14792–14793. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Gratton, S.E.A.; Ropp, P.A.; Pohlhaus, P.D.; Luft, J.C.; Madden, V.J.; Napier, M.E.; DeSimone, J.M. The effect of particle design on cellular internalization pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 11613–11618. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Wu, S.H.; Hung, Y.; Mou, C.Y. Size effect on cell uptake in well-suspended, uniform mesoporous silica nanoparticles. Small 2009, 5, 1408–1413. [Google Scholar] [CrossRef] [PubMed]
- Sorkin, A.; Von Zastrow, M. Signal transduction and endocytosis: Close encounters of many kinds. Nat. Rev. Mol. Cell Biol. 2002, 3, 600–614. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Zhang, J.; Shi, J.; Zhu, Z.; Zhang, L.; Bu, W.; Guo, L.; Chen, Y. The effect of PEGylation of mesoporous silica nanoparticles on nonspecific binding of serum proteins and cellular responses. Biomaterials 2010, 31, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yu, C. Advances in silica based nanoparticles for targeted cancer therapy. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J.; Hanahan, D. Switch to the angiogenic phenotype during tumorigenesis. Princess Takamatsu Symp. 1991, 22, 339–347. [Google Scholar] [PubMed]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 1–12. [Google Scholar] [CrossRef]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Van Furth, R.; Cohn, Z.A.; Hirsch, J.G.; Humphrey, J.H.; Spector, W.G.; Langevoort, H.L. The mononuclear phagocyte system: A new classification of macrophages, monocytes, and their precursor cells. Bull. World Health Organ. 1972, 46, 845–852. [Google Scholar] [PubMed]
- Huang, X.; Li, L.; Liu, T.; Hao, N.; Liu, H.; Chen, D.; Tang, F. The shape effect of mesoporous silica nanoparticles on biodistribution, clearance, and biocompatibility in vivo. ACS Nano 2011, 5, 5390–5399. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Liong, M.; Li, Z.; Zink, J.I.; Tamanoi, F. Biocompatibility, biodistribution, and drug-delivery efficiency of mesoporous silica nanoparticles for cancer therapy in animals. Small 2010, 6, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Malugin, A.; Ghandehari, H. Cellular uptake and toxicity of gold nanoparticles in prostate cancer cells: A comparative study of rods and spheres. J. Appl. Toxicol. 2010, 30, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Webster, P.; Davis, M.E. PEGylation significantly affects cellular uptake and intracellular trafficking of non-viral gene delivery particles. Eur. J. Cell Biol. 2004, 83, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Fang, Y.; Borchardt, L.; Kaskel, S. PEGylated hollow mesoporous silica nanoparticles as potential drug delivery vehicles. Microporous Mesoporous Mater. 2011, 141, 199–206. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Rubin, K.; Pietras, K.; Östman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Parker, N.; Turk, M.J.; Westrick, E.; Lewis, J.D.; Low, P.S.; Leamon, C.P. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal. Biochem. 2005, 338, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Yu, D.; Jia, B.; Jin, C.; Liu, X.; Zhao, X.; Zhang, G. Targeting CD133+ laryngeal carcinoma cells with chemotherapeutic drugs and siRNA against ABCG2 mediated by thermo/pH-sensitive mesoporous silica nanoparticles. Tumor Biol. 2016, 37, 2209–2217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, W.; Zhou, Y.; Jiang, Y.; Li, S. Dual Functional Mesoporous Silicon Nanoparticles Enhance the Radiosensitivity of VPA in Glioblastoma. Transl. Oncol. 2017, 10, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Nie, J.; Xu, L.; Liang, C.; Peng, Y.; Liu, G.; Wang, T.; Mei, L.; Huang, L.; Zeng, X. A pH-sensitive delivery vehicle based on folic acid-conjugated polydopamine-modified mesoporous silica nanoparticles for targeted cancer therapy. ACS Appl. Mater. Interfaces 2017. