
Upregulation of TET2 and Resistance to DNA Methyltransferase (DNMT) Inhibitors in DNMT1-Deleted Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Drugs
2.2. MTT Assay
2.3. Annexin-V Apoptosis Detection
2.4. Western Blot
2.5. Real-Time Methylation-Specific PCR
3. Results
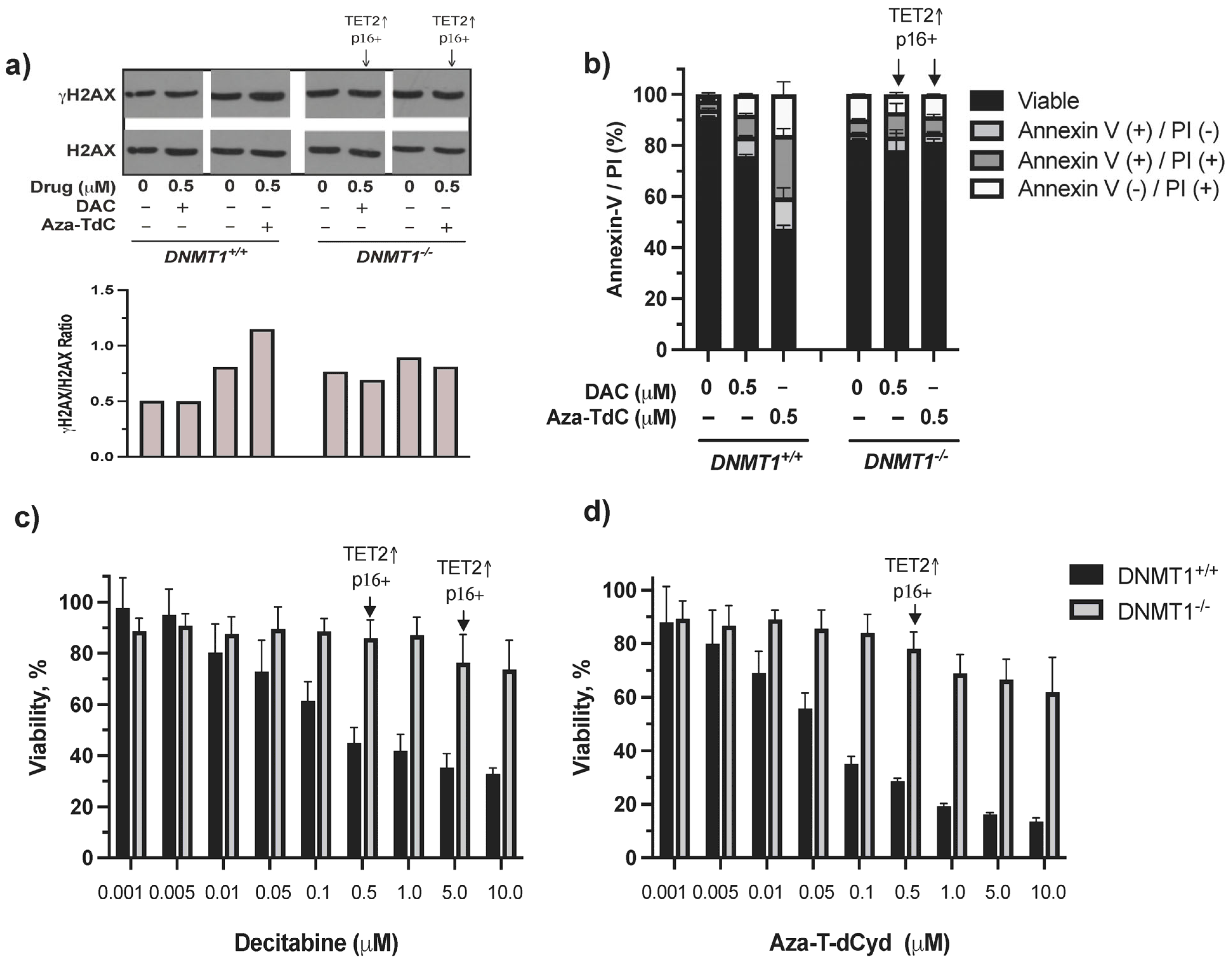
3.1. TET2 Upregulation and Tumor Suppressor Re-Expression by DNMT Inhibitors in DNMT1−/− Cells
3.2. CDKN2A Promoter Demethylation by DNMTi in HCT116 DNMT1+/+ and DNMT1–/– Cells
3.3. DNA Damage, Apoptosis, and Cytotoxic Effects of DNMTi on HCT116 DNMT1+/+ and DNMT1–/– Cells
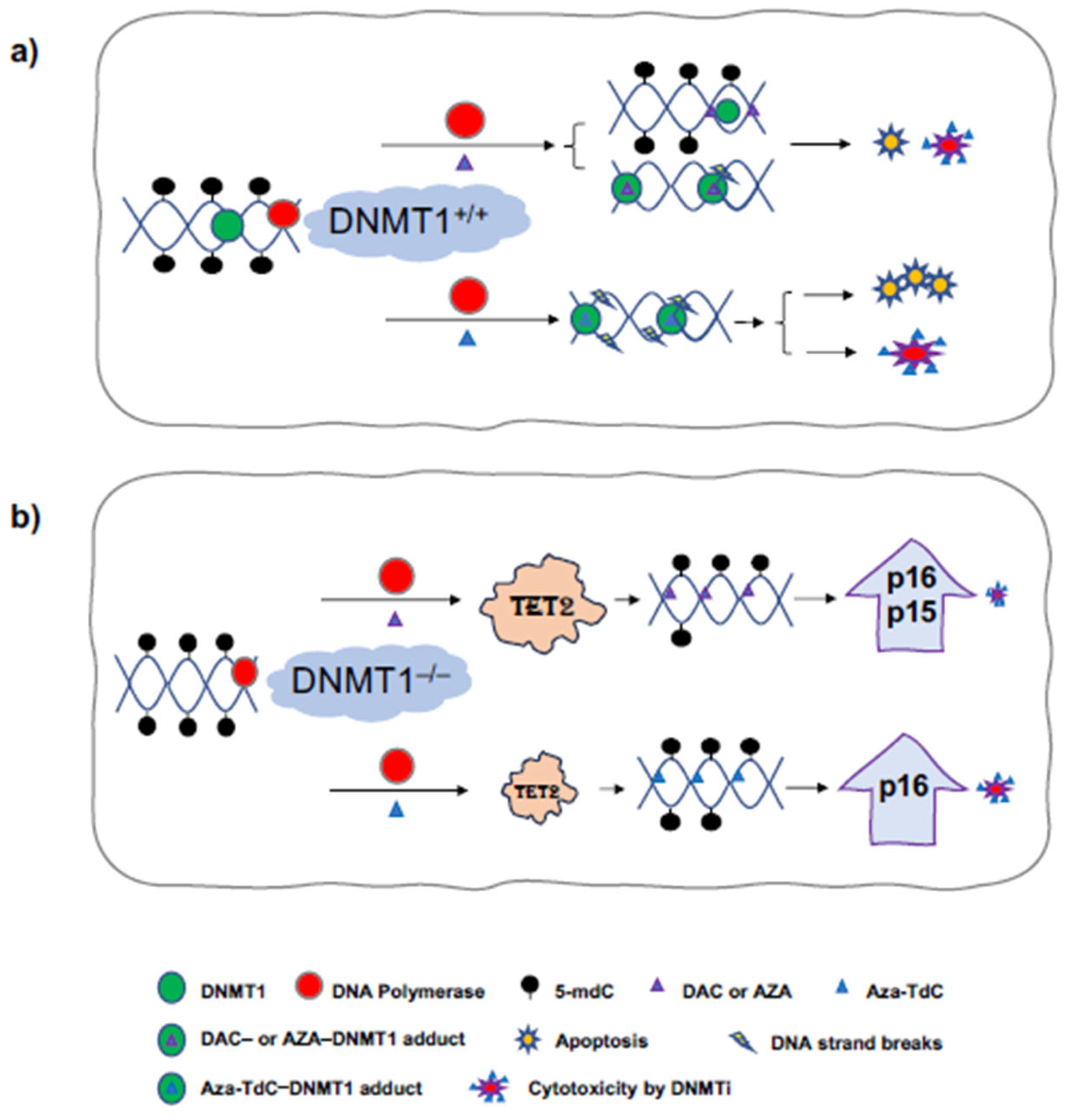
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Mabe, N.W.; Perry, J.A.; Malone, C.F.; Stegmaier, K. Pharmacological targeting of the cancer epigenome. Nat. Cancer 2024, 5, 844–865. [Google Scholar] [CrossRef] [PubMed]
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Merlo, A.; Mao, L.; Lapidus, R.G.; Issa, J.P.; Davidson, N.E.; Sidransky, D.; Baylin, S.B. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995, 55, 4525–4530. [Google Scholar] [PubMed]
- Merlo, A.; Herman, J.G.; Mao, L.; Lee, D.J.; Gabrielson, E.; Burger, P.C.; Baylin, S.B.; Sidransky, D. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat. Med. 1995, 1, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Robert, M.F.; Morin, S.; Beaulieu, N.; Gauthier, F.; Chute, I.C.; Barsalou, A.; MacLeod, A.R. DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat. Genet. 2003, 33, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P.; Kantarjian, H.M. Targeting DNA methylation. Clin. Cancer Res. 2009, 15, 3938–3946. