Abstract
Lung cancer, characterized by its heterogeneity, presents a significant challenge in therapeutic management, primarily due to the development of resistance to conventional drugs. This resistance is often compounded by the tumor’s ability to reprogram its metabolic pathways, a survival strategy that enables cancer cells to thrive in adverse conditions. This review article explores the complex link between drug resistance and metabolic reprogramming in lung cancer, offering a detailed analysis of the molecular mechanisms and treatment strategies. It emphasizes the interplay between drug resistance and changes in metabolic pathways, crucial for developing effective lung cancer therapies. This review examines the impact of current treatments on metabolic pathways and the significance of considering metabolic factors to combat drug resistance. It highlights the different challenges and metabolic alterations in non-small-cell lung cancer and small-cell lung cancer, underlining the need for subtype-specific treatments. Key signaling pathways, including PI3K/AKT/mTOR, MAPK, and AMPK, have been discussed for their roles in promoting drug resistance and metabolic changes, alongside the complex regulatory networks involved. This review article evaluates emerging treatments targeting metabolism, such as metabolic inhibitors, dietary management, and combination therapies, assessing their potential and challenges. It concludes with insights into the role of precision medicine and metabolic biomarkers in crafting personalized lung cancer treatments, advocating for metabolic targeting as a promising approach to enhance treatment efficacy and overcome drug resistance. This review underscores ongoing advancements and hurdles in integrating metabolic considerations into lung cancer therapy strategies.
1. Introduction
Cancer refers to a group of diseases characterized by the uncontrolled growth and malignancy of cells in the body. Lung cancer is among the top five most highly diagnosed cancers in the world and the second most diagnosed cancer in both men and women, as well as being the most common cause of death due to cancer. The very high mortality rate also surpasses the death occurring due to breast, colorectal, and prostate cancers combined. The primary cause of this illness is cigarette smoking, which accounts for about 80% of instances of lung cancer [1]. Lung cancer has a very high incidence rate, with an estimated 238,340 new cases (117,550 men and 120,790 women) expected be diagnosed and 127,070 deaths projected to occur due to the disease. In India, lung cancer accounts for 5.9% of all cancers and 8.1% of all cancer-related deaths [2]. As per a report, approximately 6.2% of men and 5.8% of women in the general population have the propensity to develop lung cancer in their lifetime. This means that 1 in 16 men and 1 in 17 women are at risk of developing lung cancer during their lifetime, with smokers and ex-smokers being at higher risk [3]. Most of the cases of lung cancer are diagnosed at later stages, rendering the treatment ineffective, and thus the disease becomes typically lethal. However, in the past twenty years, exciting gains in survival rate have resulted from the advancement in the screening techniques and their implementation, development of tailored treatments, and growing understanding of cancer biology. Primarily two types of lung cancer are known, non-small-cell lung cancer (NSCLC), which accounts for 80% of cases, and small-cell lung cancer (SCLC), which accounts for 15% of cases. NSCLC is further categorized as adenocarcinoma, squamous cell carcinoma, and large-cell carcinoma [4]. Adenocarcinoma mainly originates in the glands that secret mucus, whereas squamous cell carcinoma is mainly seen in the epithelium lining of lungs. In contrast to adenocarcinoma, squamous cell carcinoma is more aggressive and develops from the cell lining of the airway in the lungs. In contrast to the other two NSCLC subtypes, large-cell carcinoma is more aggressive and can arise from any part of the lung [5]. Small-cell carcinoma is the most prevalent subtype of lung cancer, followed by mixed small-cell carcinoma. SCLC is typically more aggressive than NSCLC and is slightly more prevalent in women (14%) than in males (13%). Metastasis is observed more frequently in SCLC patients than NSCLC patients, with the disease migrating outside the lungs in 94% of SCLC cases versus 70% of NSCLC cases [6].
Figure 1 depicts the significant advancements in lung cancer treatment. Despite these advances in therapeutics, resistance to drugs remains a major obstacle to long-term treatment effectiveness, eventually leading to therapeutic insensitivity, poor progression-free survival, and disease relapse [7]. Drug resistance refers to the reduction in the effectiveness of medication that occurs when cancers evolve and acquire resistance to the drugs used for treatment. Major treatment options for different types of lung cancer include surgery, chemotherapy, radiotherapy, immunotherapy, targeted therapy, etc. Major issues related to drug resistance are therapeutic insensitivity, poor progression-free survival, and disease relapse. Resistance mechanisms in lung cancer can arise from various factors, such as genetic mutations, epigenetic modifications, uncontrolled drug efflux, tumor hypoxia, changes in the tumor microenvironment, and several other cellular and molecular alterations [8].
Reprogramming of metabolic pathways is an essential characteristic of tumor cells. To meet the material, energy, and redox force requirements for fast multiplication, tumor cells remodel their metabolic pathways. Metabolic reprogramming, achieved through alterations in gene expression, cell state, and the tumor itself, causes certain metabolites to change in quantity or type both inside and outside of cells, generating an environment that is conducive for tumor growth and progression [9]. The various metabolic changes that take place in tumor cells include alteration in glucose metabolism, amino acid metabolism, and lipid metabolism. This reprogramming of tumor cells is beneficial for conferring drug resistance to many cancer cells [10,11,12].
Accordingly, this review aims to provide an overview of the currently available therapeutic approaches to treat lung cancer, as well as issues pertaining to drug resistance that persist despite prolonged treatment. This review also explores drug resistance signaling pathways and the role of tumor cell metabolic reprogramming in developing drug resistance in the tumor microenvironment. In addition, this work explores the junction of medication resistance and metabolic reprogramming to identify and create novel and effective therapeutic approaches for this persistent problem.
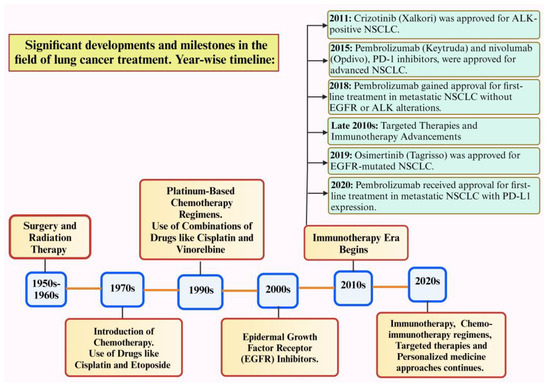
Figure 1.
Timeline for the discovery of significant advancements in lung cancer treatment [13]. Created with Bio Render.
Figure 1.
Timeline for the discovery of significant advancements in lung cancer treatment [13]. Created with Bio Render.
2. Signaling Pathways Involved in Lung Cancer
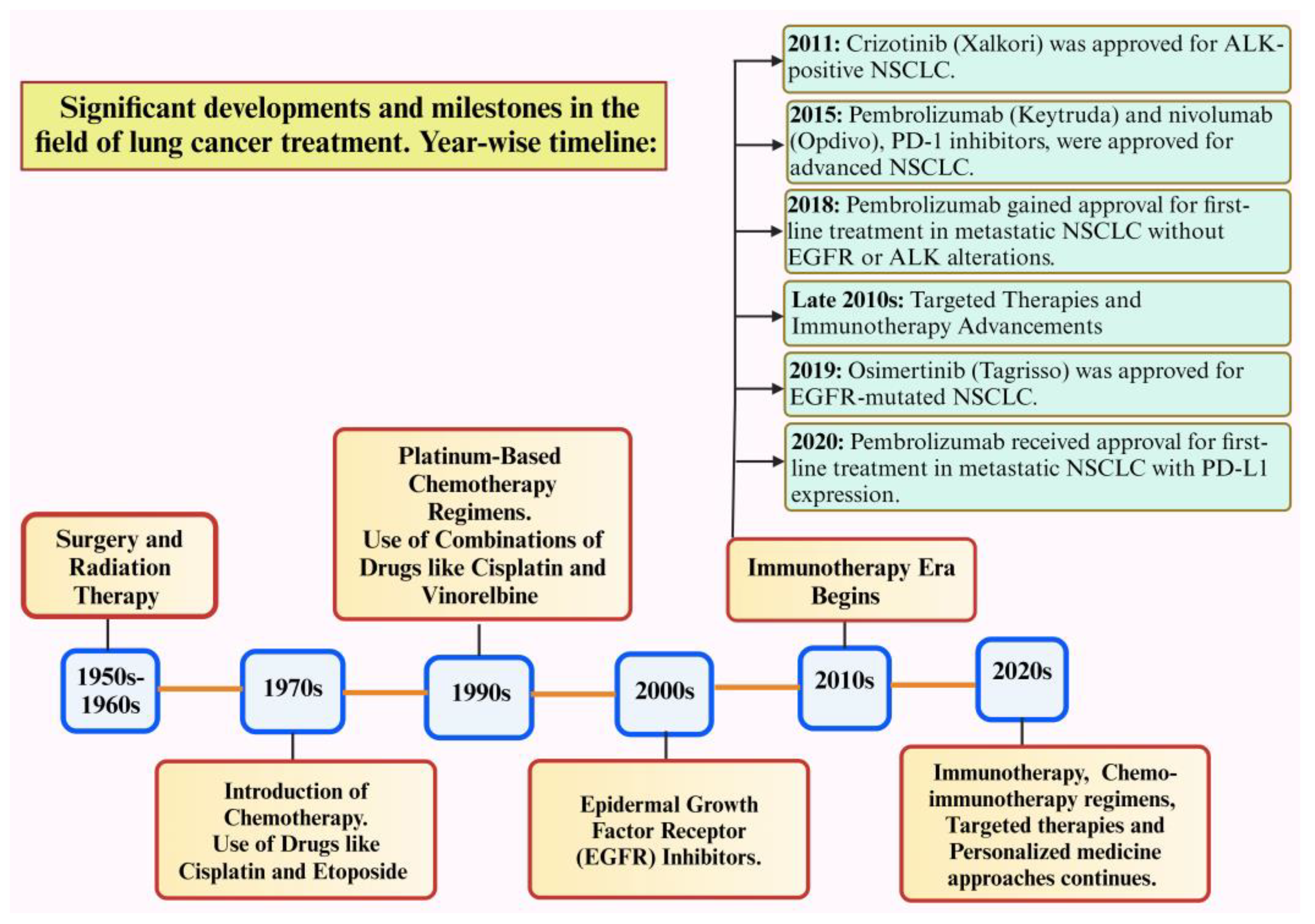
Figure 2 illustrates the various signaling pathways in the lung, where aberrations at different levels are implicated in lung cancer development and progression. The phosphatidylinositol 3-kinase (PI3K)/Akt pathway is pivotal in regulating diverse cellular processes and is frequently dysregulated in cancers, playing a significant role in the initiation and advancement of tumors. The mitogen-activated protein kinases/extracellular signal-regulated kinase or the RAS/RAF/MEK/ERK (MAPK) predominantly engages in apoptosis, pathogenesis, progression survival, and invasion. Further, there are other intracellular signaling pathways in the lung that, when downregulated, lead to cancer and serve as important targets for therapeutic treatments. These include the DNA repair pathways, inflammatory signaling pathway, anaplastic lymphoma kinase (ALK) pathway, and tumor suppressor genes. Additionally, these signaling pathways interact with one another; thereby, enhancing one pathway may either strengthen or inhibit another [14].
