
The Role of the MAPK Signaling, Topoisomerase and Dietary Bioactives in Controlling Cancer Incidence

Abstract
1. Introduction
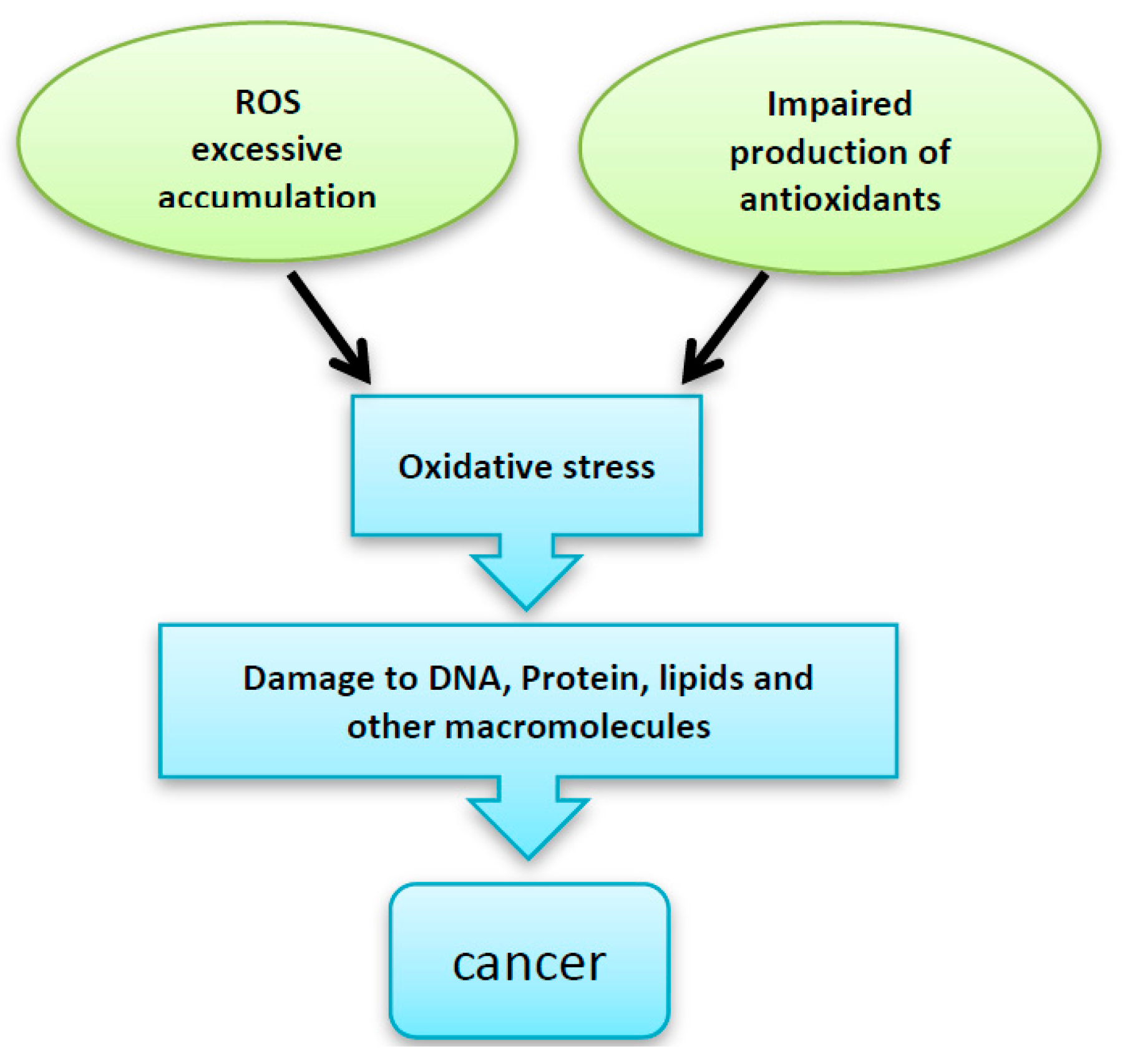
2. Disturbance in ROS Levels Causes Diseases
3. ROS and Cell Signaling
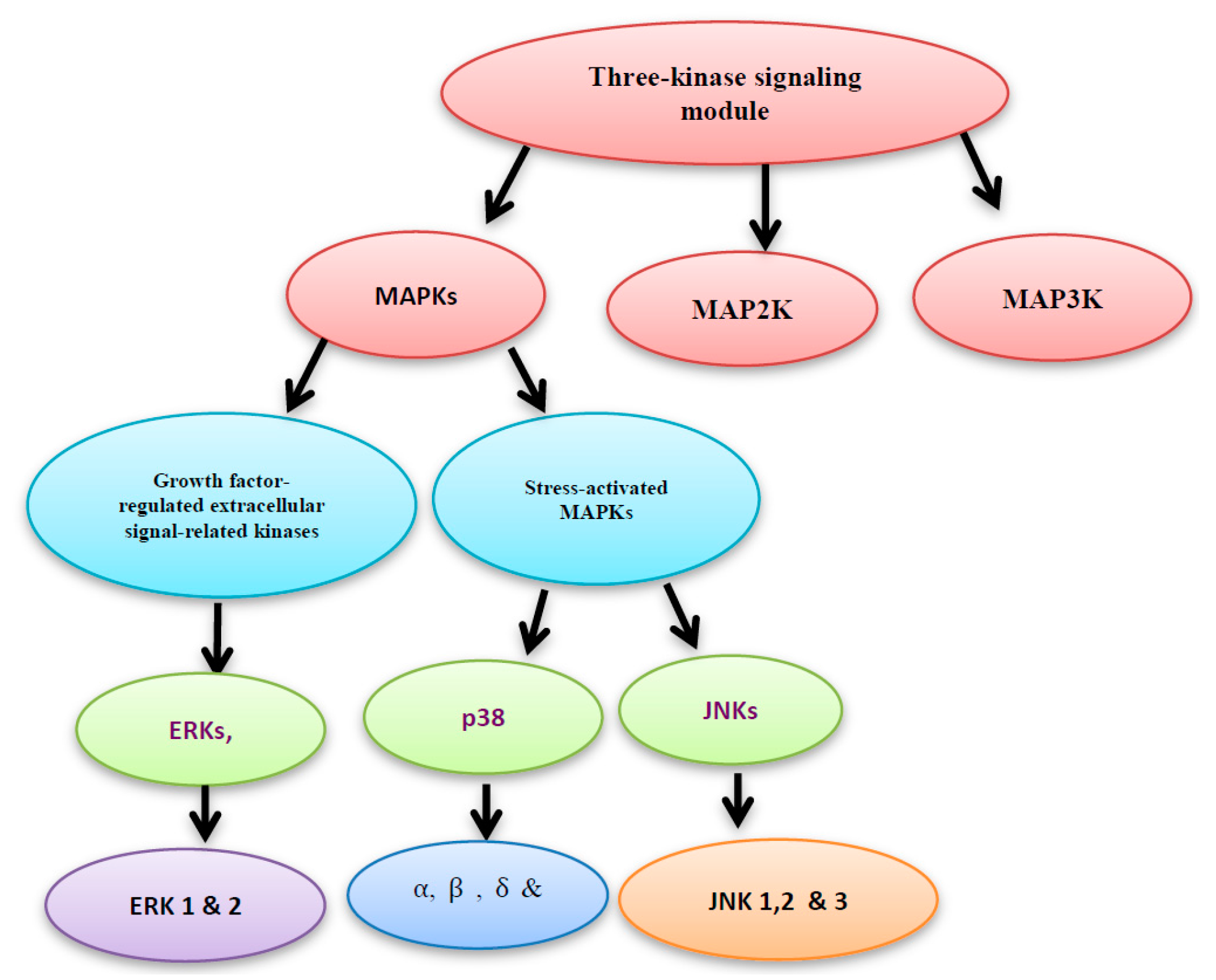
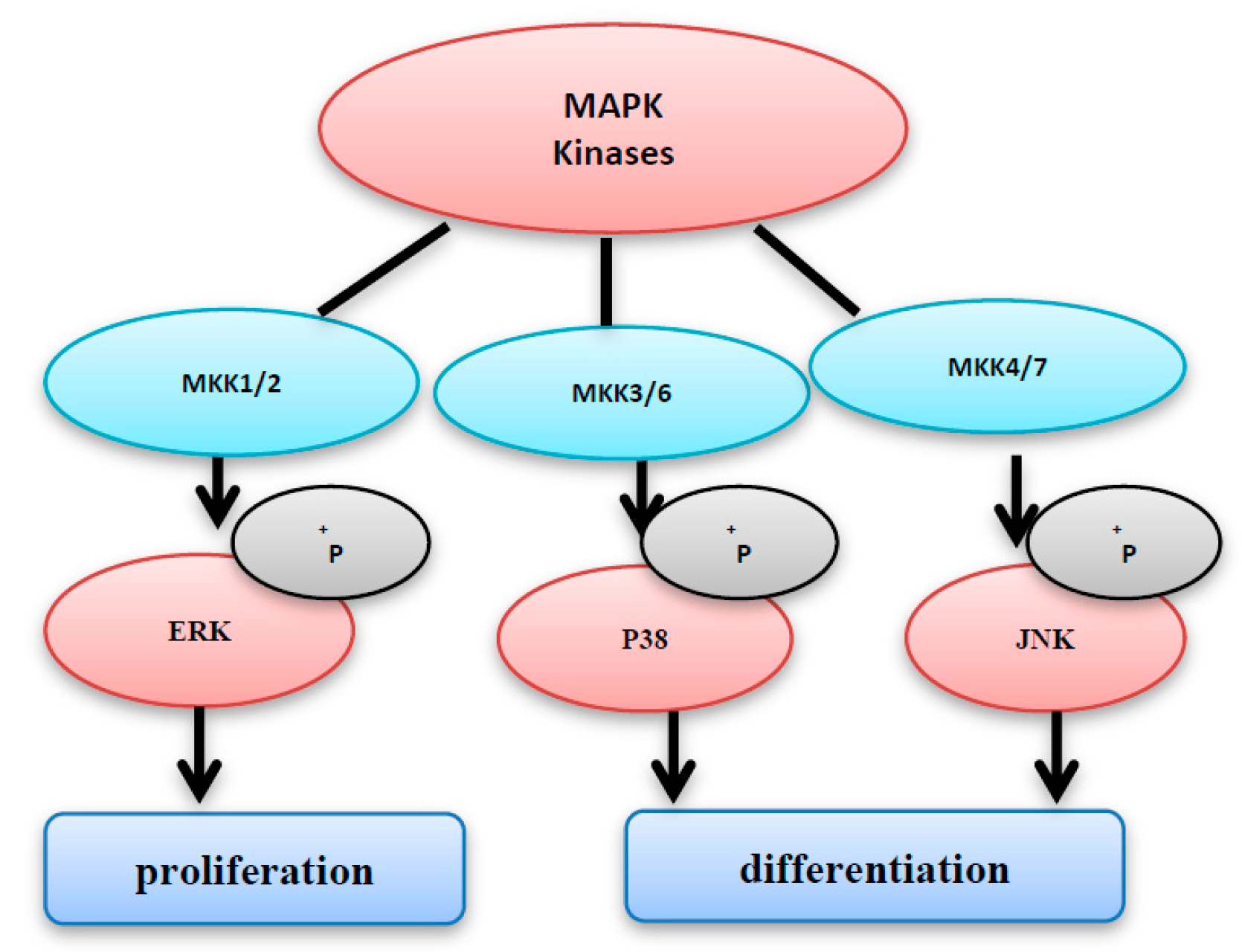
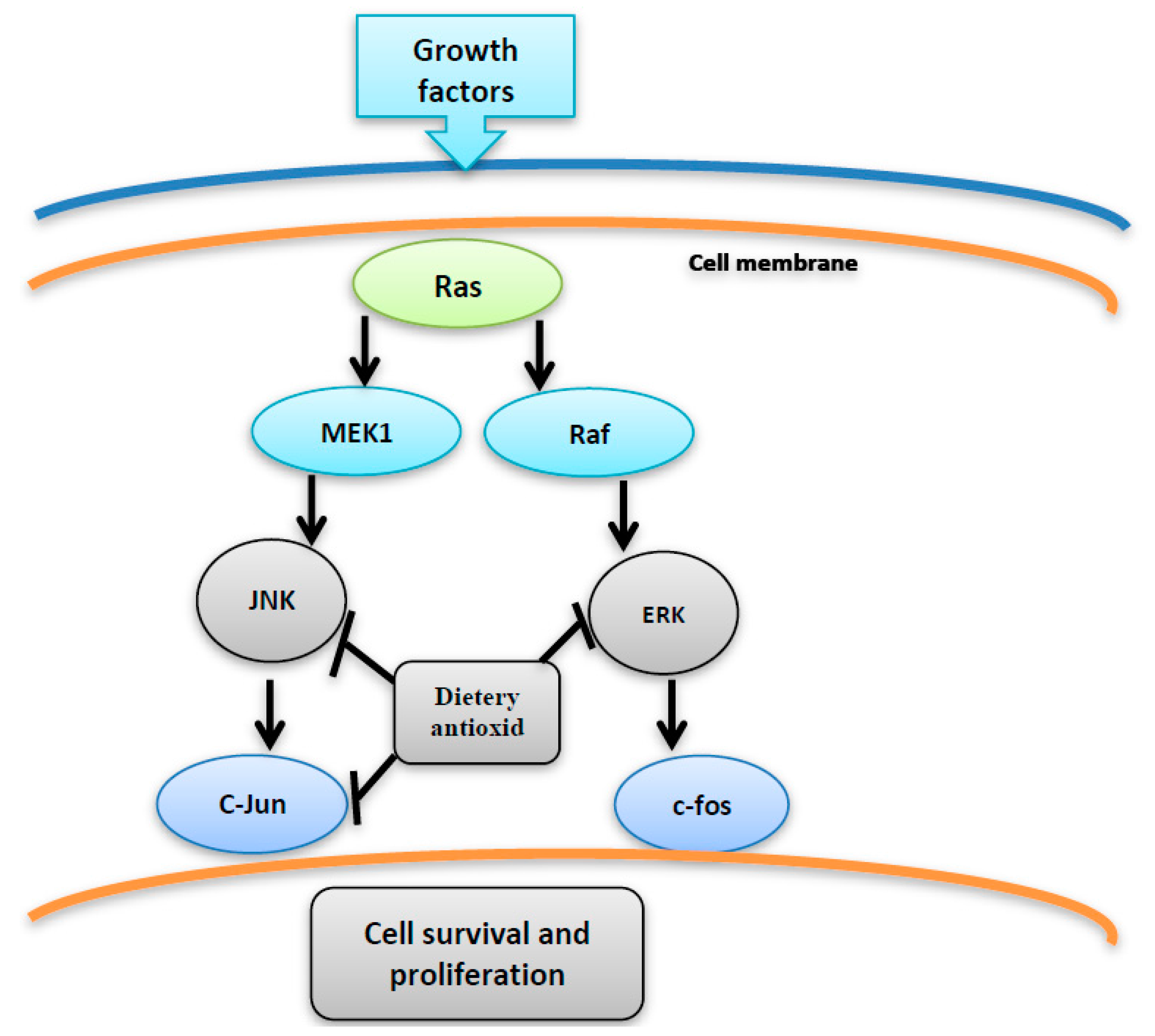
4. MAPK Activation by MAP Kinases
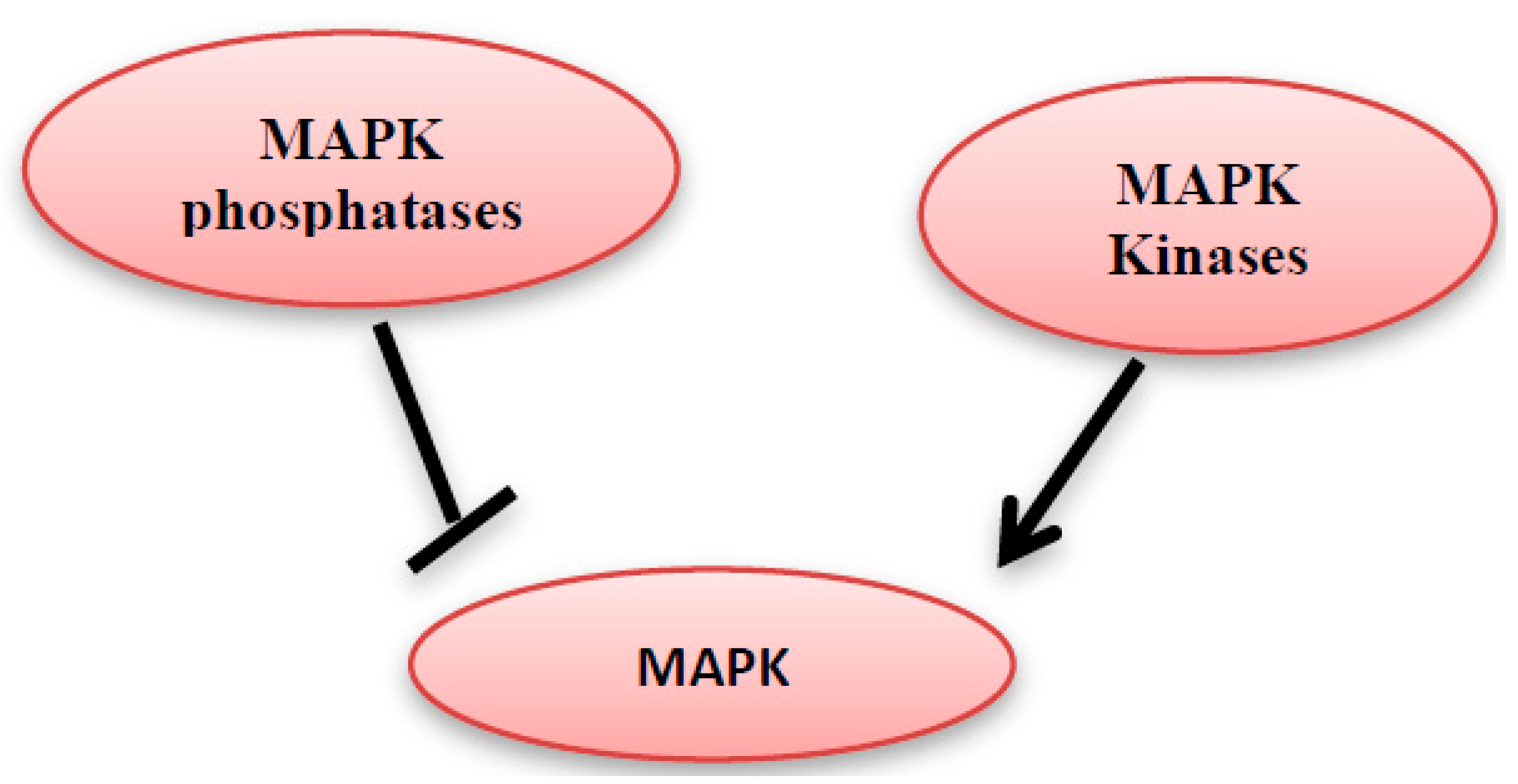
5. MAPK Inhibition by MAPK Phosphatases
6. ROS Activate MAPK Pathway
7. Topoisomerase
7.1. Classification of Topoisomerases
7.2. Topoisomerase Inhibition
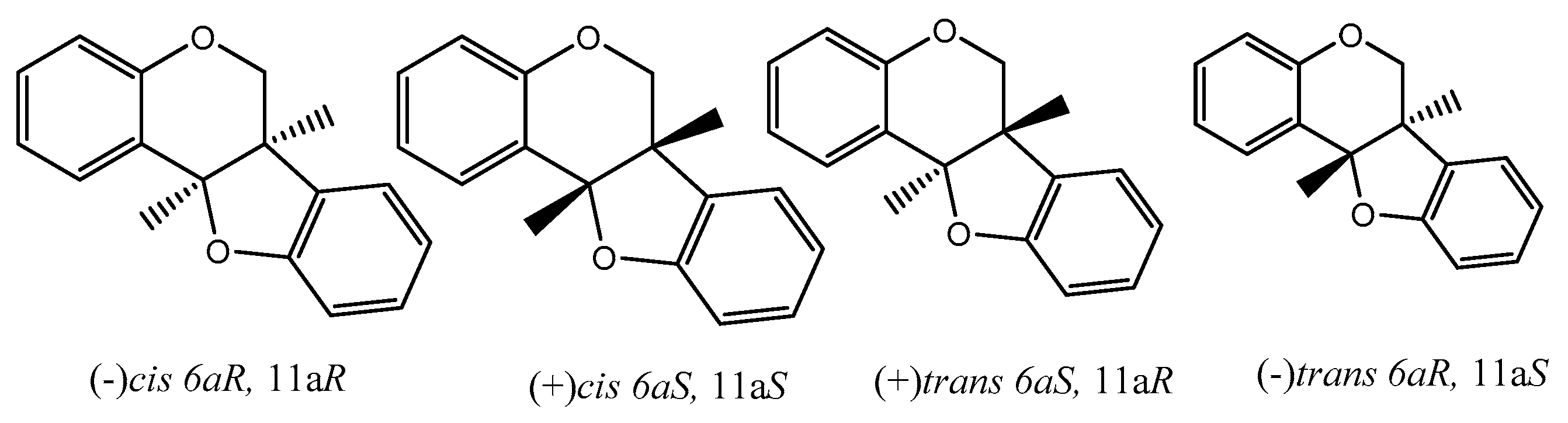
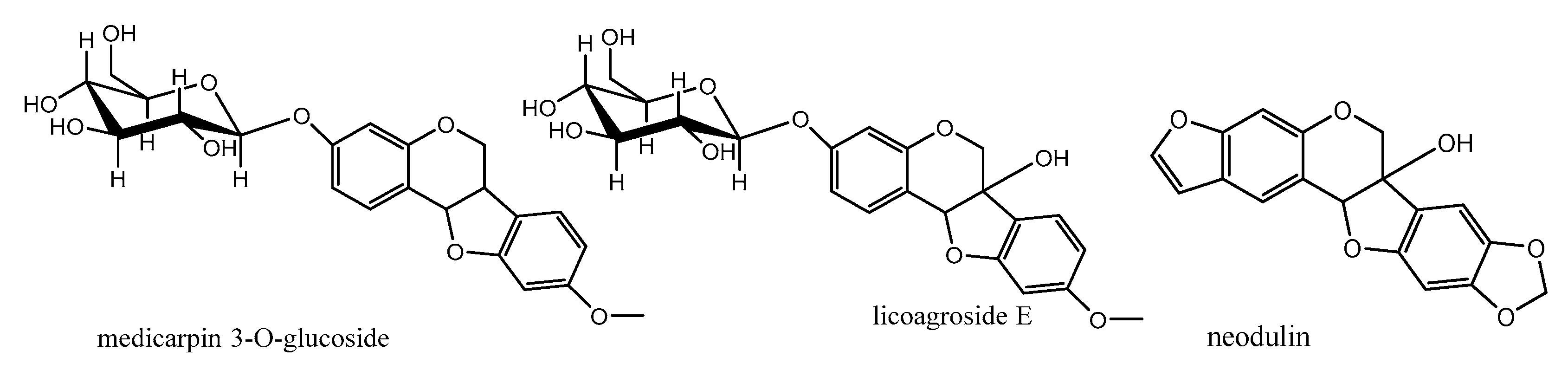
8. Pterocarpans as Anti-Oxidant/Cancer Agents
9. Conclusions
Acknowledgments
Author Contributions
References
- Benhar, M.; Engelberg, D.; Levitzki, A. ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep. 2002, 3, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Afaq, F.; Mukhtar, H. Cancerchemoprevention through dietary antioxidants: Progress and promise. Antioxid. Redox Signal. 2008, 10, 475–510. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Nikulenkov, F.; Zawacka-Pankau, J.; Li, H.; Gabdoulline, R.; Xu, J.; Eriksson, S.; Hedström, E.; Issaeva, N.; Kel, A.; et al. ROS-dependentactivation of JNKconvertsp53 into an efficient inhibitor of oncogenes leading to robust apoptosis. Cell Death Differ. 2014, 21, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal Transduct. 2011. [Google Scholar] [CrossRef] [PubMed]
- Haagenson, K.K.; Wu, G.S. The role of MAP kinases and MAP kinase phosphatase-1 in resistance to breast cancer treatment. Cancer Metast. Rev. 2010, 29, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Banerjee, P.; Nikolakaki, E.; Dai, T.; Rubie, E.A.; Ahmad, M.F.; Avruch, J.; Woodgett, J.R. The stress-activated protein kinase subfamily of c-Jun kinases. Nature 1994, 369, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Raingeaud, J.; Gupta, S.; Rogers, J.S.; Dickens, M.; Han, J.; Ulevitch, R.J.; Davis, R.J. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem. 1995, 270, 7420–7426. [Google Scholar] [PubMed]
- Zhou, J.Y.; Liu, Y.; Wu, G.S. The role of mitogen-activated protein kinase phosphatase-1 in oxidative damage-induced cell death. Cancer Res. 2006, 66, 4888–4894. [Google Scholar] [CrossRef] [PubMed]
- Koul, H.K.; Pal, M.; Koul, S. Role of p38 MAP Kinase Signal Transduction in Solid Tumors. Genes Cancer 2013, 4, 342–359. [Google Scholar] [CrossRef] [PubMed]
