
Complete Androgen Insensitivity Syndrome in a Young Girl with Primary Amenorrhea and Suspected Delayed Puberty: A Case-Based Review of Clinical Management, Surgical Follow-Up, and Oncological Risk

 ,
,  ,
,  and
and {kind=link}
Abstract
:1. Introduction
2. Case Presentation
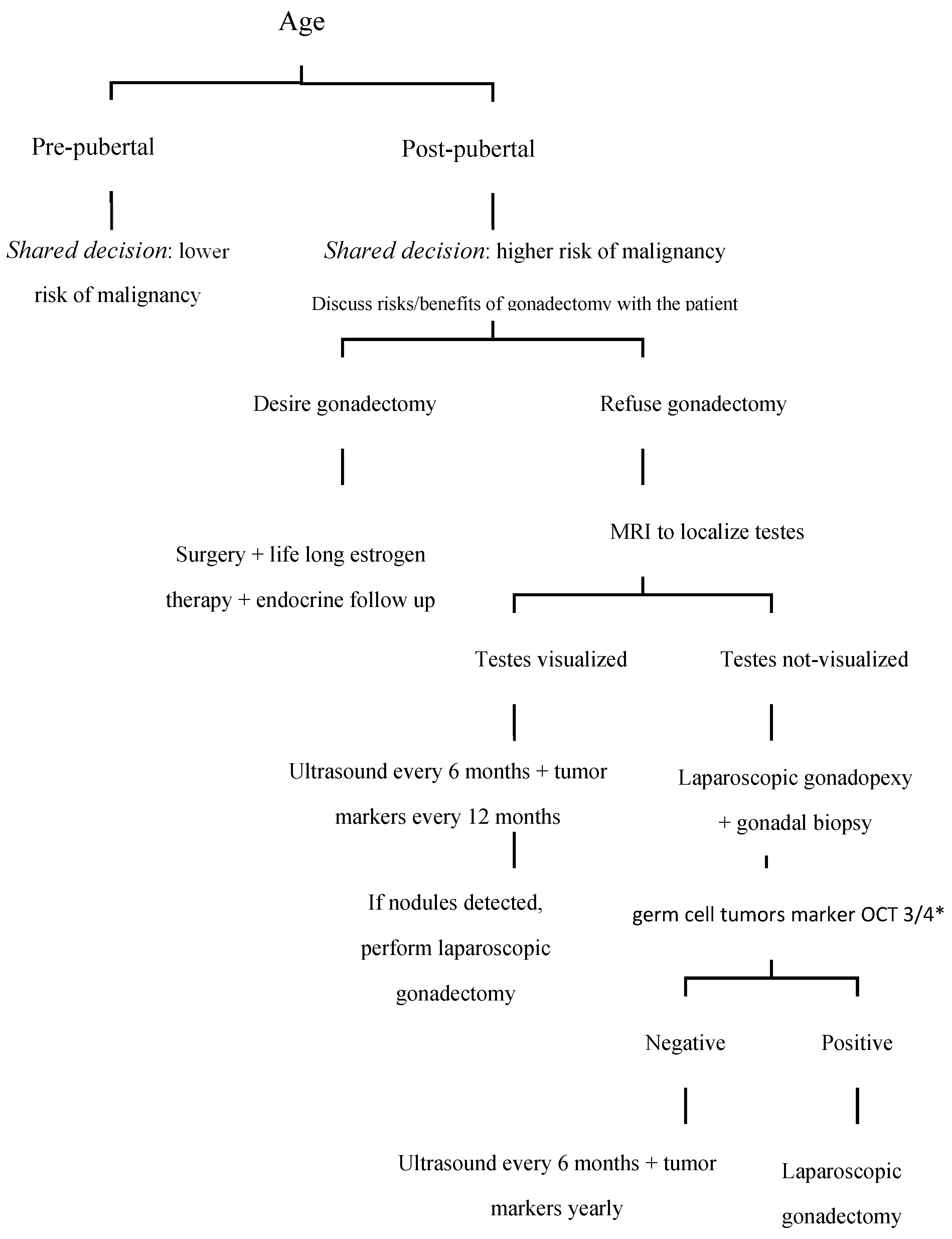
3. Discussion and the Case-Based Review of the Medical Literature
- -
- Lack of negative feedback on LH [14]: in patients with CAIS, early puberty LH levels are within the normal range for pubertal females or males, but they rise sharply during late puberty, similar to levels seen in mid-cycle females. This late but marked and continuous increase in LH is a sign that the testosterone receptor in the pituitary/hypothalamus is defective and unable to suppress LH release even though testosterone levels are extremely high.