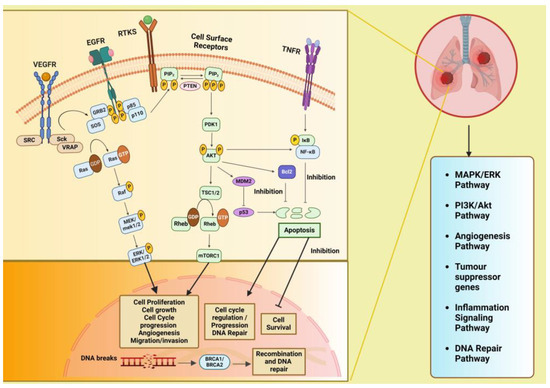
Figure 2.
Signaling pathways involved in lung cancer that have a role in metabolic reprogramming viz. PI3K/Akt pathway, MAPK/ERK pathway, inflammation signaling pathways, tumor suppressor genes, and DNA repair pathways and their cross-talk. Created with BioRender.com. Legends: EGFR: epidermal growth factor receptor; VEGFR: vascular endothelial growth factor receptor, RTK: receptor tyrosine kinase; SRC: proto-oncogene tyrosine-protein kinase Src; Sck: Shc-like protein; VRAP: VEGF receptor-associated protein; A-Raf: serine/threonine-protein kinase A-Raf; Ras: Rat sarcoma gene; MAPK/ERK: mitogen-activated protein kinase/extracellular signal-regulated kinase; PI3K: phosphatidylinositol 3-kinase; PTEN: phosphatase and tensin homolog; PIP2: phosphatidylinositol bisphosphate; PIP3: phosphatidylinositol triphosphate; PDK1: phosphoinositide-dependent kinase 1: Akt: protein kinase-B; TSC1: tuberous sclerosis complex 1: TSC2: tuberous sclerosis complex 2; Rheb: Ras homolog enriched in brain; mTOR: mammalian target of rapamycin; MDM2: mouse double minute 2 homolog; Bcl-2: B-cell lymphoma-2; TNF: tumor necrosis factor; TNFR1/2: TNF receptor 1/2; NF-κB: nuclear factor-κB; BRCA1 and BRCA2: breast cancer-associated protein receptors. Arrows represent activation, bars represent inhibition.
The PI3K/AKT/mTOR signaling pathway has cross-talk with the Ras/Raf/MAPK (MEK)/ERK pathway and tumor suppressor gene pathway: PI3K, a family of lipid kinases and class IA PI3Ks, which comprises a catalytic p110 subunit and a regulatory p85 subunit. Upon ligand binding to receptor tyrosine kinases (RTK) on the cell membrane, such as epidermal growth factor receptors (EGFRs), vascular endothelial growth factor receptors (VEGFRs), and others, the PI3Ks are activated, leading to the phosphorylation of PIP2 to PIP3 (phosphatidylinositol (4,5)–bisphosphate to phosphatidylinositol (3,4,5)–trisphosphate) catalyzed by the p110 subunit. The phosphatase and tensin homolog (PTEN) convert PIP3 to PIP2, suppressing the PIP3-dependent processes and thus restricting cell survival, growth, and proliferation. Next, phosphoinositide-dependent protein kinase (PDK1) and mTORC1 (a serine/threonine kinase) mediate the phosphorylation and activation of Akt together. It is known that the kinase mTOR is negatively regulated by the TSC1/TSC2 complex (tuberous sclerosis protein 1/2), which inhibits Ras homolog enriched in brain (Rheb) activation, a GTPase that activates mTOR. Accordingly, activated AKT kinases can phosphorylate both TSC1 and TSC2. Akt also inhibits the pro-apoptotic Bcl-2 protein family members and indirectly upregulates p53 by activating mouse double minute 2 homolog (MDM2) and NF-κB transcription factors, leading to increased expression of cell survival and anti-apoptotic signals. Dysregulation of the PI3K/Akt/mTOR pathway has been found to be involved in tumorigenesis and disease progression in NSCLC, including angiogenesis, migration, metastasis, proliferation, and apoptosis [15,16]. The VEGF/VEGFR-2 dimers mediate tumor metastasis, angiogenesis, and tumor survival through two major signaling pathways branches: Ras/MAPK and PI3K/Akt. Upon VEGF binding to VEGFR, dimerization and phosphorylation of the intracellular tyrosine kinase domain occur, followed by the activation of downstream proteins such as proto-oncogene tyrosine-protein kinase Src (c-Src), Shc-like protein (Sck), and VEGF receptor-associated protein (VRAP). Activation of c-Src involves the adaptor molecule growth factor receptor-bound protein 2 (GRB2)-associated binder 1. The adaptor proteins like GRB2 and SOS (Son of Sevenless) then recruit Ras (which is a type of small GTP-binding protein) and phosphatidylinositol 3-kinase (PI3K), leading to the formation of the signaling pathway branches Ras/MAPK and PI3K/Akt [17].
The mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK (MEK)/ERK) pathway is also known as the Ras/Raf/MAPK (MEK)/ERK pathway. After membrane receptor activation, adaptor proteins such as SOS recruit Ras proteins to activate Raf, a serine/threonine protein kinase, which becomes phosphorylated and activates MAPK kinase (MEK). This activation is followed by MEK phosphorylation and subsequent activation of ERK (extracellular signal-regulated kinase). ERK then activates its cytoplasmic and/or nuclear targets. Under normal conditions and in the cancer cell, the PI3K/AKT pathway also interacts with the MAPK/ERK node [17,18].
Inflammatory signaling pathway: Tumor necrosis factor (TNF) binds and activates TNF receptors TNFR1 or TNFR2 that, in turn, activate the inflammatory signaling pathway. Both the cytokine and its receptors TNFR1 and TNFR2 are expressed in lung cancer. TNF stimulates the activation of nuclear factor-κB (NF-κB), which is triggered by activation of inhibitor of NF-κB (IκB) kinase (IKK). NF-κB translocates into the nucleus and promotes transcription of many NF-κB target genes, many of which (such as FLIP: cellular FLICE-inhibitory protein, BCL-XL: B-cell lymphoma-extra-large, and others) have anti-apoptosis and pro-survival function [19].
DNA repair pathway: The tumor suppressor proteins, namely breast cancer-associated proteins BRCA1 and BRCA2, play an important role in DNA repair through a specific type of DNA repair pathway called double-strand break repair by homologous recombination (HR) repair. Many other genes, known as BRCA-related genes, also participate directly or indirectly. Mutations in the BRCA1/2 genes have been observed in 5–10% of NSCLC cases [20]. BRCA1 and BRCA2 regulate HR repair by facilitating the assembly of the DNA recombinase-RAD51 onto broken DNA ends at the site of double strand breaks (DSBs) and holding up replication forks [21].
The anaplastic lymphoma kinase (ALK) pathway: Fusion of ALK with other genes leads to constitutive ALK activation, promoting cell proliferation and survival through downstream signaling pathways, including PI3K/AKT and MAPK/ERK [22].