- Winter-Vann, A.M.; Johnson, G.L. Integrated activation of MAP3Ks balances cell fate in response to stress. J. Cell. Biochem. 2007, 102, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Farooq, A.; Zhou, M.M. Structure and regulation of MAPK phosphatases. Cell. Signal. 2004, 16, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, K.; Nishida, E. Regulation of MAP kinases by MAP kinase phosphatases. Biochim. Biophys. Acta 2007, 1773, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Keyse, S.M. An emerging family of dual specificity MAP Kinase phosphatases. Biochim. Biophys. Acta 1995, 1265, 152–160. [Google Scholar] [CrossRef]
- Keyse, S.M.; Emslie, E.A. Oxidative stress and heat shock induce a human gene encoding a protein-tyrosine phosphatase. Nature 1992, 359, 644–647. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gorospe, M.; Hutter, D.; Barnes, J.; Keyse, S.M.; Liu, Y. Transcriptional induction of MKP-1 in response to stress is associated with histone H3 phosphorylation acetylation. Mol. Cell Biol. 2001, 21, 8213–8224. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.M.; Keyse, S.M. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 2007, 26, 3203–3213. [Google Scholar] [CrossRef] [PubMed]
- Theodosiou, A.; Ashworth, A. MAP kinase phosphatases. Genome Biol. 2002, 3. [Google Scholar] [CrossRef]
- Sun, H.; Charles, C.H.; Lau, L.F.; Tonks, N.K. MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell 1993, 75, 487–493. [Google Scholar] [CrossRef]
- Noguchi, T.; Metz, R.; Chen, L.; Mattei, M.G.; Carrasco, D.; Bravo, R. Structure, mapping, and expression of erp, a growth factor-inducible gene encoding a nontransmembrane protein tyrosine phosphatase, and effect of ERP on cell growth. Mol. Cell Biol. 1993, 13, 5195–5205. [Google Scholar] [CrossRef] [PubMed]
- Franklin, C.C.; Kraft, A.S. Conditional expression of the mitogen-activated protein kinase (MAPK) phosphatase MKP-1 preferentially inhibits p38 MAPK and stress activated protein kinase in U937 cells. J. Biol. Chem. 1997, 272, 16917–16923. [Google Scholar] [CrossRef] [PubMed]
- Brondello, J.M.; McKenzie, F.R.; Sun, H.; Tonks, N.K.; Pouyssegur, J. Constitutive MAP kinase phosphatase (MKP-1) expression blocks G1 specific gene transcription and S-phase entry in fibroblasts. Oncogene 1995, 10, 1895–1904. [Google Scholar] [PubMed]