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sun, H.; Hu, J.; Han, X.; Liu, H.; Hu, Y. Transferrin gated mesoporous silica nanoparticles for redox-responsive and targeted drug delivery. Colloids Surf. B Biointerfaces 2017, 152, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.-T.; Souris, J.S.; Cheng, S.-H.; Chu, C.-H.; Wang, Y.-C.; Konda, V.; Dougherty, U.; Bissonnette, M.; Mou, C.-Y.; Chen, C.-T.; et al. Lectin-functionalized mesoporous silica nanoparticles for endoscopic detection of premalignant colonic lesions. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, S.K.; Luo, Y.; O’Donnell, M.A.; Assouline, J.G. Peptide-Mediated Targeting Mesoporous Silica Nanoparticles: A Novel Tool for Fighting Bladder Cancer. J. Biomed. Nanotechnol. 2017, 13, 232–242. [Google Scholar] [CrossRef]
- Hirano, Y.; Kando, Y.; Hayashi, T.; Goto, K.; Nakajima, A. Synthesis and cell attachment activity of bioactive oligopeptides: RGD, RGDS, RGDV, and RGDT. J. Biomed. Mater. Res. 1991, 25, 1523–1534. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, Y.; Shen, Y.; Wang, A.; Wang, S.; Xie, T. The functions and applications of RGD in tumor therapy and tissue engineering. Int. J. Mol. Sci. 2013, 14, 13447–13462. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Liu, J.; He, Q.; Shi, J. MSN-mediated sequential vascular-to-cell nuclear-targeted drug delivery for efficient tumor regression. Adv. Mater. 2014, 26, 6742–6748. [Google Scholar] [CrossRef] [PubMed]
- Hicke, B.J.; Stephens, A.W.; Gould, T.; Chang, Y.-F.; Lynott, C.K.; Heil, J.; Borkowski, S.; Hilger, C.-S.; Cook, G.; Warren, S.; et al. Tumor targeting by an aptamer. J. Nucl. Med. 2006, 47, 668–678. [Google Scholar] [PubMed]
- Babaei, M.; Abnous, K.; Taghdisi, S.M.; Amel Farzad, S.; Peivandi, M.T.; Ramezani, M.; Alibolandi, M. Synthesis of theranostic epithelial cell adhesion molecule targeted mesoporous silica nanoparticle with gold gatekeeper for hepatocellular carcinoma. Nanomedicine 2017. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Hu, H.; Zhang, M.G.; Song, J.; Nie, L.; Wang, S.; Niu, G.; Huang, P.; Lu, G.; Chen, X. An aptamer-targeting photoresponsive drug delivery system using “off–on” graphene oxide wrapped mesoporous silica nanoparticles. Nanoscale 2015, 7, 6304–6310. [Google Scholar] [CrossRef] [PubMed]
- Durfee, P.N.; Lin, Y.S.; Dunphy, D.R.; Muñiz, A.J.; Butler, K.S.; Humphrey, K.R.; Lokke, A.J.; Agola, J.O.; Chou, S.S.; Chen, I.M.; et al. Mesoporous Silica Nanoparticle-Supported Lipid Bilayers (Protocells) for Active Targeting and Delivery to Individual Leukemia Cells. ACS Nano 2016, 10, 8325–8345. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wu, D.; Yin, X.; Jin, X.; Zhang, X.; Zheng, S.; Wang, C.; Liu, Y. Intracellular pH-responsive and rituximab-conjugated mesoporous silica nanoparticles for targeted drug delivery to lymphoma B cells. J. Exp. Clin. Cancer Res. 2017, 36, 24. [Google Scholar] [CrossRef] [PubMed]
- Maloney, D.G.; Grillo-López, A.J.; White, C.A.; Bodkin, D.; Schilder, R.J.; Neidhart, J.A.; Janakiraman, N.; Foon, K.A.; Liles, T.-M.; Dallaire, B.K.; et al. IDEC-C2B8 (Rituximab) Anti-CD20 Monoclonal Antibody Therapy in Patients With Relapsed Low-Grade Non-Hodgkin’s Lymphoma. Blood 1997, 9, 2188–2195. [Google Scholar]
- Chen, F.; Hong, H.; Shi, S.; Goel, S.; Valdovinos, H.F.; Hernandez, R.; Theuer, C.P.; Barnhart, T.E.; Cai, W. Engineering of Hollow Mesoporous Silica Nanoparticles for Remarkably Enhanced Tumor Active Targeting Efficacy. Sci. Rep. 2014, 4, 5080. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Nayak, T.R.; Goel, S.; Valdovinos, H.F.; Hong, H.; Theuer, C.P.; Barnhart, T.E.; Cai, W. In vivo tumor vasculature targeted PET/NIRF Imaging with TRC105(Fab)-conjugated, dual-labeled mesoporous silica nanoparticles. Mol. Pharm. 2014, 11, 4007–4014. [Google Scholar] [CrossRef] [PubMed]