3. Mechanisms of Drug Resistance and Metabolic Reprogramming
3.1. Molecular Basis of Drug Resistance
One of the significant issues that reduces the efficacy of chemotherapies employed to treat cancer is drug resistance. Prior to treatment, tumors may exhibit innate resistance to chemotherapy. However, tumors that are initially responsive to chemotherapy may potentially develop drug resistance during treatment. It is estimated that more than 90% of patients with metastatic cancer experience treatment failure due to resistance to chemotherapy. Additionally, resistant micro metastatic tumor cells may also diminish the efficacy of chemotherapy in the adjuvant setting [23]. The molecular analysis of several of the most frequently mutated molecules in lung cancer includes V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS; 25%), anaplastic lymphoma kinase (ALK; 6%), epidermal growth factor receptor (EGFR), phosphoinositide-3-kinase, catalytic, alpha polypeptide (PIK3CA; 3%), v-Raf murine sarcoma viral oncogene homolog B1 (BRAF; 3%), and human epidermal growth factor receptor 2 (HER2; 1%) [24].
3.2. Metabolic Pathways and Their Alterations in Lung Cancer
Alterations in major metabolic pathways in signaling pathways involved in lung cancer progression include glycolysis, amino acid metabolic pathways (such as glutamine, serine, glycine, tryptophan), and fatty acid synthesis pathways. Metabolic reprogramming or alteration is due to downregulation or upregulation of different metabolite levels in tumor cells compared to normal cells [25,26]. Understanding these metabolic abnormalities and identifying relevant metabolites that differ from normal metabolites could facilitate the implementation and evaluation of novel treatment strategies and targeted medicines for patients with lung cancer (LC). Changes in metabolomic pathways were verified by quantitative proteomics analysis of the main enzymes found in the disrupted pathways. This analysis identified 13 distinct biomarkers linked to metabolic disruption of NSCLC morbidity, implicated in 4 major pathways: aminoacyl-tRNA biosynthesis, glycine serine and threonine metabolism, tyrosine metabolism, and sphingolipid metabolism. Important enzymes involved in these pathways, including phosphoserine phosphatase, argininosuccinic acid catenase, tyrosinase, and 3-phosphoglycerate dehydrogenase, showed distinct variations in expression, according to the proteomics study [27].
An increase in the levels of glucose transporters (GLUT) in tumor cells is responsible for altered metabolism. Both GLUT3 and GLUT5 are found to be higher in tumor cells. The elevated glucose levels inside cells lead to metabolic changes [28]. The Warberg effect is evident in lung cancer cells, where glucose is metabolized via lactic acid fermentation rather than being broken down to pyruvate as in normal cells. Tumor M2-Pyruvate Kinase (PKM2), a glycolytic enzyme, is detected to be increased in LC and several other malignancies, and it is necessary for the malignant transformation [29]. Lactate formed inside the cell is transported with the help of transporters called monocarboxylate transporters (MCTs). This lactate shuttle, mainly via MCT1 and MCT4, is an important mechanism through which cancer tissue maintains the balance of the connections between glycolytic and oxidative cells [30].
Lung cancer cells show an increased uptake of glutamine. After entering the cell, glutamine is converted into glutamate, which is further converted to α-ketoglutarate with the help of a mitochondrial enzyme glutaminase (GLS). This pathway is called glutaminolysis. The alanine-serine-cysteine-transporter-2 (ASCT2 or SLC1A5) mediates the uptake of these glutamines into the cancer cells, and it has been noted that LC patients express these transporters with greater frequency [31]. L-type amino acid transporter 1 (LAT1), also known as SLC7A5/SLC3A2, is one of the various types of transporters found in LC patients, and it plays a role of exchanging glutamine for vital amino acids like phenylalanine, valine, and methionine [32]. Another transporter, SLC38A3, controls the transfer of glutamine and histidine, which, in turn, promotes the metastasis of NSCLC by modulating the PDK1/AKT signaling cascade. HIF-1α activates SLC38A1 and provides a variety of mechanisms to alter the glutamine level in the cell, whereas HIF-2α controls and upregulates the expression of SLC1A5 [33,34].
The serine synthesis pathway is a crucial pathway for energy production in cancer cells, along with glycolysis. The precursors, namely 3-phosphoglycerate and glutamate, which are generated by the glycolysis and glutaminolysis pathways, respectively, drive the manufacturing of serine. Glycine produced during the glycolysis process can be used as a source of carbon for one-carbon metabolism [35]. An enzyme known as phosphoserine aminotransferase (PSAT1) helps oxidize and catalyze the precursor 3-phosphoglycerate into 3-phosphoserine and α-KG (alpha- ketoglutarate), which is subsequently transformed into serine with the aid of another enzyme known as 1-3-phosphosrine phosphatase [36]. It was found that nuclear factor erythroid 2–related factor 2 (NRF2) controls the expression of the critical serine synthesis enzymes phosphoglycerate dehydrogenase (PHGDH), PSAT1, and serine hydroxymethyltransferase-2 (SHMT2) by activating activating transcription factor 4 (ATF4) to promote glutathione and nucleotide formation. PSAT1 overexpression worsens the prognosis of cancer by encouraging cancer cell proliferation, metastasis, and chemoresistance [37].
Glycine decarboxylase is necessary for tumor-initiating cells. When glycine decarboxylase is inhibited in cells with elevated levels of serine hydroxy-methyltransferase 2 (SHMT2), the accumulation of glycine is converted into toxic compounds such as methylglyoxal and aminoacetone, resulting in cell growth arrest [38].
The fatty acid synthesis pathways are also affected in lung cancer cells. The de novo production of fatty acids requires the enzyme fatty acid synthase (FASN), which has been shown to be highly expressed in NSCLC patients. Additionally, FASN was discovered to play a role in the metabolism of glucose by inhibiting the AKT/ERK pathway, which modifies LC cells [39]. There are other enzymes upregulated in different types of lung cancer cells, such as ATP citrate lyase enzyme (ACLY) and AcCoA carboxylase (ACC1/2) [40]. Cholesterol synthesis is also affected, as reported in an in vitro study showing that the presence of 25-Hydroxycholesterol increased LC cell growth and invasion. A vital regulator of lung development and morphogenesis, thyroid transcription factor 1 (TTF-1), specifically targets ATP-binding cassette transporter A1 (ABCA1) in LC cells. TTF-1 may be utilized as a diagnostic marker for LC, as it was found to be overexpressed in LC samples [41].
4. Current Therapeutic Approaches and Metabolic Considerations
4.1. Overview of Current Lung Cancer Therapies
There are some differences in the treatment approach of both types of lung cancer, namely NSCLC and SCLC. SCLC is the more aggressive form of lung cancer, often diagnosed at a later stage with frequent metastases. Therefore, the primary treatment modalities used for SCLC typically include chemotherapy, radiotherapy, targeted therapy, and immunotherapy. Surgery is very rarely used in SCLC cases due to its aggressive nature. In contrast, for NSCLC, treatment options may include chemotherapy, chemoradiotherapy, radiotherapy, targeted therapy, immunotherapy, as well as surgery, depending on factors such as the stage and characteristics of the tumor.
4.1.1. Chemotherapy
Chemotherapy consistently serves as the first line of treatment for cancer, representing a standard therapeutic approach for lung cancer. Chemotherapeutic drugs are broadly classified into four categories: (1) alkylating agents, such as platinum compounds like cisplatin and carboplatin, (2) microtubule-targeting drugs, including vinorelbine, paclitaxel, and docetaxel, (3) antimetabolites, exemplified by pemetrexed and gemcitabine, and (4) topoisomerase inhibitors like etoposide [42]. Notably, in NSCLC, the principal chemotherapy components comprise gemcitabine, taxanes, pemetrexed, cisplatin, and carboplatin. Additionally, targeted therapy medications like EGFR inhibitors (e.g., erlotinib) and VEGFR inhibitors (e.g., bevacizumab) are utilized. The mechanisms of action for these drugs vary: cisplatin, carboplatin, and gemcitabine disrupt the DNA repair system, induce DNA damage, and prompt programmed cell death (apoptosis) in the cancer cell [43], while taxane-based drugs disrupt microtubule dynamics, induce cell cycle arrest, and trigger apoptosis [44]. Conversely, pemetrexed and methotrexate, classified as antifolate drugs, can lead to cell cycle arrest in the S phase [45]. In the management of metastatic SLCL, the first line of treatment includes a combination of platinum and etoposide; nevertheless, it shows a very poor survival rate, as it relapses within one year [46]. Currently, topotecan stands as the sole medication conforming to the accepted standard of care for second-line treatment of SCLC [47].
4.1.2. Radiotherapy
Radiotherapy is commonly utilized in combination with chemotherapy, especially in stage III unresectable NSCLC. The adverse effects of radiation therapy have decreased due to the development of intensity-modulated radiotherapy (IMRT), stereotactic body radiation (SBRT), and four-dimensional computed tomography (4DCT) [48]. Primarily, the mode of action of radiotherapy stems from DNA damage, which can elicit immune responses in lung cancer. As a result, the combination of radiotherapy and immunotherapy could improve the recovery in lung cancer patients [49]. Comparatively to conventional chemoradiotherapy, a phase 3 trial suggests that platinum-based chemotherapy, radiotherapy, and durvalumab (an immune checkpoint inhibitor of PD-L1) as combinational therapy for lung cancer offer greater benefits, potentially extending overall survival (up to 4 years) in patients with stage III NSCLC [50]. In the case of SCLC patients, radiotherapy in combination with chemotherapy is applicable in the initial stages of treatment but not suitable for later metastatic stages. Around 30% of newly diagnosed SCLC cases receive standard treatment with curative intent, involving four cycles of platinum-doublet chemotherapy combined with radiotherapy [51].