- Wu, G.S. The functional interactions between the p53 and MAPK signaling pathways. Cancer Biol. Ther. 2004, 3, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Franklin, C.C.; Srikanth, S.; Kraft, A.S. Conditional expression of mitogen-activated protein kinase phosphatase-1, MKP-1, is cytoprotective against UV induced apoptosis. Proc. Natl. Acad. Sci. USA 1998, 95, 3014–3019. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Perez, I.; Martinez-Gomariz, M.; Williams, D.; Keyse, S.M.; Perona, R. CL100/MKP-1 modulates JNK activation and apoptosis in response to cisplatin. Oncogene 2000, 19, 5142–5552. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Lahair, M.M.; Franklin, R.A. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid. Redox Signal. 2006, 8, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 221ra15. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Peter Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Okada, M.; Shibuya, K.; Watanabe, E.; Seino, S.; Narita, Y.; Shibui, S.; Kayama, T.; Kitanaka, C. Pivotal role for ROS activation of p38 MAPK in the control of differentiation and tumor-initiating capacity of glioma-initiating cells. Stem. Cell Res. 2014, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hirao, A.; Arai, F.; Matsuoka, S.; Takubo, K.; Hamaguchi, I.; Nomiyama, K.; Hosokawa, K.; Sakurada, K.; Nakagata, N.; et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 2004, 431, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Bigarella, C.L.; Liang, R.; Ghaffari, S. Stem cells and the impact of ROS signaling. Development 2014, 141, 4206–4218. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, C.I.; Suda, T. Regulation of reactive oxygen species in stem cells and cancer stem cells. J. Cell. Physiol. 2012, 227, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Sheweita, S.A.; Sheikh, B.Y. Can dietary antioxidants reduce the incidence of brain tumors? Curr. Drug Metab. 2011, 12, 587–593. [Google Scholar] [CrossRef] [PubMed]
- DeLorenze, G.N.; McCoy, L.; Tsai, A.-L. Daily intake of antioxidants in relation to survival among adult patients diagnosed with malignant glioma. BMC Cancer 2010, 10, 215. [Google Scholar] [CrossRef] [PubMed]
- Kimberly, K.; Weiping, Y.; Bob, G. Vitamin E: Mechanisms of Action as Tumor Cell Growth Inhibitors. J. Nutr. 2001, 131, 161S–163S. [Google Scholar]
- Wu, K.; Zhao, Y.; Li, G.C.; Yu, W.P. c-Jun N-terminal kinase is required for vitamin E succinate-induced apoptosis in human gastric cancer cells. World J. Gastroenterol. 2004, 10, 1110–1114. [Google Scholar] [PubMed]