- -
- Intact feedback from inhibin and other proteins [15]: inhibin, produced by Sertoli cells in the testes (which are present in patients with CAIS), is a protein hormone that acts by inhibiting follicle-stimulating hormone (FSH) and, more precisely, in women, it stimulates the maturation capacity of ovarian follicles (and is considered a marker of follicular reserve), whereas in men, it indirectly affects gamete development and controls spermatogenesis with a feedback mechanism on FSH secretion. Since these proteins specifically regulate FSH and are not significantly altered by the presence of androgens or their insensitivity, FSH levels tend to remain normal. Furthermore, FSH inhibition is less dependent on androgen levels than LH, thus explaining why FSH remains normal even when LH is elevated.
- -
- Aromatization of testosterone [16]: a portion of the testosterone produced by the gonads in patients with CAIS is converted into estradiol via the aromatase enzyme. This leads to estradiol levels within the lower female range. However, since the amount of testosterone converted is limited, estradiol levels do not reach the typical levels observed in women of reproductive age but fall within the lower range of the female spectrum.
4. Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hughes, I.A.; Houk, C.; Ahmed, S.F.; LWPES/ESPE Consensus Group. Consensus statement on the management of intersex disorders.Consensus statement on management of intersex disorders. Arch. Dis. Child. 2006, 91, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Cools, M.; Nordenström, A.; Robeva, R.; Hall, J.; Westerveld, P.; Flück, C.; Köhler, B.; Berra, M.; Springer, A.; Schweizer, K.; et al. Caring for individuals with a difference of sex development (DSD): A Consensus Statement. Nat. Rev. Endocrinol. 2018, 14, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Lanciotti, L.; Cofini, M.; Leonardi, A.; Bertozzi, M.; Penta, L.; Esposito, S. Different Clinical Presentations and Management in Complete Androgen Insensitivity Syndrome (CAIS). Int. J. Environ. Res. Public Health 2019, 16, 1268. [Google Scholar] [CrossRef] [PubMed]
- Deans, R.; Creighton, S.M.; Liao, L.M.; Conway, G.S. Timing of gonadectomy in adult women with complete androgen insensitivity syndrome (CAIS): Patient preferences and clinical evidence. Clin. Endocrinol. 2012, 76, 894–898. [Google Scholar] [CrossRef]
- World Health Organization. Reference values for human chorionic gonadotropin (hCG) and human menopausal gonadotropin (HMG). Int. J. Gynaecol. Obstet. 1999, 67, 119–129. [Google Scholar] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Oakes, M.B.; Eyvazzadeh, A.D.; Quint, E.; Smith, Y.R. Complete androgen insensitivity syndrome-a review. J. Pediatr. Adolesc. Gynecol. 2008, 21, 305–310. [Google Scholar] [CrossRef]
- Gulía, C.; Baldassarra, S.; Zangari, A.; Briganti, V.; Gigli, S.; Gaffi, M.; Signore, F.; Vallone, C.; Nucciotti, R.; Costantini, F.M.; et al. Androgen insensitivity syndrome. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3873–3887. [Google Scholar] [CrossRef]
- Batista, R.L.; Costa, E.M.F.; Rodrigues, A.d.S.; Gomes, N.L.; Faria, J.A.; Nishi, M.Y.; Arnhold, I.J.P.; Domenice, S.; de Mendonca, B.B. Androgen insensitivity syndrome: A review. Arch. Endocrinol. Metab. 2018, 62, 227–235. [Google Scholar] [CrossRef]
- Dey, R.; Biswas, S.C.; Chattopadhvav, N.; Gupta, D.; Roybiswas, R.; Mukhopadhyay, A. The XY Female (Androgen Insensitivity Syndrome)-Runs in the Family. J. Obstet. Gynaecol. India 2012, 62, 332–333. [Google Scholar] [CrossRef]