4.1.3. Targeted Therapy
In lung cancer, targeted therapy refers to the application of medications or other compounds that specifically target and obstruct the growth and metastasis of cancer cells. These treatments function by either focusing on certain chemicals implicated in the initiation and spread of cancer or by obstructing the signals that propel the growth of cancer cells. Anaplastic lymphoma kinase (ALK), epidermal growth factor receptor (EGFR), ROS proto-oncogene 1 (ROS1), and serine/threonine-protein kinase B-Raf (BRAF) inhibitors have become key components of lung cancer therapy. Recently, a new class of medications has emerged that can target the formerly “un-druggable” KRAS mutation [52,53]. FDA-approved drugs for targeted therapy include gefitinib, erlotinib, afatinib, dacomitinib, and osimertinib, which are used as EGFR inhibitors. ALK inhibitors include crizotinib, alectinib, brigatinib, ceritinib, and lorlatinib. ROS1 inhibitors comprise crizotinib, lorlatinib, entrectinib, and brigatinib. RET (rearranged during transfection) inhibitors such as pralsetinib and selpercatinib are available [54,55]. The FDA-approved medications have significantly improved patients’ overall survival (OS). For instance, gefitinib has improved median progression-free survival (mPFS) by approximately 10.8 months, erlotinib by approximately 14 months, afatinib by approximately 48 months, and dacomitinib by up to 14.7 months [56,57]. Some medications are currently undergoing clinical trials. For example, pyrotinib has demonstrated an extension of patients’ PFS by 6.9 months and median OS by 14.4 months in patients with advanced NSCLC with HER2 mutation [58]. Combination treatments for lung cancer, such as bevacizumab, a monoclonal antibody that targets VEGF, and erlotinib, an EGFR inhibitor, can extend the PFS in patients with NSCLC. In advanced NSCLC with an EGFR mutation, the combination of apatinib, a VEGFR inhibitor, with gefitinib, a first-generation EGFR tyrosine kinase inhibitor (TKI), may extend the mPFS for 19.2 months. However, this combination therapy has some adverse effects and lowers quality of life [59].
4.1.4. Immunotherapy
Immunotherapy represents a novel therapeutic approach for cancer treatment and holds significant appeal to researchers due to its promising result in dealing with the disease. It entails boosting the immune system’s capacity to identify and combat cancerous cells. Even while cancer cells frequently find ways to avoid detection, the immune system can identify and eliminate aberrant cells, including cancer cells. The goal of immunotherapy is to get past these defense mechanisms and improve the immune system’s capacity to recognize and destroy cancer cells [60]. This therapy mainly focuses on inhibition of immune checkpoints cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4), programmed death 1 (PD-1), and programmed death-ligand 1(PD-L1). These antibodies demonstrate potential in restoring antitumor immunity by targeting the host immune system to compel it to combat cancer cells [61]. Currently, FDA-approved immunotherapies in use include adoptive T-cell therapy, cancer vaccine, immune checkpoint inhibitors (ICIs), and cytokine modulators. Among the monoclonal antibodies utilized for treating lung cancer are anti-CTLA-4 antibody ipilimumab, anti-PD-1 antibodies pembrolizumab and nivolumab, and anti-PD-L1 antibodies atezolizumab, durvalumab, and avelumab [62]. The efficacy and overall survival outcomes of these drugs vary among patients based on their mutation. Studies have demonstrated that in metastatic NSCLC patients with high PD-L1 expression (≥50%) that lack EGFR or ALK mutation, drugs such as pembrolizumab and atezolizumab could improve PFS for approximately 10.3 months and median OS for 15.5 months [63]. In a phase III trial, durvalumab was shown to improve PFS up to 907 days and OS up to 1,420 days in patients with unresectable stage III NSCLC following failed chemotherapy and radiotherapy [64]. Immunotherapy is also used in combinational therapy along with chemotherapy and radiotherapy. Ongoing trials suggest an improvement in survival rates when immunotherapy is combined with other monoclonal antibodies, chemotherapy, or radiotherapy. For example, durvalumab, an anti-PD-L1 antibody, demonstrates better outcomes when used in combination rather than alone. One of the most prominent combinations involves radiation or chemotherapy along with an anti-CTLA-4 antibody called tremelimumab and durvalumab [64]. A phase 3 trial shows that when pembrolizumab, an anti-PD-1 antibody, is used with radiotherapy, it can enhance the overall survival of metastatic NSCLC patients [65]. Further, immunotherapy is used as a first-line treatment for SCLC in combination with chemotherapy, which also yields good outcomes in SCLC patients.
4.1.5. Antiangiogenic Therapy
Angiogenesis is a physiological process by which new blood vessels are formed. In cancer cells, there is a significant increase in angiogenesis. Within the tumor microenvironment, as cells proliferate, they require more blood vessels to supply blood, leading to an increase in this process. The major angiogenic factors are vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), tumor necrosis factor-alpha (TNF-α), transforming growth factor-beta (TGF-β), and angiopoietins (Ang) [66]. In the tumor microenvironment, VEGF may suppress the immune cells’ response, and, consequently, VEGF inhibitors may also boost immune cell capability. Therefore, antiangiogenic therapy could be combined with immunotherapy to benefit cancer patients [67]. The FDA has approved bevacizumab, a monoclonal antibody that targets VEGF, for use in the treatment of NSCLC. Bevacizumab, when combined with monoclonal antibodies such as atezolizumab, has been confirmed as a potential therapy for non-squamous NSCLC with higher PD-L1 expression (≥50%) but without EGFR/ALK/ROS1 mutations. In patients of NSCLC carrying KRAS and STK11 mutations, as well as Serine/Threonine Kinase 11 (STK11), Kelch-like ECH-associated protein 1 (KEAP1), tumor protein p53 (TP53), and/or strong PD-L1 expression (≥50%), bevacizumab in combination with atezolizumab and chemotherapy (carboplatin and paclitaxel) could serve as the first-line treatment [68].
4.2. Impact on Metabolic Pathways
Lung cancer therapeutic approaches have a significant effect on metabolic pathways of cells. Metabolism refers to the complex set of chemical processes that occurs within living organisms to maintain life. In recent time, various metabolomics studies show the difference in metabolic pathways in patients undergoing treatment. Studies have been conducted to investigate the proteomic and genomic effects of the chemotherapeutic drug anlotinib. Anlotinib is a novel oral tyrosine kinase inhibitor that targets platelet-derived growth factor receptor-α, c-Kit, fibroblast growth factor receptors 1, 2, and 3 (FGFR), and vascular endothelial growth factor receptors 1, 2, and 3 (VEGF) and suppresses the proliferation of cancer cells [69]. Anlotinib regulates some essential cancer cell metabolism like amino acids metabolism of tryptophan, threonine, glycine, serine, and phenylalanine and amino acid biosynthesis of valine, leucine, isoleucine pathways, and glyoxylate and decarboxylate pathways [70]. Additionally, certain metabolites serve as potential biomarkers to assess the efficacy of anlotinib, such as glycine-associated glycocholic and glycodeoxycholic acid [71]. Some chemotherapeutic drugs show alteration in metabolism, as it leads to a reduction in valine and lactate, ultimately showing relieved glycolysis. Drugs show their effect by reversing the Warburg effect and also affecting the high serum level of lipids, choline, and isobutyrate, etc. A549 cells (adenocarcinomic) treated with cisplatin have increased lipid levels but decreased niacinamide levels [72]. Chemotherapy causes the integrity of the cell membrane to be disrupted, which leads to cell membrane rupture and degradation. Lipids and glycoproteins are consequently more concentrated in the blood. Additionally, it has been proposed that chemotherapy raises the levels of the 3-hydroxybutyrate metabolite in sera, which, in turn, enhances lipolysis [73]. Chemotherapy is necessary for many biological functions. It alters energy metabolism, disrupts the production of phosphatidylcholine, disrupts the metabolites in lung cancer serum, and has an impact on DNA replication and microtubule function. Therefore, any chemotherapy treatment may disrupt metabolic pathways and result in the production of metabolites [74]. Patients with lung cancer are expected to have altered metabolite profiles following surgery. Metabolomic studies could offer a deeper understanding of these operation-related modifications, as a study showed higher levels of sphingolipids such as ceramide and sphingomyelin in lung cancer patients compared to controls in both pre- and post-surgery patients [75].