- Shim, M.; Eling, T.E. Vitamin E succinate induces NAG-1 expression in a p38 kinase-dependent mechanism. Mol. Cancer Ther. 2008, 7, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Khan, W.A.; Wang, Z.Y.; Athar, M.; Bickers, D.R.; Mukhtar, H. Inhibition of the skin tumorigenicity of (±)-7β,8α-dihydroxy-9α,10α-epoxy-7,8,9,10-tetrahydrobenz[α]pyrene by tannic acid, green tea polyphenols and quercetin in Sencar mice. Cancer Lett. 1988, 42, 7–12. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Huang, M.T.; Ferraro, T.; Wong, C.Q.; Lou, Y.R.; Reuhl, K.; Iatropoulos, M.; Yang, C.S.; Conney, A.H. Inhibitory effect of green tea in the drinking water on tumorigenesis by ultraviolet light and 12-O-tetradecanoylphorbol-13-acetate in the skin of SKH-1 mice. Cancer Res. 1992, 52, 1162–1170. [Google Scholar] [PubMed]
- Wang, Z.Y.; Huang, M.T.; Ho, C.T.; Chang, R.; Ma, W.; Ferraro, T.; Reuhl, K.; Yang, C.S.; Conney, A.H. Inhibitory effect of green tea onthe growth of established skin papillomas in mice. Cancer Res. 1992, 52, 6657–6665. [Google Scholar] [PubMed]
- Conney, A.H.; Lu, Y.P.; Lou, Y.R.; Xie, J.G.; Huang, M.T. Inhibitory effect of green and black tea on tumor growth. Proc. Soc. Exp. Biol. Med. 1999, 220, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Soni, K.B.; Lahiri, M.; Chackradeo, P.; Bhide, S.V.; Kuttan, R. Protective effect of food additives on aflatoxin-induced mutagenicity and hepatocarcinogenicity. Cancer Lett. 1997, 115, 129–133. [Google Scholar] [CrossRef]
- Deguchi, H.; Fujii, T.; Nakagawa, S.; Koga, T.; Shirouzu, K. Analysis of cell growth inhibitory effects of catechin through MAPK in human breast cancer cell line T47D. Int. J. Oncol. 2002, 21, 1301–1305. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, R.K.; Singh, A.K.; Gaddipati, J.; Srimal, R.C. Multiple biological activities of curcumin: A short review. Life Sci. 2006, 78, 2081–2087. [Google Scholar] [CrossRef] [PubMed]
- Dorai, T.; Gehani, N.; Katz, A. Therapeutic potential of curcumin in human prostate cancer-I. curcumin induces apoptosis in both androgen-dependent and androgen-independent prostate cancer cells. Prostate Cancer Prostatic Dis. 2000, 3, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Qiao, Z.; Wang, H.; Zhu, L.; Zhang, L. Flavonoids: Promising anticancer agents. Med. Res. Rev. 2003, 23, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Whitsett, T.; Carpenter, M.; Lamartiniere, C.A. Resveratrol, but not EGCG, in the diet suppresses DMBA-induced mammary cancer in rats. J. Carcinog. 