Synthesis of Reduced Graphene Oxide with Adjustable Microstructure Using Regioselective Reduction in the Melt of Boric Acid: Relationship Between Structural Properties and Electrochemical Performance




 and
and 
Abstract
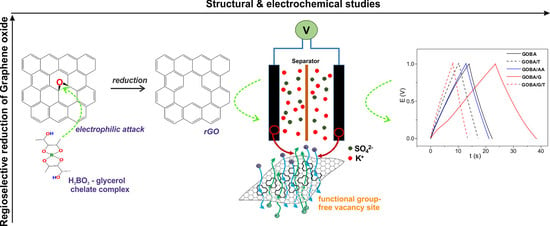
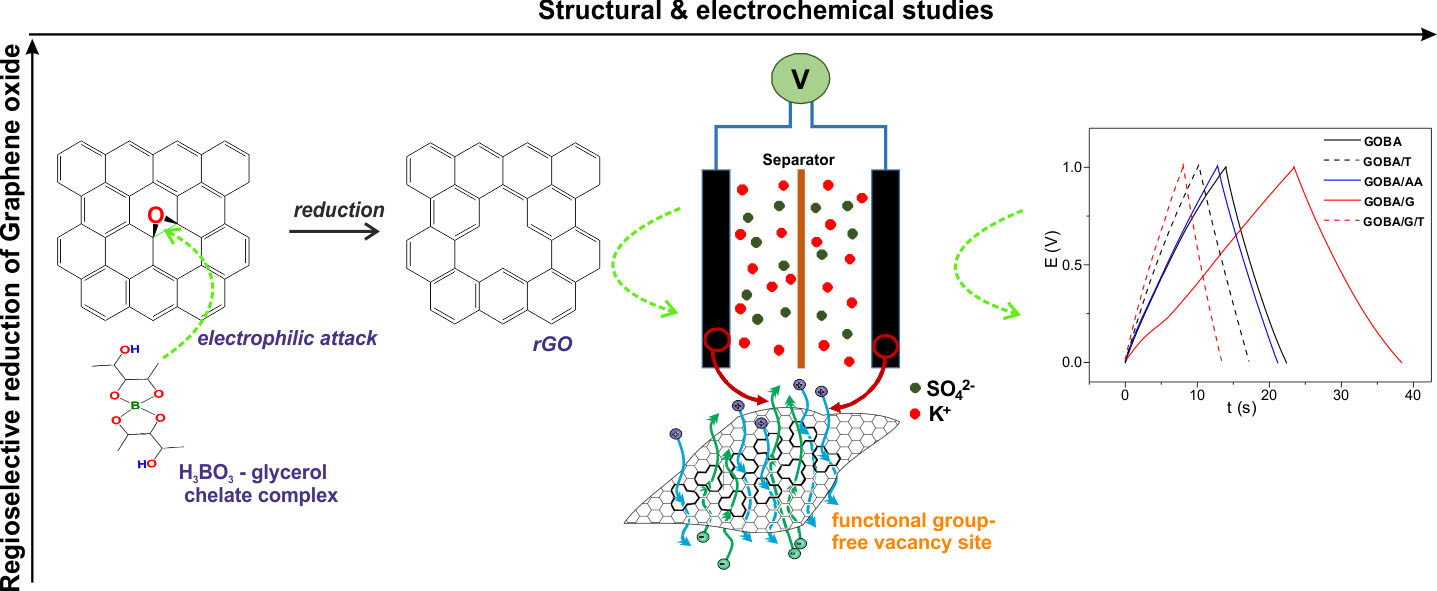
:
1. Introduction
2. Materials and Methods
2.1. Synthesis of GO
2.2. GO Reduction in the Melt of Boric Acid
2.3. Thermal Treatment of MrGO Samples
2.4. Materials Characterization
2.5. Electrochemical Measurements
3. Results and Discussion
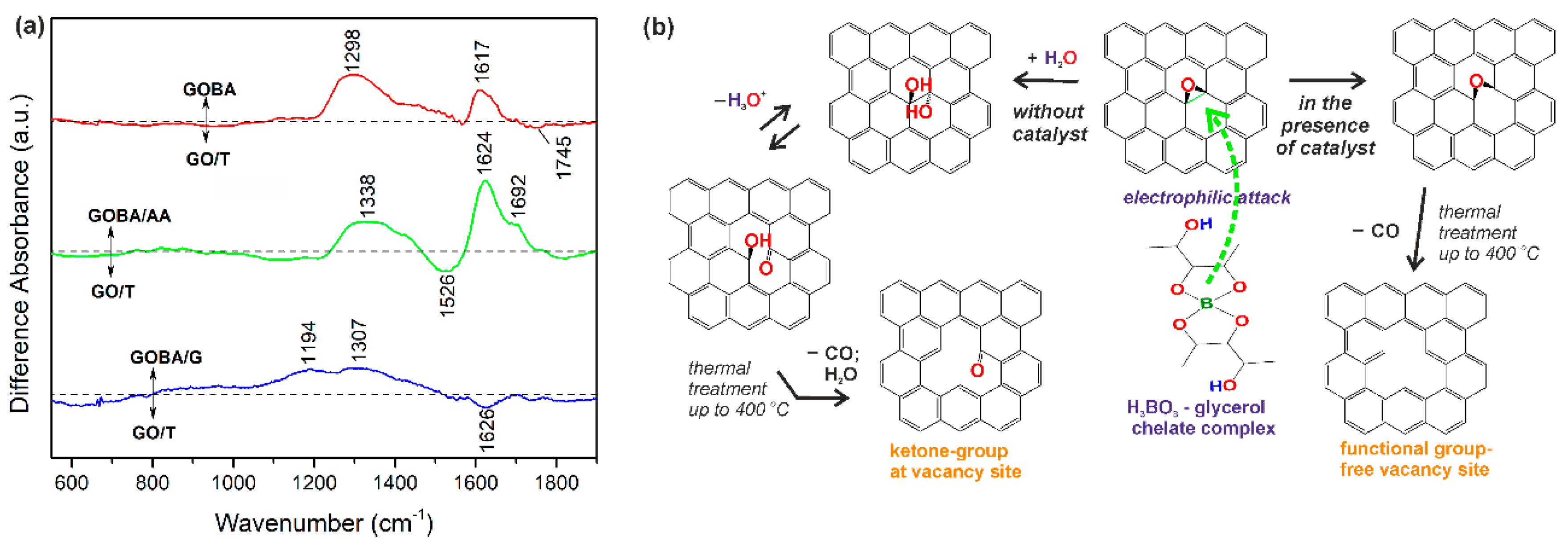
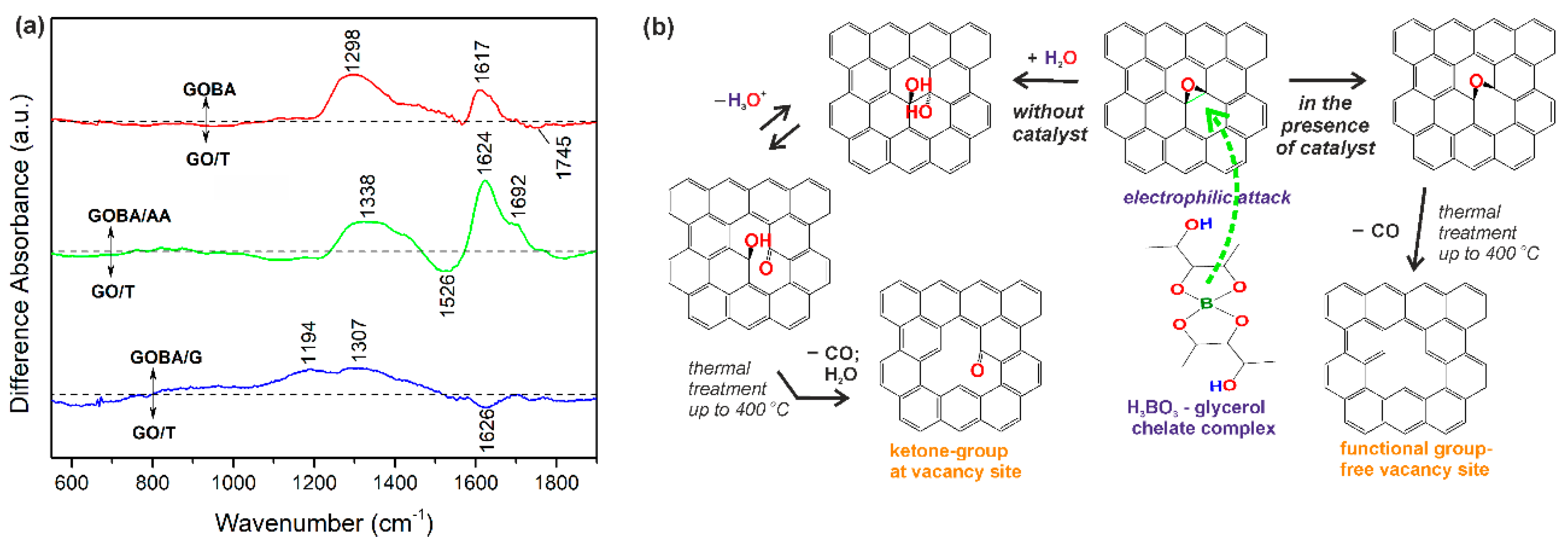
3.1. Investigation of the Reduction Process of GO in the Melt of Boric Acid
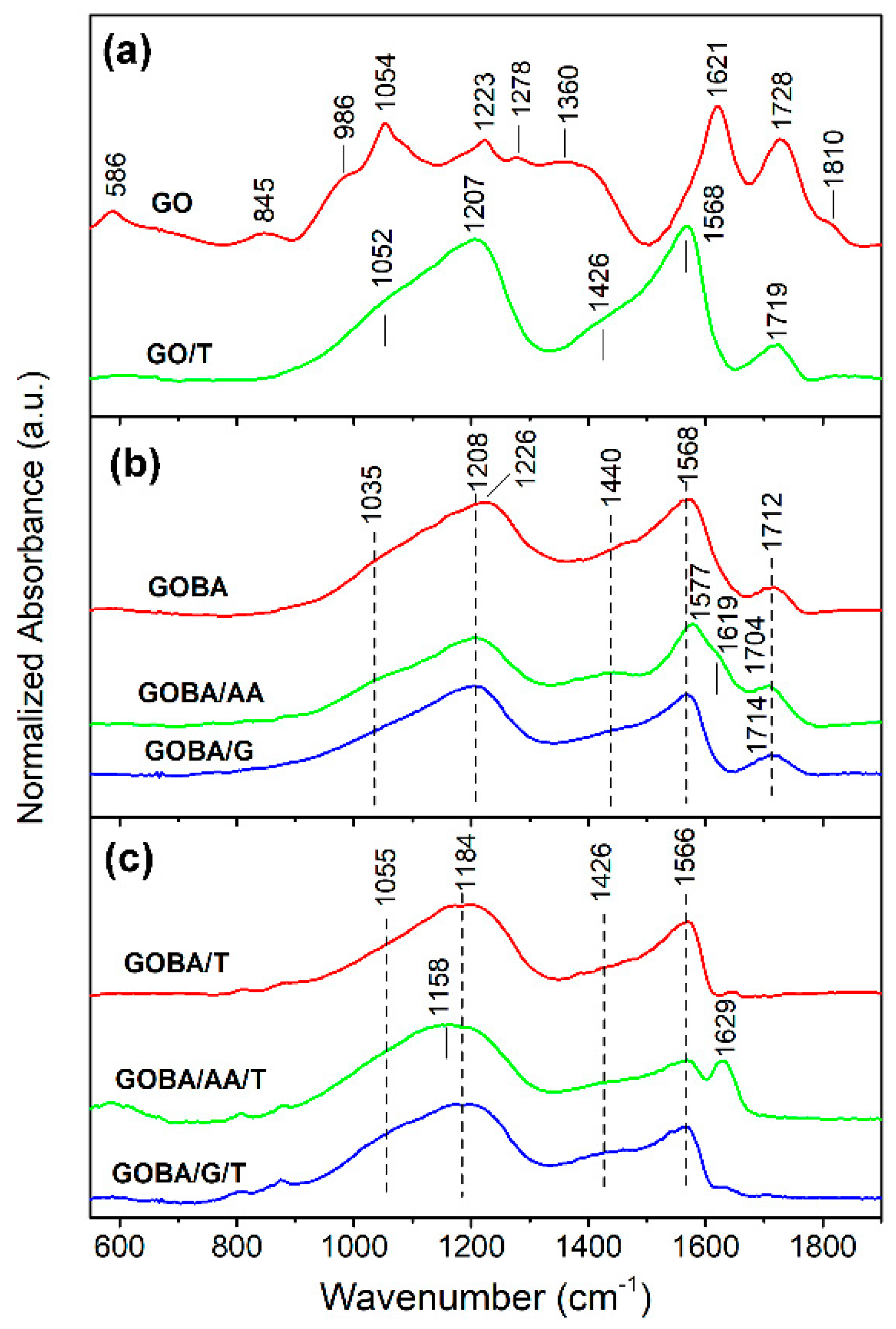
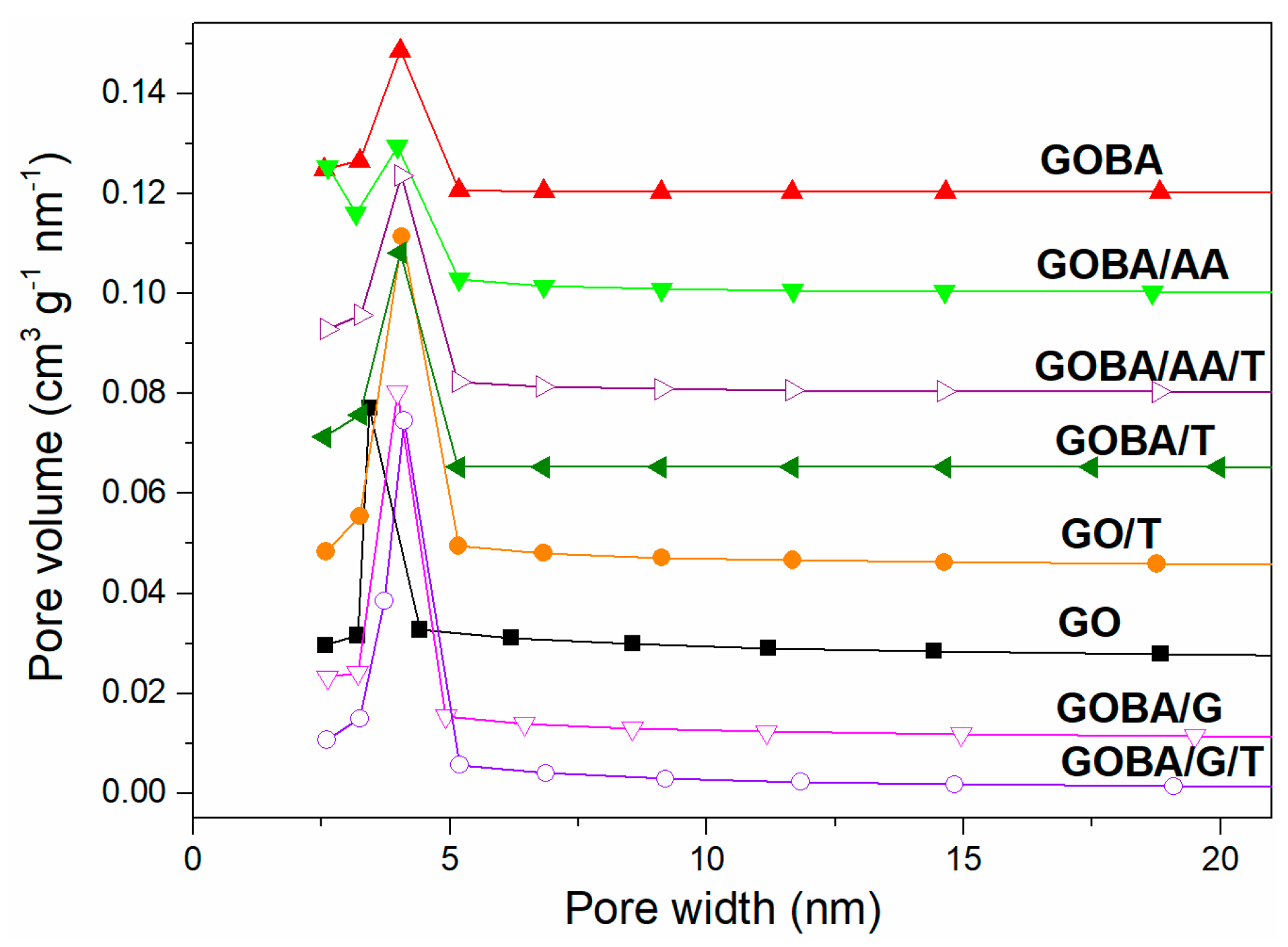
3.2. Structural Characterization of GO Reduction Products
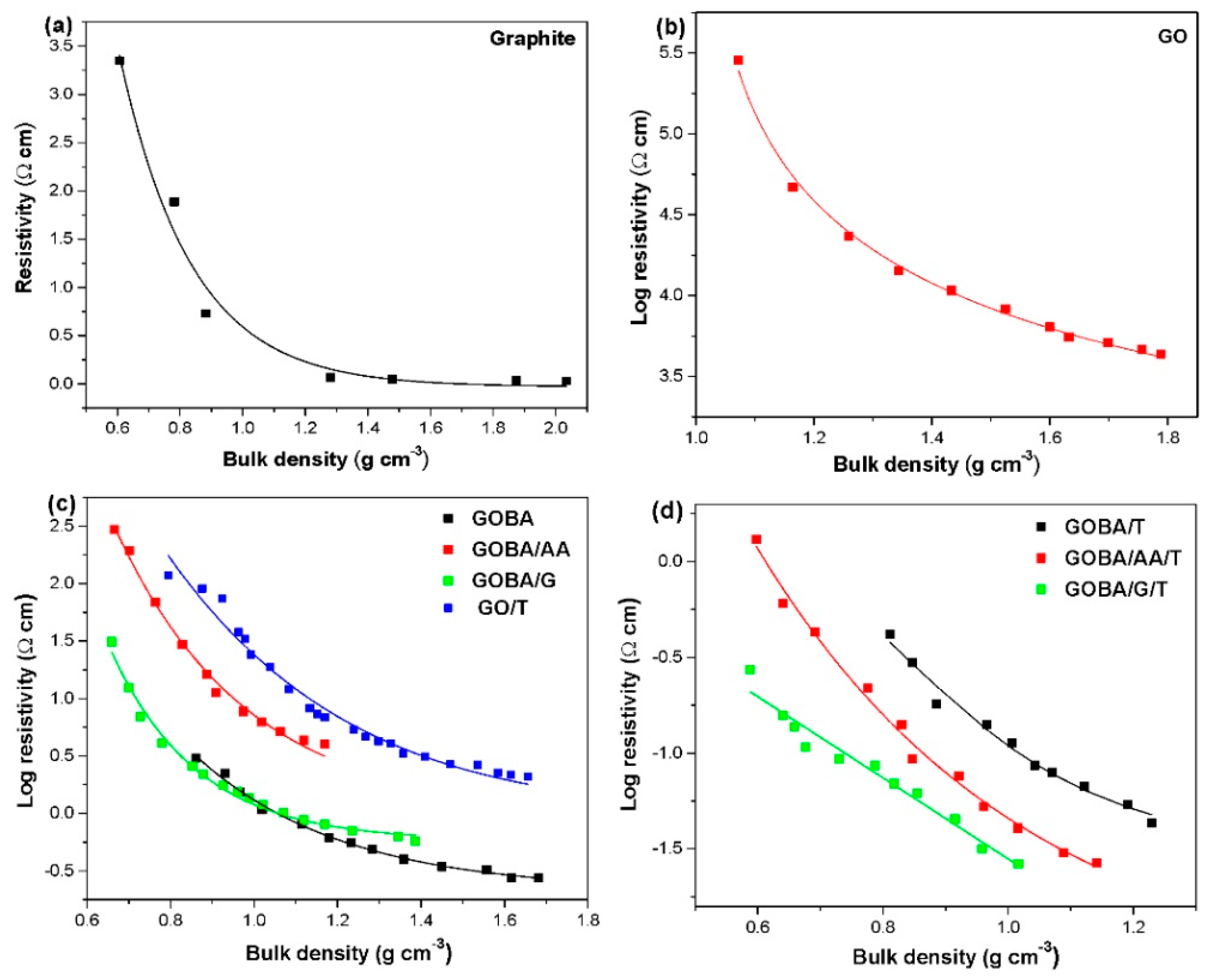
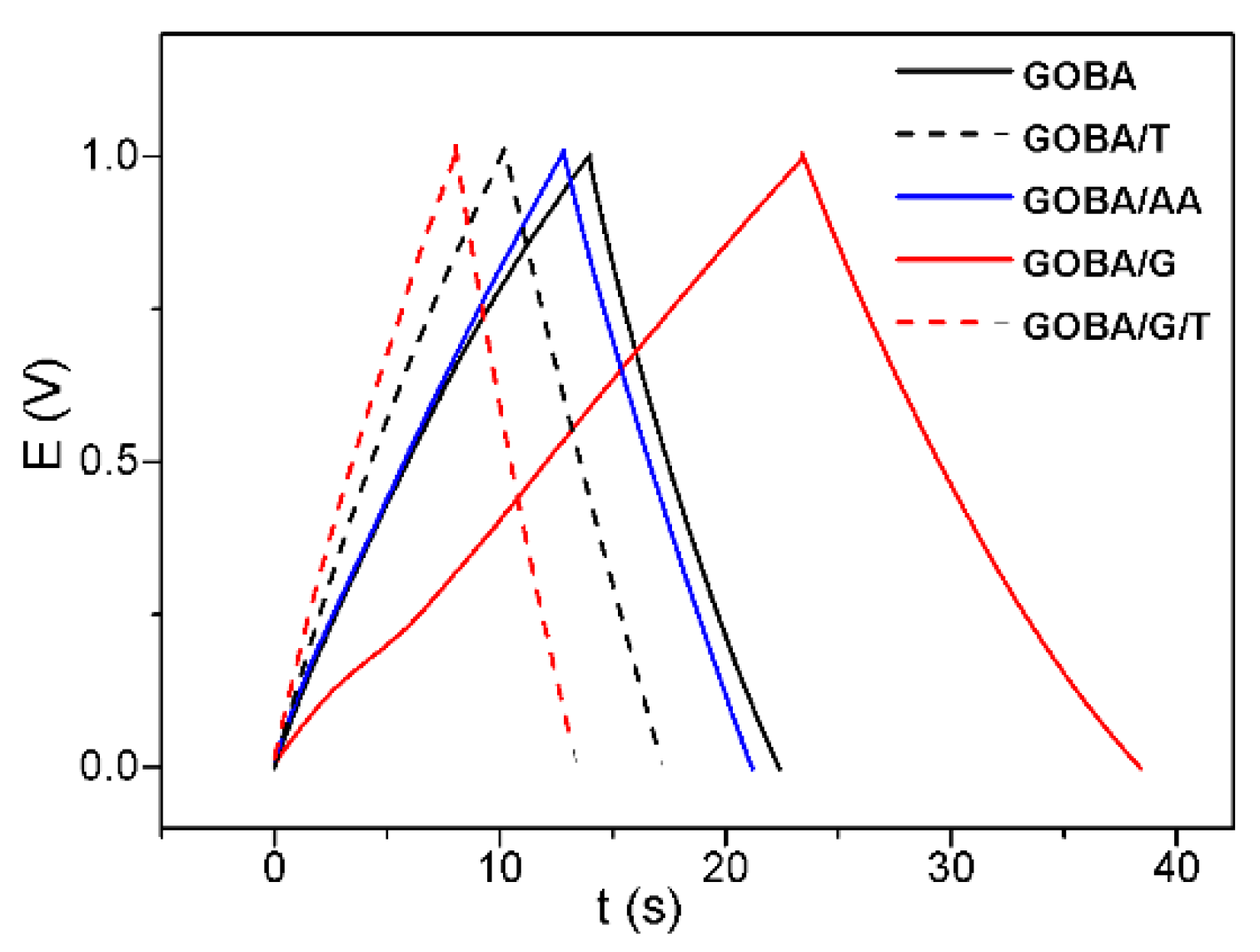
3.3. Electrochemical Characterization
3.4. Impact of Structural Properties on Electrochemical Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Antink, W.H.; Choi, Y.; Seong, K.; Kim, J.M.; Piao, Y. Recent progress in porous graphene and reduced graphene oxide-based nanomaterials for electrochemical energy storage devices. Adv. Mater. Interfaces 2018, 5, 1701212. [Google Scholar] [CrossRef]
- Singh, D.P.; Herrera, C.E.; Singh, B.; Singh, S.; Singh, R.K.; Kumar, R. Graphene oxide: An efficient material and recent approach for biotechnological and biomedical applications. Mater. Sci. Eng. C 2018, 86, 173–197. [Google Scholar] [CrossRef] [PubMed]
- Sturala, J.; Luxa, J.; Pumera, M.; Sofer, Z. Chemistry of graphene derivatives: Synthesis, applications and perspectives. Chem. Eur. J. 2018, 24, 5992–6006. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Kurihara, S.; Tateishi, H.; Hatakeyama, K.; Koinuma, M.; Yokoi, H.; Hara, M.; Ishikawa, H.; Matsumoto, Y. pH-driven, reversible epoxy ring opening/closing in graphene oxide. Carbon 2015, 84, 560–566. [Google Scholar] [CrossRef]