4.3. Considerations for Overcoming Drug Resistance
Drug resistance in lung cancer is a significant challenge in the management of the disease. It refers to the ability of cancer cells to survive and continue growing despite the presence of drugs designed to eliminate or control them. There are various mechanisms through which lung cancer cells can develop resistance to different types of treatment, including chemotherapy, targeted therapy, and immunotherapy. Lung cancer exhibits drug resistance through various key mechanisms, including multidrug resistance (MDR), DNA repair mechanisms, activation of alternative pathways, changes in tumor microenvironment, intertumoral heterogeneity, etc. [76]. To address these challenges, targeted and multifunctional nanoscale drug constructions have been developed as a result of nanotechnology advancements. These constructs offer promising methods to overcome important biological barriers, such as encapsulating different active treatments, surface functionalization of nanomedicine components, and potential regulation of these components. These characteristics improve the transport of various therapeutic drugs directly to the tumor microenvironment (TME), which reverses the anticancer treatment-resistant state of LC [77]. Some of the newer nanoparticle-based approaches include applying magnetic nanoparticles (NPs), namely the commercially available ferrofluids, as intravenous medication delivery systems. After injection, the tumor site is exposed to an external magnetic field, which causes the NPs to accumulate there and decreases the drug’s systemic toxicity. Moreover, NPs have the potential to serve as carriers of several anticancer agents, such as radionuclides, cancer-specific antibodies, and genes. On the other hand, within the past 10 years, photodynamic therapy (PDT) has become a viable therapeutic strategy for the treatment of cancer. As most photosensitizers exhibit hydrophobic properties, suitable delivery mechanisms are necessary [78]. Delivering siRNAs via NPs is a further novel approach to stop DR. Different siRNAs could be delivered in tandem to silence many genes, including the DR-related genes. The physicochemical characteristics of siRNAs hinder their cellular uptake due to their difficulty in navigating phospholipid membranes. Thus, the creation of novel RNAi technologies and suitable carriers is necessary. The technology of genome editing could offer a foundation for the creation of fresher, more effective strategies; another recent approach is the use of biocompatible compounds, and the use of biopolymers, such as tamarind seed polysaccharide PST, to prepare Paclitaxel (PTX)-loaded NPs through epichlorohydrin crosslinking [79]. Using nanoparticles to deliver therapy to cure drug-resistant lung cancer is a growing area of research to deal with the drug resistance issue.
5. Emerging Therapeutic Strategies Targeting Metabolism
In addition to the present therapeutic approaches, there are many new emerging treatment options under development, which mainly target the metabolic changes that occur due to cancer progression. Targeting metabolism has been an area of interest in the development of therapeutic strategies for lung cancer. Some major metabolic processes that can be targeted are glucose metabolism inhibition, mitochondria, fatty acid, amino acid metabolism, targeting oncogenic pathways, and immunometabolism [80]. Exciting developments in molecular targeted therapy and immunotherapy in combination with new surgical, pathological, radiographical, and radiation techniques have resulted in spectacular improvement in patients’ survival. KRAS is nearly exclusively seen in adenocarcinomas, while TP53 is the most frequently mutated gene in non-small-cell lung cancers (NSCLCs). RAF-MEK-MAPK and PI3K-Akt-mTOR are the two signaling pathways that are most frequently altered in lung cancer [81]. In NSCLC tumors, TP53 is mutated in approximately 46% of cases. This mutation leads to reduced apoptosis, primarily due to the suppression of TP53-induced glycolysis and apoptosis regulator (TIGAR). Further, there is enhanced glycolysis and glucose transport attributed to the upregulation of phosphoglucomutase (PGM). Additionally, TP53 is no longer capable of inhibiting the activity of glucose 6 phosphate dehydrogenase (G6PD) [82]. In addition to controlling glucose levels, the PI3K-Akt-mTOR signal transduction pathway triggers cancerous mutations that have a variety of negative side effects, including angiogenesis, proliferation, and differentiation. As a result, it establishes a clear connection between metabolic targeting, treatment response, and carcinogenesis [80]. Most of the cancer signaling pathways mutated in lung cancer, like RAS-RAF-MEK-MAPK pathway, are mutated in 58% of all NSCLC tumor’s and 76% of lung adenocarcinomas [83], and KRAS and EGFR pathways, KEAP1/NRF2 pathway, etc., have a great impact on major metabolic pathways like glucose metabolism, amino acid metabolism pathways, and reactive oxygen species (ROS) regulation [84]. Therefore, proper targeting of these metabolic pathways gives us a better approach to develop new therapeutic options. There are various drugs under development that target glucose metabolism. Glucose transporter (GLUT) inhibitors like fasentin, WZB117 (2-fluoro-6-(m-hydroxybenzoyloxy) phenyl m-hydroxybenzoate), DRB18 (a pan-GLUT inhibitor), STF31 (selective glucose transporter GLUT1 inhibitor), etc., are under pre-clinical trials. These drugs are mainly targeting the transport of glucose. WZB117 and DRB18 demonstrated inhibition under in vitro and in vivo conditions in NSCLC cancer models [85]. Shikonin, a drug used for late-stage lung cancer treatment, is a pyruvate kinase M2 (PKM2) inhibitor that is given to patients not undergoing surgery, radiotherapy, or chemotherapy. Activation of pyruvate kinase M2 (PKM2) appears to be inactivated in cancer and raises the ratio of glycolysis to glucose oxidation [86]. Some other drugs for lung cancer therapy under trial include dichloroacetate (DCA), which is a pyruvate dehydrogenase kinase 1 (PDK1) inhibitor. Also, PSTMB targets lactate dehydrogenase A (LDHA) [87].
Major targets for mutation in lung cancer include EGFR mutations, ALK rearrangements, ROS1 rearrangements, RET rearrangements, neurotrophic tyrosine receptor kinase (NTRK) fusions, mesenchymal–epithelial transition factor (MET) exon 14 skipping mutation, KRAS G12C mutation, BRAF V600E mutation, and ERBB2 (HER2) mutation. FDA-approved drugs are available for targeting these mutations; still, further translational research is exploring novel driver mutations, converting traditionally undruggable mutations into therapeutic targets [88]. Several drugs are currently under phase 1/2 clinical trials with different mechanisms of action compared to existing drugs. For example, Patritumabderuxtecan is a monotherapy drug under phase 1 clinical trial; it is an anti-HER3 antibody–drug conjugate targeting mutated EGFR and NSCLC that has progressed after tyrosine kinase inhibitors (TKI) showing an objective response rate (ORR) of 39%. Other monotherapy drugs under trial include Pemigatinib (aFGFR1-3 inhibitor), Telisotuzumabvedotin (anti-MET antibody–drug conjugate), Taletrectinib (ROS1 inhibitor), and Trastuzumabderuxtecan (anti-HER2 antibody–drug conjugate) [89,90].
Combinational therapy is a recent approach that targets multiple pathways associated with cancer development, such as PI3K/AKT/mTOR and RAF/MEK/ERK, by focusing on tumor cell receptors. There are certain drugs under different phases of clinical trials, like Anlotinib (multi targeting TKI), and used in combination with Osimertinibis as a first line of therapy for mutated EGFR and NSCLC [91]. Other drugs under clinical trials are Alisertib (Aurora kinase inhibitor), Sapanisertib (mTOR inhibitor), and Repotrectinib (ROS1/TRK/ALK inhibitor); all of these are used in combination with Osimertinib [92,93]. Some of the current therapeutic approaches under development or in clinical trials are listed in Table 1 below.

Table 1.
Current therapeutic approaches for lung cancer treatment.
7. Conclusions
The fight against lung cancer, a leading global health challenge, necessitates a multifaceted approach due to the complex interplay of drug resistance, metabolic reprogramming, and the genetic landscape of the disease. This review has highlighted the critical role of targeting pathways, mutations, and genetic alterations, alongside the importance of understanding cancer metabolism for effective therapeutic strategies. The advancements in targeted therapies, immunotherapies, and combinational treatments, complemented by emerging options like metabolic inhibitors and nanocarriers, underscore the potential to significantly improve patient outcomes. Moreover, the integration of mathematical modeling and systems biology approaches offers promising avenues for enhanced detection and personalized treatment strategies. The adoption of AI-ML technologies and innovative techniques such as hyperthermia, cryoablation, and photodynamic therapy further expands our arsenal against lung cancer. Through a comprehensive understanding of the disease’s underlying mechanisms and leveraging interdisciplinary approaches and cutting-edge technologies, we can pave the way for more effective treatments and ultimately improve the survival rates and quality of life for lung cancer patients.