2006, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Lian, F.; Li, Y.; Bhuiyan, M.; Sarkar, F.H. p53-independent apoptosis induced by genistein in lung cancer cells. Nutr. Cancer 1999, 33, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Champoux, J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed]
- Bandele, O.J.; Osheroff, N. (−)-EpigallocatechinGallate, A Major Constituent of Green Tea, Poisons Human Type II Topoisomerases. Chem. Res. Toxicol. 2008, 21, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Leone, S.; Basso, E.; Polticelli, F.; Cozzi, R. Resveratrol acts as a topoisomerase II poison in human glioma cells. Int. J. Cancer 2012, 131, E173–E178. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; Kerns, R.J.; Osheroff, N. Mechanism of Quinolone Action and Resistance. Biochemistry 2014, 53, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Kumar, A.; Raghuvanshi, A. Synthesis, stereochemistry, structural classification, and chemical reactivity of natural pterocarpans. Chem. Rev. 2012, 113, 1614–1640. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Lutterodt, H.; Cheng, Z.; Yu, L. Anti-inflammatory and antiproliferative activities of trifolirhizin, a flavonoid from Sophoraflavescens roots. J. Agric. Food Chem. 2009, 57, 4580–4585. [Google Scholar] [CrossRef] [PubMed]
- Aratanechemuge, Y.; Hibasami, H.; Katsuzaki, H.; Imai, K.; Komiya, T. Induction of apoptosis by maackiain and trifolirhizin (maackiain glycoside) isolated from sanzukon (SophoraSubprostrate Chen et T. Chen) in human promyelotic leukemia HL-60 cells. Oncol. Rep. 2004, 12, 1183–1188. [Google Scholar] [PubMed]
- Lu, X.; Ma, J.; Qiu, H.; Yang, L.; Cao, L.; Shen, J. Anti-proliferation effects of trifolirhizin on MKN45 cells and possible mechanism. Oncol. Rep. 2016, 36, 2785–2792. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Shishodia, S.; Sandur, S.K.; Pandey, M.K.; Sethi, G. Inflammation and cancer: How hot is the link? Biochem. Pharmacol. 2006, 72, 1605–1621. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.J.L.; Lamb, J.H.; Verschoyle, R.D.; Howells, L.M.; Butterworth, M.; Lim, C.K.; Ferry, D.; Farmer, P.B.; Gescher, A.J. Characterization of metabolites of the putative cancer chemopreventive agent quercetin and their effect on cyclo-oxygenase activity. Br. J. Cancer 2004, 91, 1213–1219. [Google Scholar] [PubMed]