- Pei, S.; Cheng, H.M. The reduction of graphene oxide. Carbon 2012, 50, 3210–3228. [Google Scholar] [CrossRef]
- Larciprete, R.; Fabris, S.; Sun, T.; Lacovig, P.; Baraldi, A.; Lizzit, S. Dual path mechanism in the thermal reduction of graphene oxide. J. Am. Chem. Soc. 2011, 133, 17315–17321. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Collin, F.; Hurt, R.H.; Külaots, I. Thermochemistry and kinetics of graphite oxide exothermic decomposition for safety in large-scale storage and processing. Carbon 2016, 96, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Tang, J.; Zhang, K.; Yuan, J.; Li, J.; Zhu, D.M.; Ozawa, K.; Qin, L.C. Comparison of reduction products from graphite oxide and graphene oxide for anode applications in lithium-ion batteries and sodium-ion batteries. Nanoscale 2017, 9, 2585–2595. [Google Scholar] [CrossRef] [PubMed]
- Brycht, M.; Leniart, A.; Zavasnik, J.; Nosal-Wiercinska, A.; Wasinski, K.; Polrolniczak, P.; Skrzypek, S.; Kalcher, K. Synthesis and characterization of the thermally reduced graphene oxide in argon atmosphere and ist application to construct graphene paste electrode as a naptalam electrochemical sensor. Anal. Chim. Acta 2018, 1035, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Kovtyukhova, N.I.; Wang, Y.; Berkdemir, A.; Cruz-Silva, R.; Terrones, M.; Crespi, V.H.; Mallouk, T.E. Non-oxidative intercalation and exfoliation of graphite by Brønsted acids. Nat. Chem. 2014, 6, 957. [Google Scholar] [CrossRef] [PubMed]
- Elango, M.; Parthasarathi, R.; Subramanian, V.; Sathyamurthy, N. Bowls, Balls and sheets of boric acid clusters: The role of pentagon and hexagon motifs. J. Phys. Chem. A 2005, 109, 8587–8593. [Google Scholar] [CrossRef] [PubMed]
- Mikhal´chenko, L.V.; Leibzon, V.N.; Leonova, M.Y.; Gultyai, V.P. Effect of medium acidity on the efficiency of oxidation of 2,4,6-trinitrotoluene to 2,4,6-trinitrobenzoic acid. Russ. Chem. Bull. 2016, 65, 2216–2219. [Google Scholar] [CrossRef]
- Köse, D.A.; Zümreoglu-Karan, B. Complexation of boric acid with vitamin C. New J. Chem. 2009, 33, 1874–1881. [Google Scholar] [CrossRef]