Author Contributions
Conceptualization, H.K.V. and L.V.K.S.B.; methodology, P.M., R.A. and B.P.; validation; H.K.V. and L.V.K.S.B.; resources and data curation, P.M., R.A. and B.P.; writing—original draft preparation and review and editing; P.M., B.P. and H.K.V.; supervision, H.K.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Islami, F.; Goding Sauer, A.; Miller, K.D.; Siegel, R.L.; Fedewa, S.A.; Jacobs, E.J.; McCullough, M.L.; Patel, A.V.; Ma, J.; Soerjomataram, I.; et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J. Clin. 2018, 68, 31–54. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Agrawal, S.; Jiwnani, S.; Khosla, D.; Malik, P.S.; Mohan, A.; Penumadu, P.; Prasad, K.T. Lung Cancer in India. J. Thorac. Oncol. 2021, 16, 1250–1266. [Google Scholar] [CrossRef]
- Thun, M.J.; Carter, B.D.; Feskanich, D.; Freedman, N.D.; Prentice, R.; Lopez, A.D.; Hartge, P.; Gapstur, S.M. 50-year trends in smoking-related mortality in the United States. N. Engl. J. Med. 2013, 368, 351–364. [Google Scholar] [CrossRef]
- Lemjabbar-Alaoui, H.; Hassan, O.U.; Yang, Y.-W.; Buchanan, P. Lung cancer: Biology and treatment options. Biochim. Biophys. Acta 2015, 1856, 189–210. [Google Scholar] [CrossRef]
- Anusewicz, D.; Orzechowska, M.; Bednarek, A.K. Lung squamous cell carcinoma and lung adenocarcinoma differential gene expression regulation through pathways of Notch, Hedgehog, Wnt, and ErbB signalling. Sci. Rep. 2020, 10, 21128. [Google Scholar] [CrossRef]
- Rudin, C.M.; Brambilla, E.; Faivre-Finn, C.; Sage, J. Small-cell lung cancer. Nat. Rev. Dis. Prim. 2021, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, A.; Akter, Z.; Modareszadeh, P.; Modareszadeh, P.; Berisha, E.; Alemi, P.S.; Castro, M.d.C.C.; Deese, A.R.; Zhang, L. Current Landscape of Therapeutic Resistance in Lung Cancer and Promising Strategies to Overcome Resistance. Cancers 2022, 14, 4562. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Li, X.; Liu, M.; Liu, H.; Chen, J. Tumor metabolic reprogramming in lung cancer progression. Oncol. Lett. 2022, 24, 287. [Google Scholar] [CrossRef]
- Kalyanaraman, B. Teaching the basics of cancer metabolism: Developing antitumor strategies by exploiting the differences between normal and cancer cell metabolism. Redox Biol. 2017, 12, 833–842. [Google Scholar] [CrossRef]
- Verma, H.K.; Kampalli, P.K.; Lakkakula, S.; Chalikonda, G.; Bhaskar, L.V.; Pattnaik, S. A Retrospective Look at Anti-EGFR Agents in Pancreatic Cancer Therapy. Curr. Drug Metab. 2019, 20, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Verma, H.K.; Falco, G.; Bhaskar, L.V.K.S. Molecular Signaling Pathways Involved in Gastric Cancer Chemoresistance. In Theranostics Approaches to Gastric and Colon Cancer; Raju, G.S.R., Bhaskar, L.V.K.S., Eds.; Springer: Singapore, 2020; pp. 117–134. [Google Scholar]
- Tan, W.-L.; Jain, A.; Takano, A.; Newell, E.W.; Iyer, N.G.; Lim, W.-T.; Tan, E.-H.; Zhai, W.; Hillmer, A.M.; Tam, W.L.; et al. Novel therapeutic targets on the horizon for lung cancer. Lancet Oncol. 2016, 17, e347–e362. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Shao, X.; Li, Y.; Gihu, R.; Xie, H.; Zhou, J.; Yan, H. Targeting Signaling Pathway Networks in Several Malignant Tumors: Progresses and Challenges. Front. Pharmacol. 2021, 12, 675675. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Shcherba, M.; Pendurti, G.; Liang, Y.; Piperdi, B.; Perez-Soler, R.; Shrivastav, A.; Murphy, L.; Zhou, Q.; Lui, V.W.; et al. Targeting the PI3K/AKT/mTOR pathway: Potential for lung cancer treatment. Lung Cancer Manag. 2014, 3, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Sanaei, M.-J.; Razi, S.; Pourbagheri-Sigaroodi, A.; Bashash, D. The PI3K/Akt/mTOR pathway in lung cancer; oncogenic alterations, therapeutic opportunities, challenges, and a glance at the application of nanoparticles. Transl. Oncol. 2022, 18, 101364. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, R.; Singhvi, G.; Dubey, S.K.; Gupta, G.; Dua, K. MAPK pathway: A potential target for the treatment of non-small-cell lung carcinoma. Future Med. Chem. 2019, 11, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Gong, K.; Guo, G.; Beckley, N.; Zhang, Y.; Yang, X.; Sharma, M.; Habib, A.A. Tumor necrosis factor in lung cancer: Complex roles in biology and resistance to treatment. Neoplasia 2021, 23, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Venugopala, K.N. Targeting the DNA Damage Response Machinery for Lung Cancer Treatment. Pharmaceuticals 2022, 15, 1475. [Google Scholar] [CrossRef]
- Mamdani, H.; Jalal, S.I. DNA repair in lung cancer: Potential not yet reached. Lung Cancer Manag. 2016, 5, 5–8. [Google Scholar] [CrossRef]
- Ou, S.I.; Shirai, K. Anaplastic Lymphoma Kinase (ALK) Signaling in Lung Cancer. Adv. Exp. Med. Biol. 2016, 893, 179–187. [Google Scholar]
- Longley, D.; Johnston, P. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef]
- Kannampuzha, S.; Mukherjee, A.G.; Wanjari, U.R.; Gopalakrishnan, A.V.; Murali, R.; Namachivayam, A.; Renu, K.; Dey, A.; Vellingiri, B.; Madhyastha, H.; et al. A Systematic Role of Metabolomics, Metabolic Pathways, and Chemical Metabolism in Lung Cancer. Vaccines 2023, 11, 381. [Google Scholar] [CrossRef]
- Ratre, Y.K.; Verma, H.K.; Mehta, A.; Soni, V.K.; Sonkar, S.C.; Shukla, D.; Ekka, A.; Prajapati, S.K.; Mahilkar, S.; Vishvakarma, N.K. Therapeutic Targeting of Glutamine Metabolism in Colorectal Cancer. In Colon Cancer Diagnosis and Therapy: Volume 2; Vishvakarma, N.K., Nagaraju, G.P., Shukla, D., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 333–356. [Google Scholar]
- Xu, H.-D.; Luo, W.; Lin, Y.; Zhang, J.; Zhang, L.; Zhang, W.; Huang, S.-M. Discovery of potential therapeutic targets for non-small cell lung cancer using high-throughput metabolomics analysis based on liquid chromatography coupled with tandem mass spectrometry. RSC Adv. 2019, 9, 10905–10913. [Google Scholar] [CrossRef]
- Pezzuto, A.; D’Ascanio, M.; Ricci, A.; Pagliuca, A.; Carico, E. Expression and role of p16 and GLUT1 in malignant diseases and lung cancer: A review. Thorac. Cancer 2020, 11, 3060–3070. [Google Scholar] [CrossRef]
- Zahra, K.; Dey, T.; Ashish; Mishra, S.P.; Pandey, U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef]
- Mohamed, A.; Deng, X.; Khuri, F.R.; Owonikoko, T.K. Altered glutamine metabolism and therapeutic opportunities for lung cancer. Clin. Lung Cancer 2013, 15, 7–15. [Google Scholar] [CrossRef]
- Hassanein, M.; Hoeksema, M.D.; Shiota, M.; Qian, J.; Harris, B.K.; Chen, H.; Clark, J.E.; Alborn, W.E.; Eisenberg, R.; Massion, P.P. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin. Cancer Res. 2013, 19, 560–570. [Google Scholar] [CrossRef]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and Cancer Metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef]
- Morotti, M.; Bridges, E.; Valli, A.; Choudhry, H.; Sheldon, H.; Wigfield, S.; Gray, N.; Zois, C.E.; Grimm, F.; Jones, D.; et al. Hypoxia-induced switch in SNAT2/SLC38A2 regulation generates endocrine resistance in breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 12452–12461. [Google Scholar] [CrossRef]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.-K.; Heo, Y.; Lee, H.S.; Lee, M.Y.; et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab. 2020, 31, 267–283.e12. [Google Scholar] [CrossRef]
- Sowers, M.L.; Herring, J.; Zhang, W.; Tang, H.; Ou, Y.; Gu, W.; Zhang, K. Analysis of glucose-derived amino acids involved in one-carbon and cancer metabolism by stable-isotope tracing gas chromatography mass spectrometry. Anal. Biochem. 2019, 566, 1–9. [Google Scholar] [CrossRef]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef]
- Yang, S.; Kobayashi, S.; Sekino, K.; Kagawa, Y.; Miyazaki, H.; Shil, S.K.; Umaru, B.A.; Wannakul, T.; Owada, Y. Fatty acid-binding protein 5 controls lung tumor metastasis by regulating the maturation of natural killer cells in the lung. FEBS Lett. 2021, 595, 1797–1805. [Google Scholar] [CrossRef]