- Mekaru, H.; Lu, J.; Tamanoi, F. Development of mesoporous silica-based nanoparticles with controlled release capability for cancer therapy. Adv. Drug Deliv. Rev. 2015, 95, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Xue, M.; Xia, T.; Zhao, Y.L.; Tamanoi, F.; Stoddart, J.F.; Zink, J.I.; Nel, A.E. Autonomous in vitro anticancer drug release from mesoporous silica nanoparticles by pH-sensitive nanovalves. J. Am. Chem. Soc. 2010, 132, 12690–12697. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Bi, J.; Tang, Y.; Qiao, S.Z. Magnetic Core-Shell Silica Nanoparticles with Large Radial Mesopores for siRNA Delivery. Small 2016, 12, 4735–4742. [Google Scholar] [CrossRef] [PubMed]
- Popat, A.; Liu, J.; Lu, G.Q.M.; Qiao, S.Z. A pH-responsive drug delivery system based on chitosan coated mesoporous silica nanoparticles. J. Mater. Chem. 2012, 22, 11173–11178. [Google Scholar] [CrossRef]
- Liu, J.; Luo, Z.; Zhang, J.; Luo, T.; Zhou, J.; Zhao, X.; Cai, K. Hollow mesoporous silica nanoparticles facilitated drug delivery via cascade pH stimuli in tumor microenvironment for tumor therapy. Biomaterials 2016, 83, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Estrela, J.M.; Ortega, A.; Obrador, E. Glutathione in Cancer Biology and Therapy. Crit. Rev. Clin. Lab. Sci. 2006, 43, 143–181. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, S.; Park, C.; Lee, H.; Park, H.J.; Kim, C. Glutathione-induced intracellular release of guests from mesoporous silica nanocontainers with cyclodextrin gatekeepers. Adv. Mater. 2010, 22, 4280–4283. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhao, X.; Wu, T.; Feng, P. Tunable Redox-Responsive Hybrid Nanogated Ensembles. J. Am. Chem. Soc. 2008, 130, 14418–14419. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hei, M.; Xu, Y.; Qian, X.; Zhu, W. Ammonium salt modified mesoporous silica nanoparticles for dual intracellular-responsive gene delivery. Int. J. Pharm. 2016, 511, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Mondragón, L.; Mas, N.; Ferragud, V.; de la Torre, C.; Agostini, A.; Martínez-Máñez, R.; Sancenón, F.; Amorós, P.; Pérez-Payá, E.; Orzáez, M. Enzyme-responsive intracellular-controlled release using silica mesoporous nanoparticles capped with ε-poly-L-lysine. Chemistry 2014, 20, 5271–5281. [Google Scholar] [CrossRef] [PubMed]
- Bernardos, A.; Mondragón, L.; Aznar, E.; Marcos, M.D.; Martínez-Mánez, R.; Sancenón, F.; Soto, J.; Barat, J.M.; Pérez-Payá, E.; Guillem, C.; et al. Enzyme-responsive intracellular controlled release using nanometric silica mesoporous supports capped with “saccharides”. ACS Nano 2010, 4, 6353–6368. [Google Scholar] [CrossRef] [PubMed]
- Guardado-Alvarez, T.M.; Sudha Devi, L.; Russell, M.M.; Schwartz, B.J.; Zink, J.I. Activation of snap-top capped mesoporous silica nanocontainers using two near-infrared photons. J. Am. Chem. Soc. 2013, 135, 14000–14003. [Google Scholar] [CrossRef] [PubMed]
- Croissant, J.; Maynadier, M.; Gallud, A.; Peindy N’Dongo, H.; Nyalosaso, J.L.; Derrien, G.; Charnay, C.; Durand, J.O.; Raehm, L.; Serein-Spirau, F.; et al. Two-photon-triggered drug delivery in cancer cells using nanoimpellers. Angew. Chem. Int. Ed. 2013, 52, 13813–13817. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-J.; Hu, S.-H.; Hsiao, C.-S.; Chen, Y.-Y.; Liu, D.-M.; Chen, S.-Y. Multifunctional magnetically removable nanogated lids of Fe3O4–capped mesoporous silica nanoparticles for intracellular controlled release and MR imaging. J. Mater. Chem. 2011, 21, 2535. [Google Scholar] [CrossRef]
- Thomas, C.R.; Ferris, D.P.; Lee, J.H.; Choi, E.; Cho, M.H.; Kim, E.S.; Stoddart, J.F.; Shin, J.S.; Cheon, J.; Zink, J.I. Noninvasive remote-controlled release of drug molecules in vitro using magnetic actuation of mechanized nanoparticles. J. Am. Chem. Soc. 2010, 132, 10623–10625. [Google Scholar] [CrossRef] [PubMed]