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Taylor, S.M. Cellular differentiation, cytidine analogs and DNA methylation. Cell 1980, 20, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Thottassery, J.V.; Sambandam, V.; Allan, P.W.; Maddry, J.A.; Maxuitenko, Y.Y.; Tiwari, K.; Hollingshead, M.; Parker, W.B. Novel DNA methyltransferase-1 (DNMT1) depleting anticancer nucleosides, 4′-thio-2′-deoxycytidine and 5-aza-4′-thio-2′-deoxycytidine. Cancer Chemother. Pharmacol. 2014, 74, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Laranjeira, A.B.A.; Hollingshead, M.G.; Nguyen, D.; Kinders, R.J.; Doroshow, J.H.; Yang, S.X. DNA damage, demethylation and anticancer activity of DNA methyltransferase (DNMT) inhibitors. Sci. Rep. 2023, 13, 5964. [Google Scholar] [CrossRef] [PubMed]
- Schermelleh, L.; Spada, F.; Easwaran, H.P.; Zolghadr, K.; Margot, J.B.; Cardoso, M.C.; Leonhardt, H. Trapped in action: Direct visualization of DNA methyltransferase activity in living cells. Nat. Methods 2005, 2, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Maslov, A.Y.; Lee, M.; Gundry, M.; Gravina, S.; Strogonova, N.; Tazearslan, C.; Bendebury, A.; Suh, Y.; Vijg, J. 5-aza-2′-deoxycytidine-induced genome rearrangements are mediated by DNMT1. Oncogene 2012, 31, 5172–5179. [Google Scholar] [CrossRef] [PubMed]
- Palii, S.S.; Van Emburgh, B.O.; Sankpal, U.T.; Brown, K.D.; Robertson, K.D. DNA methylation inhibitor 5-Aza-2′-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol. Cell. Biol. 2008, 28, 752–771. [Google Scholar] [CrossRef] [PubMed]
- Pastor, W.A.; Aravind, L.; Rao, A. TETonic shift: Biological roles of TET proteins in DNA demethylation and transcription. Nat. Rev. Mol. Cell Biol. 2013, 14, 341–356. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.X.; Hollingshead, M.; Rubinstein, L.; Nguyen, D.; Larenjeira, A.B.A.; Kinders, R.J.; Difilippantonio, M.; Doroshow, J.H. TET2 and DNMT3A mutations and exceptional response to 4′-thio-2′-deoxycytidine in human solid tumor models. J. Hematol. Oncol. 2021, 14, 83. [Google Scholar] [CrossRef] [PubMed]