- Schwarcz, R.; Stone, T.W. The kynurenine pathway and the brain: Challenges, controversies and promises. Neuropharmacology 2017, 112 Pt B, 237–247. [Google Scholar] [CrossRef]
- Chang, L.; Fang, S.; Chen, Y.; Yang, Z.; Yuan, Y.; Zhang, J.; Ye, L.; Gu, W. Inhibition of FASN suppresses the malignant biological behavior of non-small cell lung cancer cells via deregulating glucose metabolism and AKT/ERK pathway. Lipids Health Dis. 2019, 18, 118. [Google Scholar] [CrossRef]
- Li, E.Q.; Zhao, W.; Zhang, C.; Qin, L.Z.; Liu, S.J.; Feng, Z.Q.; Wen, X.; Chen, C.P. Synthesis and anti-cancer activity of ND-646 and its derivatives as acetyl-CoA carboxylase 1 inhibitors. Eur. J. Pharm. Sci. 2019, 137, 105010. [Google Scholar] [CrossRef]
- Lai, S.-C.; Phelps, C.A.; Short, A.M.; Dutta, S.M.; Mu, D. Thyroid transcription factor 1 enhances cellular statin sensitivity via perturbing cholesterol metabolism. Oncogene 2018, 37, 3290–3300. [Google Scholar] [CrossRef]
- Olaussen, K.; Postel-Vinay, S. Predictors of chemotherapy efficacy in non-small-cell lung cancer: A challenging landscape. Ann. Oncol. 2016, 27, 2004–2016. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Baxevanos, P.; Mountzios, G. Novel chemotherapy regimens for advanced lung cancer: Have we reached a plateau? Ann. Transl. Med. 2018, 6, 139. [Google Scholar] [CrossRef]
- Visentin, M.; Zhao, R.; Goldman, I.D. The antifolates. Hematol. Oncol. Clin. N. Am. 2012, 26, 629–648.ix. [Google Scholar] [CrossRef]
- Hiddinga, B.I.; Raskin, J.; Janssens, A.; Pauwels, P.; Van Meerbeeck, J.P. Recent developments in the treatment of small cell lung cancer. Eur. Respir. Rev. 2021, 30, 210079. [Google Scholar] [CrossRef]
- Horita, N.; Yamamoto, M.; Sato, T.; Tsukahara, T.; Nagakura, H.; Tashiro, K.; Shibata, Y.; Watanabe, H.; Nagai, K.; Inoue, M.; et al. Topotecan for Relapsed Small-cell Lung Cancer: Systematic Review and Meta-Analysis of 1347 Patients. Sci. Rep. 2015, 5, 15437. [Google Scholar] [CrossRef]
- Vinod, S.K.; Hau, E. Radiotherapy treatment for lung cancer: Current status and future directions. Respirology 2020, 25 (Suppl. S2), 61–71. [Google Scholar] [CrossRef]
- Miyasaka, Y.; Sato, H.; Okano, N.; Kubo, N.; Kawamura, H.; Ohno, T. A Promising Treatment Strategy for Lung Cancer: A Combination of Radiotherapy and Immunotherapy. Cancers 2021, 14, 203. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; De Wit, M.; et al. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. N. Engl. J. Med. 2018, 379, 2342–2350. [Google Scholar] [CrossRef]
- Corso, C.D.; Rutter, C.E.; Park, H.S.; Lester-Coll, N.H.; Kim, A.W.; Wilson, L.D.; Husain, Z.A.; Lilenbaum, R.C.; Yu, J.B.; Decker, R.H. Role of chemoradiotherapy in elderly patients with limited-stage small-cell lung cancer. J. Clin. Oncol. 2015, 33, 4240–4246. [Google Scholar] [CrossRef]
- Drosten, M.; Barbacid, M. Targeting KRAS mutant lung cancer: Light at the end of the tunnel. Mol. Oncol. 2022, 16, 1057–1071. [Google Scholar] [CrossRef]
- Díaz-Serrano, A.; Gella, P.; Jiménez, E.; Zugazagoitia, J.; Rodríguez, L.P.-A. Targeting EGFR in Lung Cancer: Current Standards and Developments. Drugs 2018, 78, 893–911. [Google Scholar] [CrossRef]
- Chevallier, M.; Borgeaud, M.; Addeo, A.; Friedlaender, A. Oncogenic driver mutations in non-small cell lung cancer: Past, present and future. World J. Clin. Oncol. 2021, 12, 217–237. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhang, T.; Xu, Q. Therapeutic advances in non-small cell lung cancer: Focus on clinical development of targeted therapy and immunotherapy. Medcomm 2021, 2, 692–729. [Google Scholar] [CrossRef]
- Nagano, T.; Tachihara, M.; Nishimura, Y. Dacomitinib, a second-generation irreversible epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI) to treat non-small cell lung cancer. Drugs Today 2019, 55, 231–236. [Google Scholar] [CrossRef]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or chemotherapy for non–small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef]
- Villalobos, P.; Wistuba, I.I. Lung Cancer Biomarkers. Hematol. Oncol. Clin. N. Am. 2017, 31, 13–29. [Google Scholar] [CrossRef]
- Zhao, H.; Yao, W.; Min, X.; Gu, K.; Yu, G.; Zhang, Z.; Cui, J.; Miao, L.; Zhang, L.; Yuan, X.; et al. Apatinib Plus Gefitinib as First-Line Treatment in Advanced EGFR-Mutant NSCLC: The Phase III ACTIVE Study (CTONG1706). J. Thorac. Oncol. 2021, 16, 1533–1546. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, J. Current status and future directions of cancer immunotherapy. J. Cancer 2018, 9, 1773–1781. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef]
- Nasser, N.J.; Gorenberg, M.; Agbarya, A. First line Immunotherapy for Non-Small Cell Lung Cancer. Pharmaceuticals 2020, 13, 373. [Google Scholar] [CrossRef]
- Santini, F.C.; Rudin, C.M. Atezolizumab for the treatment of non-small cell lung cancer. Expert Rev. Clin. Pharmacol. 2017, 10, 935–945. [Google Scholar] [CrossRef]
- Heymach, J.V.; Harpole, D.; Mitsudomi, T.; Taube, J.M.; Galffy, G.; Hochmair, M.; Winder, T.; Zukov, R.; Garbaos, G.; Gao, S.; et al. Perioperative Durvalumab for Resectable Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2023, 389, 1672–1684. [Google Scholar] [CrossRef]
- Johnson, M.L.; Cho, B.C.; Luft, A.; Alatorre-Alexander, J.; Geater, S.L.; Laktionov, K.; Kim, S.-W.; Ursol, G.; Hussein, M.; Lim, F.L.; et al. Durvalumab With or Without Tremelimumab in Combination With Chemotherapy as First-Line Therapy for Metastatic Non–Small-Cell Lung Cancer: The Phase III POSEIDON Study. J. Clin. Oncol. 2023, 41, 1213–1227. [Google Scholar] [CrossRef]
- Theelen, W.S.; Chen, D.; Verma, V.; Hobbs, B.P.; Peulen, H.M.; Aerts, J.G.; Bahce, I.; Niemeijer, A.L.N.; Chang, J.Y.; de Groot, P.M.; et al. Pembrolizumab with or without radiotherapy for metastatic non-small-cell lung cancer: A pooled analysis of two randomised trials. Lancet Respir. Med. 2021, 9, 467–475. [Google Scholar] [CrossRef]
- Ucuzian, A.A.; Gassman, A.A.; East, A.T.; Greisler, H.P. Molecular mediators of angiogenesis. J. Burn. Care Res. 2010, 31, 158–175. [Google Scholar] [CrossRef]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: Current status and future directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- West, H.J.; McCleland, M.; Cappuzzo, F.; Reck, M.; Mok, T.S.; Jotte, R.M.; Nishio, M.; Kim, E.; Morris, S.; Zou, W.; et al. Clinical efficacy of atezolizumab plus bevacizumab and chemotherapy in KRAS-mutated non-small cell lung cancer with STK11, KEAP1, or TP53 comutations: Subgroup results from the phase III IMpower150 trial. J. Immunother. Cancer 2022, 10, e003027. [Google Scholar] [CrossRef]
- Xie, C.; Wan, X.; Quan, H.; Zheng, M.; Fu, L.; Li, Y.; Lou, L. Preclinical characterization of anlotinib, a highly potent and selective vascular endothelial growth factor receptor-2 inhibitor. Cancer Sci. 2018, 109, 1207–1219. [Google Scholar]
- Pan, X.; Chen, W.; Nie, M.; Liu, Y.; Xiao, Z.; Zhang, Y.; Zhang, W.; Zou, X. A Serum Metabolomic Study Reveals Changes in Metabolites During the Treatment of Lung Cancer-Bearing Mice with Anlotinib. Cancer Manag. Res. 2021, 13, 6055–6063. [Google Scholar] [CrossRef]
- Hu, T.; An, Z.; Sun, Y.; Wang, X.; Du, P.; Li, P.; Chi, Y.; Liu, L. Longitudinal Pharmacometabonomics for Predicting Malignant Tumor Patient Responses to Anlotinib Therapy: Phenotype, Efficacy, and Toxicity. Front. Oncol. 2020, 10, 548300. [Google Scholar] [CrossRef]
- Duarte, I.F.; Ladeirinha, A.F.; Lamego, I.; Gil, A.M.; Carvalho, L.; Carreira, I.M.; Melo, J.B. Potential Markers of Cisplatin Treatment Response Unveiled by NMR Metabolomics of Human Lung Cells. Mol. Pharm. 2013, 10, 4242–4251. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Koussounadis, A.; Langdon, S.P.; Harrison, D.J.; Smith, V.A. Chemotherapy-induced dynamic gene expression changes in vivo are prognostic in ovarian cancer. Br. J. Cancer 2014, 110, 2975–2984. [Google Scholar] [CrossRef]