- Papadimitriou, D.T.; Linglart, A.; Morel, Y.; Chaussain, J.L. Puberty in subjects with complete androgen insensitivity syndrome. Horm. Res. 2006, 65, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Zachmann, M.; Prader, A.; Sobel, E.H.; Crigler, J.F.; Ritzén, E.M.; Atarés, M.; Ferrandez, A. Pubertal growth in patients with androgen insensitivity: Indirect evidence for the importance of estrogens in pubertal growth of girls. J. Pediatr. 1986, 108 Pt 1, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Danilovic, D.L.S.; Correa, P.H.S.; Costa, E.M.F.; Melo, K.F.S.; Mendonca, B.B.; Arnhold, I.J.P. Height and bone mineral density in androgen insensitivity syndrome with mutations in the androgen receptor gene. Osteoporos. Int. 2007, 18, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, S. Gonadotropin regulation during puberty in complete androgen insensitivity syndrome with testicles in situ. Horm. Res. 1994, 42, 253–256. [Google Scholar] [CrossRef]
- Baird, D.T. Inhibin and oestradiol in the control of FSH secretion in the sheep. J. Reprod. Fertil. Suppl. 1991, 43, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Doehnert, U. Characteristic features of reproductive hormone profiles in late adolescent and adult females with complete androgen insensitivity syndrome. Sex. Dev. 2015, 9, 69–74. [Google Scholar] [CrossRef]
- Griffin, J.E.; Edwards, C.; Madden, J.D.; Harrod, M.J.; Wilson, J.D. Congenital absence of the vagina. The Mayer-Rokitansky-Kuster-Hauser syndrome. Ann. Intern. Med. 1976, 85, 224–236. [Google Scholar] [CrossRef]
- Hughes, I.A.; Houk, C.; Ahmed, S.F.; Lee, P.A.; Lawson Wilkins Pediatric Endocrine Society/European Society for Paediatric Endocrinology Consensus Group. Consensus statement on management of intersex disorders. J. Pediatr. Urol. 2006, 2, 148–162. [Google Scholar] [CrossRef]
- Hughes, I.A.; Deeb, A. Androgen resistance. Best Pract. Res. Clin. Endocrinol. Metab. 2006, 20, 577–598. [Google Scholar] [CrossRef]
- Cools, M.; Drop, S.L.S.; Wolffenbuttel, K.P.; Oosterhuis, J.W.; Looijenga, L.H.J. Germ cell tumors in the intersex gonad: Old paths, new directions, moving frontiers. Endocr. Rev. 2006, 27, 468–484. [Google Scholar] [CrossRef]
- Cools, M.; van Aerde, K.; Kersemaekers, A.M.; Boter, M.; Drop, S.L.S.; Wolffenbuttel, K.P.; Steyerberg, E.W.; Oosterhuis, J.W.; Looijenga, L.H.J. Morphological and immunohistochemical differences between gonadal maturation delay and early germ cell neoplasia in patients with undervirilization syndromes. J. Clin. Endocrinol. Metab. 2005, 90, 5295–5303. [Google Scholar] [CrossRef] [PubMed]
- Hughes, I.A.; Davies, J.D.; Bunch, T.I.; Pasterski, V.; Mastroyannopoulou, K.; MacDougall, J. Androgen insensitivity syndrome. Lancet 2012, 380, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.; Hanif, F.; Hanif, S.M.; Abdullah, F.E.; Shamim, M.S. Complete Androgen Insensitivity Syndrome. J. Coll. Physicians Surg. Pak. 2008, 18, 442–444. [Google Scholar]
- Hannema, S.E.; Scott, I.S.; Rajpert-De Meyts, E.; Skakkebaek, N.E.; Coleman, N.; Hughes, I.A. Testicular development in the complete androgen insensitivity syndrome. J. Pathol. 2006, 208, 518–527. [Google Scholar] [CrossRef]
- Nakhal, R.S.; Hall-Craggs, M.; Freeman, A.; Kirkham, A.; Conway, G.S.; Arora, R.; Woodhouse, C.R.J.; Wood, D.N.; Creighton, S.M. Evaluation of retained testes in adolescent girls and women with complete androgen insensitivity syndrome. Radiology 2013, 268, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Wunsch, L.; Holterhus, P.M.; Wessel, L.; Hiort, O. Patients with disorders of sex development (DSD) at risk of gonadal tumour development: Management based on laparoscopic biopsy and molecular diagnosis. BJU Int. 2012, 110 Pt C, E958–E965. [Google Scholar] [CrossRef]
- Sessa, L.; Rotunno, G.; Sodero, G.; Pane, L.C.; Rendeli, C.; Maresca, G.; Rigante, D.; Cipolla, C. Predictive value of transabdominal pelvic ultrasonography for the diagnosis of central precocious puberty: A single-center observational retrospective study. Clin. Pediatr. Endocrinol. 2024, 2024-0025. [Google Scholar] [CrossRef]
- Warne, G.L.; Grover, S.; Zajac, J.D. Hormonal therapies for individuals with intersex conditions: Protocol for use. Treat. Endocrinol. 2005, 4, 19–29. [Google Scholar] [CrossRef]