- Freeman, E.C.; Weiland, L.M.; Meng, W.S. Modeling the proton sponge hypothesis: examining proton sponge effectiveness for enhancing intracellular gene delivery through multiscale modeling. J. Biomater. Sci. Polym. Ed. 2013, 24, 398–416. [Google Scholar] [CrossRef] [PubMed]
- Finlay, J.; Roberts, C.M.; Dong, J.; Zink, J.I.; Tamanoi, F.; Glackin, C.A. Mesoporous silica nanoparticle delivery of chemically modified siRNA against TWIST1 leads to reduced tumor burden. Nanomedicine 2015, 11, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Ma, D. Enhancing endosomal escape for nanoparticle mediated siRNA delivery. Nanoscale 2014, 6, 6415–6425. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, D.; Martin, O.C. Transbilayer redistribution of phosphatidylethanolamine during fusion of phospholipid vesicles. Dependence on fusion rate, lipid phase separation, and formation of nonbilayer structures. Biochemistry 1982, 21, 6097–6103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, F.; Guo, S.; Chen, X.; Wang, X.; Li, J.; Gan, Y. Biofunctionalized polymer-lipid supported mesoporous silica nanoparticles for release of chemotherapeutics in multidrug resistant cancer cells. Biomaterials 2014, 35, 3650–3665. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, Y.; Wang, M.; Ma, Y.; Xia, W.; Gu, H. A mesoporous silica nanoparticle—PEI—Fusogenic peptide system for siRNA delivery in cancer therapy. Biomaterials 2013, 34, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Gu, L.; von Maltzahn, G.; Ruoslahti, E.; Bhatia, S.N.; Sailor, M.J. Biodegradable luminescent porous silicon nanoparticles for in vivo applications. Nat. Mater. 2009, 8, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Meng, Q.; Chen, Y.; Du, Y.; Zhang, L.; Li, Y.; Zhang, L.; Shi, J. Large-pore ultrasmall mesoporous organosilica nanoparticles: Micelle/precursor co-templating assembly and nuclear-targeted gene delivery. Adv. Mater. 2015, 27, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Yu, Y.; Li, Y.; Yu, Y.; Li, Y.; Zhou, X.; Huang, P.; Sun, Z. Toxic effect of silica nanoparticles on endothelial cells through DNA damage response via Chk1-dependent G2/M checkpoint. PLoS ONE 2013, 8, e62087. [Google Scholar] [CrossRef] [PubMed]
- Heikkilä, T.; Santos, H.A.; Kumar, N.; Murzin, D.Y.; Salonen, J.; Laaksonen, T.; Peltonen, L.; Hirvonen, J.; Lehto, V.P. Cytotoxicity study of ordered mesoporous silica MCM-41 and SBA-15 microparticles on Caco-2 cells. Eur. J. Pharm. Biopharm. 2010, 74, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Tarn, D.; Ashley, C.E.; Xue, M.; Carnes, E.C.; Zink, J.I.; Brinker, C.J. Mesoporous silica nanoparticle nanocarriers: Biofunctionality and biocompatibility. Acc. Chem. Res. 2013, 46, 792–801. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Zhang, Z.; Gao, F.; Li, Y.; Shi, J. In vivo biodistribution and urinary excretion of mesoporous silica nanoparticles: Effects of particle size and PEGylation. Small 2011, 7, 271–280. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watermann, A.; Brieger, J. Mesoporous Silica Nanoparticles as Drug Delivery Vehicles in Cancer. Nanomaterials 2017, 7, 189. https://doi.org/10.3390/nano7070189
Watermann A, Brieger J. Mesoporous Silica Nanoparticles as Drug Delivery Vehicles in Cancer. Nanomaterials. 2017; 7(7):189. https://doi.org/10.3390/nano7070189
Chicago/Turabian StyleWatermann, Anna, and Juergen Brieger. 2017. "Mesoporous Silica Nanoparticles as Drug Delivery Vehicles in Cancer" Nanomaterials 7, no. 7: 189. https://doi.org/10.3390/nano7070189
APA StyleWatermann, A., & Brieger, J. (2017). Mesoporous Silica Nanoparticles as Drug Delivery Vehicles in Cancer. Nanomaterials, 7(7), 189. https://doi.org/10.3390/nano7070189