- Russo, P.; Hu, A.; Compagnini, G. Synthesis, properties and potential applications of porous graphene: A review. Nano-Micro Lett. 2013, 5, 260–273. [Google Scholar] [CrossRef]
- Niu, L.; Li, Z.; Hong, W.; Sun, J.; Wang, Z.; Ma, L.; Wang, J.; Yang, S. Pyrolytic synthesis of boron-doped graphene and ist application as electrode material for supercapacitors. Electrochim. Acta 2013, 108, 666–673. [Google Scholar] [CrossRef]
- Yan, X.; Chen, J.; Yang, J.; Xue, Q.; Miele, P. Fabrication of free-standing, electrochemically active and biocompatible graphene oxide−polyaniline and graphene−polyaniline hybrid papers. ACS Appl. Mater. Interfaces 2010, 2, 2521–2529. [Google Scholar] [CrossRef] [PubMed]
- Hummers, W.S.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Halimehjani, A.Z.; Gholami, H.; Saidi, M.R. Boric acid/glycerol as an efficient catalyst for regioselective epoxide ring opening by aromatic amines in water. Green Chem. Lett. Rev. 2012, 5, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Chua, C.K.; Pumera, M. Chemical reduction of graphene oxide: a synthetic chemistry viewpoint. Chem. Soc. Rev. 2014, 43, 291–312. [Google Scholar] [CrossRef] [PubMed]
- Cançado, L.G.; Takai, K.; Enoki, T.; Endo, M.; Kim, Y.A.; Mizusaki, H.; Jorio, A.; Coelho, L.N.; Magalhães-Paniago, R.; Pimenta, M.A. General equation for the determination of the crystallite size La of nanographite by Raman spectroscopy. Appl. Phys. Lett. 2006, 88, 163106. [Google Scholar] [CrossRef]
- Rattanaweeranon, S.; Limsuwan, P.; Thongpool, V.; Piriyawong, V.; Asanithi, P. Influence of bulk graphite density on electrical conductivity. Procedia Eng. 2012, 32, 1100–1106. [Google Scholar] [CrossRef]
- Zhang, S.; Pan, N. Supercapacitors performance evaluation. Adv. Energy Mater. 2015, 5, 1401401. [Google Scholar] [CrossRef]
- Zhang, L.; Liang, J.; Huang, Y.; Ma, Y.; Wang, Y.; Chen, Y. Size-controlled synthesis of graphene oxide sheets on a large scale using chemical exfoliation. Carbon 2009, 47, 3365–3368. [Google Scholar] [CrossRef]