- Bertelloni, S.; Dati, E.; Baroncelli, G.I.; Hiort, O. Hormonal management of complete androgen insensitivity syndrome from adolescence onward. Horm. Res. Paediatr. 2011, 76, 428–433. [Google Scholar] [CrossRef]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef]
- Villa, P.; Cipolla, C.; Amar, I.; Sodero, G.; Pane, L.C.; Ingravalle, F.; Pontecorvi, A.; Scambia, G. Bone mineral density and body mass composition measurements in premenopausal anorexic patients: The impact of lean body mass. J. Bone Miner. Metab. 2024, 42, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Hiort, O.; Reinecke, S.; Thyen, U.; Jürgensen, M.; Holterhus, P.-M.; Schön, D.; Richter-Appelt, H. Puberty in disorders of somatosexual differentiation. J. Pediatr. Endocrinol. Metab. 2003, 16 (Suppl. 2), 297–306. [Google Scholar] [PubMed]
- Arnhold, I.J.P.; Melo, K.; Costa, E.M.F.; Danilovic, D.; Inacio, M.; Domenice, S.; Mendonca, B.B. 46,XY disorders of sex development (46,XY DSD) due to androgen receptor defects: Androgen insensitivity syndrome. Adv. Exp. Med. Biol. 2011, 707, 59–61. [Google Scholar] [CrossRef]
- Cipolla, C.; Sodero, G.; Pane, L.C.; Mariani, F.; Di Sarno, L.; Rigante, D.; Candelli, M. Auxological and Metabolic Parameters of Children Undergoing the Gonadotropin-Releasing Hormone Stimulation Test: Correlations with the Final Diagnosis of Central Precocious Puberty in a Single-Center Study. Biomedicines 2023, 11, 1678. [Google Scholar] [CrossRef]
- Patel, V.; Casey, R.K.; Gomez-Lobo, V. Timing of Gonadectomy in Patients with Complete Androgen Insensitivity Syndrome-Current Recommendations and Future Directions. J. Pediatr. Adolesc. Gynecol. 2016, 29, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, P.; Lyu, Y.; Wang, Y.; Gao, K.; Wang, J.; Lan, F.; Chen, F. Molecular genetics and general management of androgen insensitivity syndrome. Intractable Rare Dis. Res. 2023, 12, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yin, X.; Li, P. Clinical, hormonal and genetic characteristics of androgen insensitivity syndrome in 39 Chinese patients. Reprod. Biol. Endocrinol. 2020, 18, 34. [Google Scholar] [CrossRef]
- He, J.; Qi, S.; Zhang, H.; Guo, J.; Chen, S.; Zhang, Q.; Zhu, B. Clinical and genetic characterization of six cases with complete androgen insensitivity syndrome in China. J. Genet. 2017, 96, 695–700. [Google Scholar] [CrossRef]
- Fulare, S.; Deshmukh, S.; Gupta, J. Androgen Insensitivity Syndrome: A rare genetic disorder. Int. J. Surg. Case Rep. 2020, 71, 371–373. [Google Scholar] [CrossRef]
- Jääskeläinen, J. Molecular biology of androgen insensitivity. Mol. Cell. Endocrinol. 2012, 352, 4–12. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fraccascia, B.; Sodero, G.; Pane, L.C.; Malavolta, E.; Gola, C.; Pane, L.; Paradiso, V.F.; Nanni, L.; Rigante, D.; Cipolla, C. Complete Androgen Insensitivity Syndrome in a Young Girl with Primary Amenorrhea and Suspected Delayed Puberty: A Case-Based Review of Clinical Management, Surgical Follow-Up, and Oncological Risk. Diseases 2024, 12, 235. https://doi.org/10.3390/diseases12100235
Fraccascia B, Sodero G, Pane LC, Malavolta E, Gola C, Pane L, Paradiso VF, Nanni L, Rigante D, Cipolla C. Complete Androgen Insensitivity Syndrome in a Young Girl with Primary Amenorrhea and Suspected Delayed Puberty: A Case-Based Review of Clinical Management, Surgical Follow-Up, and Oncological Risk. Diseases. 2024; 12(10):235. https://doi.org/10.3390/diseases12100235
Chicago/Turabian StyleFraccascia, Barbara, Giorgio Sodero, Lucia Celeste Pane, Elena Malavolta, Caterina Gola, Luigi Pane, Valentina Filomena Paradiso, Lorenzo Nanni, Donato Rigante, and Clelia Cipolla. 2024. "Complete Androgen Insensitivity Syndrome in a Young Girl with Primary Amenorrhea and Suspected Delayed Puberty: A Case-Based Review of Clinical Management, Surgical Follow-Up, and Oncological Risk" Diseases 12, no. 10: 235. https://doi.org/10.3390/diseases12100235