- Militao, G.C.; Bezerra, D.P.; Pessoa, C.; de Moraes, M.O.; da Ponte, F.A.; Lima, M.A.S.; Silveira, E.R.; Costa-Lotufo, L.V. Comparative cytotoxicity of 2,3,9-trimethoxypterocarpan in leukemia cell lines (HL-60, Jurkat, Molt-4, and K562) and human peripheral blood mononuclear cells. J. Nat. Med. 2007, 61, 196–199. [Google Scholar] [CrossRef]
- Militao, G.C.; Dantas, I.N.; Pessoa, C.; Falcão, M.J.C.; Silveira, E.R.; Lima, M.A.S.; Curi, R.; Lima, T.; Moraes, M.O.; Costa-Lotufo, L.V. Induction of apoptosis by pterocarpans from Platymiscium floribundum in HL-60 human leukemia cells. Life Sci. 2006, 78, 2409–2417. [Google Scholar] [CrossRef] [PubMed]
- Militao, G.C.; Pinheiro, S.M.; Dantas, I.N.; Pessoa, C.; de Moraes, M.O.; Lima, M.A.S.; Silveira, E.R. Bioassay-guided fractionation of pterocarpans from roots of Harpalyce brasiliana Benth. Bioorg. Med. Chem. 2007, 15, 6687–6691. [Google Scholar] [CrossRef] [PubMed]
- Falcão, M.J.; Pouliquem, Y.B.; Lima, M.A.; Gramosa, N.V.; Costa-Lotufo, L.V.; Militão, G.C.; Pessoa, C.; de Moraes, M.O.; Silveira, E.R. Cytotoxic flavonoids from Platymiscium floribundum. J. Nat. Prod. 2005, 68, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, R.; Maurya, R.; Mishra, D.P. Medicarpin, a legume phytoalexin sensitizes myeloid leukemia cells to TRAIL-induced apoptosis through the induction of DR5 and activation of the ROS-JNK-CHOP pathway. Cell Death Dis. 2014, 5, e1465. [Google Scholar] [CrossRef] [PubMed]
- Militao, G.C.; Jimenez, P.C.; Wilke, D.V.; Pessoa, C.; Falcão, M.J.; Lima, M.A.S.; Silveira, E.R.; de Moraes, M.O.; Costa-Lotufo, L.V. Antimitotic properties of pterocarpans isolated from Platymiscium floribundum on sea urchin eggs. Plantamedica 2005, 71, 683–685. [Google Scholar]
- Militao, G.C.; Prado, M.P.; Pessoa, C.; de Moraes, M.O.; Silveira, E.R.; Lima, M.A.S.; Veloso, P.A.; Costa-Lotufo, L.V.; Machado-Santelli, G.M. Pterocarpans induce tumor cell death through persistent mitotic arrest during prometaphase. Biochimie 2014, 104, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Maurich, T.; Iorio, M.; Chimenti, D.; Turchi, G. Erybraedin C and bitucarpin A, two structurally related pterocarpans purified from Bituminaria bituminosa, induced apoptosis in human colon adenocarcinoma cell lines MMR- and p53-proficient and -deficient in a dose-, time-, and structure-dependent fashion. Chem. Biol. Interact. 