- Hatakeyama, K.; Awaya, K.; Koinuma, M.; Shimizu, Y.; Hakuta, Y.; Matsumoto, Y. Production of water-dispersible reduced graphene oxide without stabilizers using liquid-phase photoreduction. Soft Matter 2017, 13, 8353–8356. [Google Scholar] [CrossRef] [PubMed]
- Freyhardt, C.C.; Wiebcke, M.; Felsche, J. The monoclinic and cubic phases of metaboric acid (precise redeterminations). Acta Crystallogr. Sect. C Struct. Chem. 2000, 56, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, J.; Winandy, J.E. Chemical mechanism of fire retardance of boric acid on wood. Wood Sci. Technol. 2004, 38, 375–389. [Google Scholar] [CrossRef]
- Juhász, M.; Kitahara, Y.; Takahashi, S.; Fujii, T. Thermal stability of vitamin C: Thermogravimetric analysis and use of total ion monitoring chromatograms. J. Pharm. Biomed. Anal. 2012, 59, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Dou, B.; Dupont, V.; Williams, P.T.; Chen, H.; Ding, Y. Thermogravimetric kinetics of crude glycerol. Bioresour. Technol. 2009, 100, 2613–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trusovas, R.; Račiukaitis, G.; Niaura, G.; Barkauskas, J.; Valušis, G.; Pauliukaite, R. Recent advances in laser utilization in the chemical modification of graphene oxide and its applications. Adv. Opt. Mater. 2016, 4, 37–65. [Google Scholar] [CrossRef]
- Yin, D.; Lu, N.; Li, Z.; Yang, J. A computational infrared spectroscopic study of graphene oxide. J. Chem. Phys. 2013, 139, 084704. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, K.; Hayashi, Y.; Yoshimura, T.; Yoshimura, Y.; Hatakeyama, K.; Taniguchi, T.; Koinuma, M.; Yoshimoto, S.; Matsumoto, Y. Electrochemical reaction of graphene oxide at Au electrode surface monitored by surface enhanced infrared absorption spectroscopy. e-J. Surf. Sci. Nanotechnol. 2015, 13, 413–416. [Google Scholar] [CrossRef]
- Zhang, C.; Dabbs, D.M.; Liu, L.-M.; Aksay, I.A.; Car, R.; Selloni, A. Combined effects of functional groups, lattice defects and edges in the infrared spectra of graphene oxide. J. Phys. Chem. C 2015, 119, 18167–18176. [Google Scholar] [CrossRef]