- Yang, D.; Yang, X.; Li, Y.; Zhao, P.; Fu, R.; Ren, T.; Hu, P.; Wu, Y.; Yang, H.; Guo, N. Clinical significance of circulating tumor cells and metabolic signatures in lung cancer after surgical removal. J. Transl. Med. 2020, 18, 243. [Google Scholar] [CrossRef]
- Haider, M.; Elsherbeny, A.; Pittalà, V.; Consoli, V.; Alghamdi, M.A.; Hussain, Z.; Khoder, G.; Greish, K. Nanomedicine Strategies for Management of Drug Resistance in Lung Cancer. Int. J. Mol. Sci. 2022, 23, 1853. [Google Scholar] [CrossRef]
- Alexiou, C.; Schmid, R.J.; Jurgons, R.; Kremer, M.; Wanner, G.; Bergemann, C.; Huenges, E.; Nawroth, T.; Arnold, W.; Parak, F.G. Targeting cancer cells: Magnetic nanoparticles as drug carriers. Eur. Biophys. J. 2006, 35, 446–450. [Google Scholar] [CrossRef]
- Deng, X.; Song, Q.; Zhang, Y.; Liu, W.; Hu, H.; Zhang, Y. Tumour microenvironment-responsive nanoplatform based on biodegradable liposome-coated hollow MnO2 for synergistically enhanced chemotherapy and photodynamic therapy. J. Drug Target. 2022, 30, 334–347. [Google Scholar] [CrossRef]
- Reshma, P.; Unnikrishnan, B.; Preethi, G.; Syama, H.; Archana, M.; Remya, K.; Shiji, R.; Sreekutty, J.; Sreelekha, T. Overcoming drug-resistance in lung cancer cells by paclitaxel loaded galactoxyloglucan nanoparticles. Int. J. Biol. Macromol. 2019, 136, 266–274. [Google Scholar] [CrossRef]
- Vanhove, K.; Graulus, G.-J.; Mesotten, L.; Thomeer, M.; Derveaux, E.; Noben, J.-P.; Guedens, W.; Adriaensens, P. The Metabolic Landscape of Lung Cancer: New Insights in a Disturbed Glucose Metabolism. Front. Oncol. 2019, 9, 1215. [Google Scholar] [CrossRef]
- Dhawan, A.; Pifer, P.M.; Sandulache, V.C.; Skinner, H.D. Metabolic targeting, immunotherapy and radiation in locally advanced non-small cell lung cancer: Where do we go from here? Front. Oncol. 2022, 12, 1016217. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef]
- Kim, D.; Hwang, H.-Y.; Ji, E.S.; Kim, J.Y.; Yoo, J.S.; Kwon, H.J. Activation of mitochondrial TUFM ameliorates metabolic dysregulation through coordinating autophagy induction. Commun. Biol. 2021, 4, 1. [Google Scholar] [CrossRef]
- Itoh, K.; Ye, P.; Matsumiya, T.; Tanji, K.; Ozaki, T. Emerging functional cross-talk between the Keap1-Nrf2 system and mitochondria. J. Clin. Biochem. Nutr. 2015, 56, 91–97. [Google Scholar] [CrossRef]
- Shriwas, P.; Roberts, D.; Li, Y.; Wang, L.; Qian, Y.; Bergmeier, S.; Hines, J.; Adhicary, S.; Nielsen, C.; Chen, X. A small-molecule pan-class I glucose transporter inhibitor reduces cancer cell proliferation in vitro and tumor growth in vivo by targeting glucose-based metabolism. Cancer Metab. 2021, 9, 14. [Google Scholar] [CrossRef]
- Chen, J.; Xie, J.; Jiang, Z.; Wang, B.; Wang, Y.; Hu, X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 2011, 30, 4297–4306. [Google Scholar] [CrossRef]
- Kim, E.-Y.; Chung, T.-W.; Han, C.W.; Park, S.Y.; Park, K.H.; Jang, S.B.; Ha, K.-T. A Novel Lactate Dehydrogenase Inhibitor, 1-(Phenylseleno)-4-(Trifluoromethyl) Benzene, Suppresses Tumor Growth through Apoptotic Cell Death. Sci. Rep. 2019, 9, 3969. [Google Scholar] [CrossRef]
- Abughanimeh, O.; Kaur, A.; El Osta, B.; Ganti, A.K. Novel targeted therapies for advanced non-small lung cancer. Semin. Oncol. 2022, 49, 326–336. [Google Scholar] [CrossRef]
- Janne, P.A.; Baik, C.S.; Su, W.-C.; Johnson, M.L.; Hayashi, H.; Nishio, M.; Kim, D.-W.; Koczywas, M.; Gold, K.A.; Steuer, C.E.; et al. Efficacy and Safety of Patritumab Deruxtecan (HER3-DXd) in EGFR Inhibitor-Resistant, EGFR-Mutated Non-Small Cell Lung Cancer. Cancer Discov. 2022, 12, 74–89. [Google Scholar] [CrossRef]
- Li, S.; Correia, G.S.d.C.; Wang, J.; Manochakian, R.; Zhao, Y.; Lou, Y. Emerging Targeted Therapies in Advanced Non-Small-Cell Lung Cancer. Cancers 2023, 15, 2899. [Google Scholar] [CrossRef]
- Han, B.; Yan, B.; Gu, A.; Chu, T.; Zhang, W.; Wang, H.; Zhong, H.; Shi, C.; Zhang, X. Phase Ib/IIa study evaluating the safety and clinical activity of osimeritinib com-bined with anlotinib in EGFRm, treatment-naive advanced NSCLC patients (AUTOMAN). J. Clin. Oncol. 2022, 40, e21140. [Google Scholar] [CrossRef]
- Elamin, Y.Y.; Negrao, M.V.; Fossella, F.V.; Byers, L.A.; Zhang, J.; Gay, C.M.; Tu, J.C.; Pozadzides, J.V.; Tran, H.T.; Lu, C.; et al. Results of a phase 1b study of osimertinib plus sapanisertib or alisertib for osimertinib-resistant, EGFR-mutant non–small cell lung cancer (NSCLC). J. Clin. Oncol. 2022, 40, 9105. [Google Scholar] [CrossRef]
- Cho, B.C.; Doebele, R.C.; Lin, J.; Nagasaka, M.; Baik, C.; Van Der Wekken, A.; Velcheti, V.; Lee, K.H.; Liu, S.; Solomon, B.; et al. MA11. 07 phase 1/2 TRIDENT-1 study of repotrectinib in patients with ROS1+ or NTRK+ advanced solid tumors. J. Thorac. Oncol. 2021, 16, S174–S175. [Google Scholar] [CrossRef]
- Viktorsson, K.; Lewensohn, R.; Zhivotovsky, B. Systems biology approaches to develop innovative strategies for lung cancer therapy. Cell Death Dis. 2014, 5, e1260. [Google Scholar] [CrossRef] [PubMed]
- Ocak, S.; Sos, M.L.; Thomas, R.K.; Massion, P.P. High-throughput molecular analysis in lung cancer: Insights into biology and potential clinical applications. Eur. Respir. J. 2009, 34, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Yang, Y.; Li, M.; Song, J.; Gao, Y.; Cheng, S.; Xiao, T. Systems biology analysis of the lung cancer-related secretome. Oncol. Rep. 2018, 40, 1103–1118. [Google Scholar] [CrossRef] [PubMed]
- Parvin, S.; Sedighian, H.; Sohrabi, E.; Mahboobi, M.; Rezaei, M.; Ghasemi, D.; Rezaei, E. Prediction of Genes Involved in Lung Cancer with a Systems Biology Approach Based on Comprehensive Gene Information. Biochem. Genet. 2022, 60, 1253–1273. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.; Khan, A.A.; Ansari, H.R.; Haque, A. A Systems Biology and LASSO-Based Approach to Decipher the Transcriptome–Interactome Signature for Predicting Non-Small Cell Lung Cancer. Biology 2022, 11, 1752. [Google Scholar] [CrossRef] [PubMed]
- La’ah, A.S.; Chiou, S.-H. Cutting-Edge Therapies for Lung Cancer. Cells 2024, 13, 436. [Google Scholar] [CrossRef]
- Lin, P.-C.; Tsai, Y.-S.; Yeh, Y.-M.; Shen, M.-R. Cutting-Edge AI Technologies Meet Precision Medicine to Improve Cancer Care. Biomolecules 2022, 12, 1133. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Agrawal, A. Cutting-Edge Nanotechnological Approaches for Lung Cancer Therapy. Curr. Drug Res. Rev. 2022, 14, 171–187. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Z.; Miao, L.; Li, Y. Boron Neutron Capture Therapy: Current Status and Challenges. Front. Oncol. 2022, 12, 788770. [Google Scholar] [CrossRef]
- Han, Y. Current status of proton therapy techniques for lung cancer. Radiat. Oncol. J. 2019, 37, 232–248. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Xu, K.; Mu, F. Cryosurgery for lung cancer. J. Thorac. Dis. 2012, 4, 408–419. [Google Scholar] [PubMed]
- DeNicola, G.M.; Shackelford, D.B. Metabolic Phenotypes, Dependencies, and Adaptation in Lung Cancer. Cold Spring Harb. Perspect. Med. 2021, 11, a037838. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.-L.; Shen, M.-O.; Han, N.; Xu, H.-Z.; Peng, X.-C.; Li, Q.-R.; Yu, T.-T.; Li, L.-G.; Xu, X.; Liu, B.; et al. Chlorin e6 mediated photodynamic therapy triggers resistance through ATM-related DNA damage response in lung cancer cells. Photodiagnosis Photodyn. Ther. 2022, 37, 102645. [Google Scholar] [CrossRef]
- Dunne, M.; Regenold, M.; Allen, C. Hyperthermia can alter tumor physiology and improve chemo- and radio-therapy efficacy. Adv. Drug Deliv. Rev. 2020, 163–164, 98–124. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).