2006, 159, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Kuete, V.; Sandjo, L.P.; Djeussi, D.E.; Zeino, M.; Kwamou, G.M.; Ngadjui, B.; Efferth, T. Cytotoxic flavonoids and isoflavonoids from Erythrina sigmoidea towards multi-factorial drug resistant cancer cells. Investig. New Drugs 2014, 32, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Kuete, V.; Sandjo, L.P.; Kwamou, G.M.; Wiench, B.; Nkengfack, A.E.; Efferth, T. Activity of three cytotoxic isoflavonoids from Erythrina excelsa and Erythrina senegalensis (neobavaisoflavone, sigmoidin H and isoneorautenol) toward multi-factorial drug resistant cancer cells. Phytomedicine 2014, 21, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Le, T.V.T.; Thuong, P.T.; Dao, T.T.; Ndinteh, D.T.; Mbafor, J.T.; Kang, K.W.; Oh, W.K. Cytotoxic and PTP1B inhibitory activities from Erythrina abyssinica. Bioorg. Med. Chem. Lett. 2009, 19, 6745–6749. [Google Scholar] [CrossRef] [PubMed]
- Wätjen, W.; Kulawik, A.; Suckow-Schnitker, A.; Chovolou, Y.; Rohrig, R.; Ruhl, S.; Kampkötter, A.; Addae-Kyereme, J.; Wright, C.; Passreiter, C. Pterocarpansphaseollin and neorautenol isolated from Erythrina addisoniae induce apoptotic cell death accompanied by inhibition of ERK phosphorylation. Toxicology 2007, 242, 71–79. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Method | Type of Cells | Incubation Period | Results | Ref. |
|---|---|---|---|---|---|
| Trifolirhizin | MTT | The A2780 ovarian cancer and H23 lung cancer cells | 24 h | Significant antiproliferation was achieved with concentrations up to 100 μM against A2780 ovarian cancer cells. However, a significant antiproliferative effect was observed only with a concentration of 250 μM for H23 lung cancer cells. | [54] |
| Trifolirhizin | Morphological changes was observed with epifluorescence microscope | Human myeloid leukemia (HL-60) | 3 days | Trifolirhizin suppressed human myeloid leukemia (HL-60) through induction of apoptosis | [55] |
| Trifolirhizin | MTT assay for cell viability. Hoechst 33342 staining and TUNEL staining for detection of apoptosis. Western blotting was used to investigate the levels of apoptotic and related signaling pathway proteins. | MKN45, L02, HEK293 cells | 2 days | A concentration- and time-dependent suppression of MKN45 cell viability with IC50 33.27 ± 2.06 μg/mL was observed. The apoptosis was mediated via EGFR-MAPK pathways. Trifolirhizin also arrested the G/M cycle through impact on Cdc2/cyclin B complex. | [56] |