- Acik, M.; Lee, G.; Mattevi, C.; Pirkle, A.; Wallace, R.M.; Chhowalla, M.; Cho, K.; Chabal, Y. The role of oxygen during thermal reduction of graphene oxide studied by infrared absorption spectroscopy. J. Phys. Chem. C 2011, 115, 19761–19781. [Google Scholar] [CrossRef]
- Taniguchi, T.; Yokoi, H.; Nagamine, M.; Tateishi, H.; Funatsu, A.; Hatakeyama, K.; Ogata, C.; Ichida, M.; Ando, H.; Koinuma, M.; et al. Correlated optical and magnetic properties in photoreduced graphene oxide. J. Phys. Chem. C 2014, 118, 28258–28265. [Google Scholar] [CrossRef]
- Novacek, M.; Jankovsky, O.; Luxa, J.; Sedmidubsky, D.; Pumera, M.; Fila, V.; Lhotka, M.; Klimova, K.; Matejkova, S.; Sofer, Z. Tuning of graphene oxide composition by multiple oxidations for carbon dioxide storage and capture of toxic metals. J. Mater. Chem. A 2017, 5, 2739–27478. [Google Scholar] [CrossRef]
- Spano, S.F.; Isgro, G.; Russo, P.; Fragala, M.E.; Compagnini, G. Tunable properties of graphene oxide reduced by laser irradiation. Appl. Phys. A 2014, 117, 19–23. [Google Scholar] [CrossRef]
- Dimiev, A.M.; Polson, T.A. Contesting the two-component structural model of graphene oxide and reexamining the chemistry of graphene oxide in basic media. Carbon 2015, 93, 544–554. [Google Scholar] [CrossRef]
- Pimenta, M.A.; Dresselhaus, G.; Dresselhaus, M.S.; Cançado, L.G.; Jorio, A.; Saito, R. Studying disorder in graphite-based systems by Raman spectroscopy. Phys. Chem. Chem. Phys. 2007, 9, 1276–1290. [Google Scholar] [CrossRef] [PubMed]
- Díez-Betriu, X.; Álvarez-García, S.; Botas, C.; Álvarez, P.; Sánchez-Marcos, J.; Prieto, C.; Menéndez, R.; de Andrés, A. Raman spectroscopy for the study of reduction mechanisms and optimization of conductivity in graphene oxide thin films. J. Mater. Chem. C 2013, 1, 6905–6912. [Google Scholar] [CrossRef]
- Vallerot, J.-M.; Bourrat, X.; Mouchon, A.; Chollon, G. Quantitative structural and textural assessment of laminar pyrocarbons through Raman spectroscopy, electron diffraction and few other techniques. Carbon 2006, 44, 1833–1844. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Robinson, J.T.; Diankov, G.; Dai, H. Nanocrystal growth on graphene with various degrees of oxidation. J. Am. Chem. Soc. 2010, 132, 3270–3271. [Google Scholar] [CrossRef] [PubMed]