- Rhee, I.; Jair, K.W.; Yen, R.W.; Lengauer, C.; Herman, J.G.; Kinzler, K.W.; Vogelstein, B.; Baylin, S.B.; Schuebel, K.E. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature 2000, 404, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Graff, J.R.; Myohanen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Gu, J.; Huang, M.; Wu, X.; Hildebrandt, M.A. Epigenetic analysis of microRNA genes in tumors from surgically resected lung cancer patients and association with survival. Mol. Carcinog. 2015, 54 (Suppl. S1), E45–E51. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Seong, H.; Liu, Y.; Yu, X.; Xu, S.; Li, Y. Ten-eleven translocation proteins (TETs): Tumor suppressors or tumor enhancers? Front. Biosci. (Landmark Ed.) 2021, 26, 895–915. [Google Scholar] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laranjeira, A.B.A.; Nguyen, D.; Pelosof, L.C.; Doroshow, J.H.; Yang, S.X. Upregulation of TET2 and Resistance to DNA Methyltransferase (DNMT) Inhibitors in DNMT1-Deleted Cancer Cells. Diseases 2024, 12, 163. https://doi.org/10.3390/diseases12070163
Laranjeira ABA, Nguyen D, Pelosof LC, Doroshow JH, Yang SX. Upregulation of TET2 and Resistance to DNA Methyltransferase (DNMT) Inhibitors in DNMT1-Deleted Cancer Cells. Diseases. 2024; 12(7):163. https://doi.org/10.3390/diseases12070163
Chicago/Turabian StyleLaranjeira, Angelo B. A., Dat Nguyen, Lorraine C. Pelosof, James H. Doroshow, and Sherry X. Yang. 2024. "Upregulation of TET2 and Resistance to DNA Methyltransferase (DNMT) Inhibitors in DNMT1-Deleted Cancer Cells" Diseases 12, no. 7: 163. https://doi.org/10.3390/diseases12070163