- Sohail, M.; Saleem, M.; Ullah, S.; Saeed, N.; Afridi, A.; Khan, M.; Arif, M. Modified and improved Hummer’s synthesis of graphene oxide for capacitors applications. Mod. Electron. Mater. 2017, 3, 110–116. [Google Scholar] [CrossRef]
- Serrano, D.P.; Botas, J.A.; Guil-Lopez, R. H2 production from methane pyrolysis over commercial carbon catalysts: Kinetic and deactivation study. Int. J. Hydrogen Energy 2009, 34, 4488–4494. [Google Scholar] [CrossRef]
- Chen, K.; Song, S.; Liu, F.; Xue, D. Structural design of graphene for use in electrochemical energy storage devices. Chem. Soc. Rev. 2015, 44, 6230–6257. [Google Scholar] [CrossRef] [PubMed]
- Gadipelli, S.; Guo, Z.X. Graphene-based materials: Synthesis and gas sorption, storage and separation. Prog. Mater. Sci. 2015, 69, 1–60. [Google Scholar] [CrossRef]
- Szabó, T.; Tombácz, E.; Illés, E.; Dékány, I. Enhanced acidity and pH-dependent surface charge characterization of successively oxidized graphite oxides. Carbon 2006, 44, 537–545. [Google Scholar] [CrossRef]
- Ruiz, V.; Blanco, C.; Granda, M.; Menéndez, R.; Santamaría, R. Effect of the thermal treatment of carbon-based electrodes on the electrochemical performance of supercapacitors. J. Electroanal. Chem. 2008, 618, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Fan, Z.; Gu, L.; Bao, X.; Wang, C. Enhanced capacitance of manganese oxide via confinement inside carbon nanotubes. Chem. Commun. 2010, 46, 3905–3907. [Google Scholar] [CrossRef] [PubMed]
- Celiešiūtė, R.; Trusovas, R.; Niaura, G.; Švedas, V.; Račiukaitis, G.; Ruželė, Ž.; Pauliukaite, R. Influence of the laser irradiation on the electrochemical and spectroscopic peculiarities of graphene-chitosan composite film. Electrochim. Acta 2014, 132, 265–276. [Google Scholar] [CrossRef]
- Luo, G.; Liu, L.; Zhang, J.; Li, G.; Wang, B.; Zhao, J. Hole Defects and nitrogen doping in graphene: implication for supercapacitor applications. ACS Appl. Mater. Interfaces 2013, 5, 11184–11193. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, H.; Koinuma, M.; Miyamoto, S.; Kamei, Y.; Hatakeyama, K.; Ogata, C.; Taniguchi, T.; Funatsu, A.; Matsumoto, Y. Effect of the electrochemical oxidation/reduction cycle on the electrochemical capacitance of graphite oxide. Carbon 2014, 76, 40–45. [Google Scholar] [CrossRef]
- Minzae, L.; Gil-Pyo, K.; Hyeon Don, S.; Soomin, P.; Jongheop, Y. Preparation of energy storage material derived from a used cigarette filter for a supercapacitor electrode. Nanotechnology 2014, 25, 345601. [Google Scholar] [CrossRef]
- Luque, N.B.; Schmickler, W. The electric double layer on graphite. Electrochim. Acta 2012, 71, 82–85. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raman Analysis | XRD Characterization | ||||||
|---|---|---|---|---|---|---|---|---|
| D-band | ID/IG | AD/AG | Lα (nm) | Crystallite Size (nm) | 2θ100max (degrees) | I100/I002 | ||
| νD (cm–1) | FWHM (cm–1) | |||||||
| GO | 1344.7 | 133.5 | 1.12 | 1.62 | 24 | 6.961 | 42.25 | 0.381 |
| GO/T | 1342.6 | 137.8 | 0.96 | 1.76 | 22 | 1.577 | 42.96 | 0.170 |
| GOBA | 1336.1 | 111.2 | 1.10 | 1.98 | 19 | 3.078 | 42.97 | 0.099 |
| GOBA/AA | 1340.2 | 121.7 | 0.91 | 1.54 | 25 | 2.833 | 43.12 | 0.107 |
| GOBA/G | 1339.6 | 124.3 | 0.96 | 1.67 | 23 | 2.019 | 42.90 | 0.183 |
| GOBA/T | 1329.6 | 89.0 | 1.48 | 2.33 | 16 | 2.145 | 43.07 | 0.156 |
| GOBA/AA/T | 1329.4 | 114.4 | 1.21 | 2.10 | 18 | 2.455 | 43.00 | 0.141 |
| GOBA/G/T | 1329.9 | 97.1 | 1.29 | 2.10 | 18 | 2.934 | 43.09 | 0.135 |
| Sample | SBET (m2 g–1) | Sext (m2 g–1) | Vtot (cm3 g–1) | Vμ (cm3 g–1) | Average Pore Width (nm) |
|---|---|---|---|---|---|
| GO | 46 | 41 | 0.17 | 0.00 | 15.13 |
| GO/T | 97 | 68 | 0.13 | 0.01 | 5.49 |
| GOBA | 66 | 46 | 0.07 | 0.01 | 3.99 |
| GOBA/AA | 336 | 188 | 0.20 | 0.07 | 2.41 |
| GOBA/G | 155 | 114 | 0.19 | 0.02 | 4.80 |
| GOBA/T | 107 | 68 | 0.09 | 0.02 | 3.42 |
| GOBA/AA/T | 229 | 118 | 0.15 | 0.05 | 5.68 |
| GOBA/G/T | 138 | 100 | 0.18 | 0.02 | 5.27 |
| Sample | Cdl for Different rGO Load (μF cm–2) | ||
|---|---|---|---|
| 0.25 mg mL–1 | 0.50 mg mL–1 | 1.00 mg mL–1 | |
| GOBA | 449 | 470 | 684 |
| GOBA/AA | 168 | 202 | 247 |
| GOBA/G | 107 | 183 | 260 |
| GOBA/T | 8.07 | 3.97 | 41.1 |
| GOBA/AA/T | 56.2 | 120 | 172 |
| GOBA/G/T | 208 | 794 | 3011 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaidukevič, J.; Pauliukaitė, R.; Niaura, G.; Matulaitienė, I.; Opuchovič, O.; Radzevič, A.; Astromskas, G.; Bukauskas, V.; Barkauskas, J. Synthesis of Reduced Graphene Oxide with Adjustable Microstructure Using Regioselective Reduction in the Melt of Boric Acid: Relationship Between Structural Properties and Electrochemical Performance. Nanomaterials 2018, 8, 889. https://doi.org/10.3390/nano8110889
Gaidukevič J, Pauliukaitė R, Niaura G, Matulaitienė I, Opuchovič O, Radzevič A, Astromskas G, Bukauskas V, Barkauskas J. Synthesis of Reduced Graphene Oxide with Adjustable Microstructure Using Regioselective Reduction in the Melt of Boric Acid: Relationship Between Structural Properties and Electrochemical Performance. Nanomaterials. 2018; 8(11):889. https://doi.org/10.3390/nano8110889
Chicago/Turabian StyleGaidukevič, Justina, Rasa Pauliukaitė, Gediminas Niaura, Ieva Matulaitienė, Olga Opuchovič, Aneta Radzevič, Gvidas Astromskas, Virginijus Bukauskas, and Jurgis Barkauskas. 2018. "Synthesis of Reduced Graphene Oxide with Adjustable Microstructure Using Regioselective Reduction in the Melt of Boric Acid: Relationship Between Structural Properties and Electrochemical Performance" Nanomaterials 8, no. 11: 889. https://doi.org/10.3390/nano8110889
APA StyleGaidukevič, J., Pauliukaitė, R., Niaura, G., Matulaitienė, I., Opuchovič, O., Radzevič, A., Astromskas, G., Bukauskas, V., & Barkauskas, J. (2018). Synthesis of Reduced Graphene Oxide with Adjustable Microstructure Using Regioselective Reduction in the Melt of Boric Acid: Relationship Between Structural Properties and Electrochemical Performance. Nanomaterials, 8(11), 889. https://doi.org/10.3390/nano8110889