| 2,3,9-trimethoxypterocarpan, Homocarpin, Medicarpin and Vesticarpin | MTT | B16 (murine melanoma), HCT-8 (human colon), MCF-7 (human breast), CEM and HL-60 | 3, 6, 12, 24, 36, 48, 60, and 72 h | 2,3,9-trimethoxypterocarpan was the most active compound against all human cancer cell lines with IC50 2.9, 0.6, 0.7, 0.6, 0.1 μg/mL, respectively. | [62] |
| Medicarpin | Determination of cell viability and LDH Release Cell cycle and cell death analysis Measurement of ROS and the mitochondrial ROS Real-time PCR Cloning of the DR5 promoter and luciferase assay staining with phycoerythrin-conjugated mouse monoclonal anti-human DR5 or DR4 for analysis of cell surface expression of DR4 and DR5 | The cell lines K562, LAMA-84 (chronic myeloid leukemia cell lines), U937, OCIAML-3 (the AML cell lines) | 48 h | A trail-induced apoptosis at a dose 20 μM was observed. The result revealed the possibility of involvement of JNK activation. | [63] |
| 2,3,9-trimethoxypterocarpan | The Trypan blue dye exclusion test | HL-60, K562, Jurkat, and Molt-4 | 3, 6, 12, 24, 36, 48, 60 and 72 h | After 24 h, Jurkat and Molt-4 showed less sensitivity (IC50 > 10 and 5.9 ± 1.1 g/mL, respectively) while HL-60 (IC50 2.5 ± 0.3 g/mL) and K562 cells showed (IC50 2.8 ± 0.67 g/mL). After 36 h, the IC50 values ranged from 0.5 to 1.1 g/mL, without significant difference among the cell lines. Maximum activity was observed after 48 h of incubation, with K562 being the most resistant cell line (IC50 0.8 ± 0.1 g/mL), followed by Molt-4 and HL-60 (both with IC50 of 0.4 g/mL), and Jurkat (IC50 0.1 ± 0.03 g/mL). | [59] |
| 2,3,9-trimethoxypterocarpan | Cell cycle analysis and measurement of the mitochondrial transmembrane potential | Breast cancer cell lines MCF7, T47d and HS578T | 24 and 48 h | The cell cycle arrest was induced in the all tested cell lines at concentration 8 µM in time-dependent manner where 24 h incubation period followed by 24 h recovery period in medium free pterocarpan led to a reversible effect while persistent mitotic inhibition followed by apoptosis was noticed after a 48 h exposure period despite the pterocarpan free medium recovery period. Mitosis was also inhibited during the prometa phase, in a crucial step where the separation of duplicated centrosomes was blocked followed by cell cycle arrest, and the persistent prometaphase arrest resulted in apoptosis of treatment, the IC50 values ranged from 0.3 to 1.6 mM | [65] |
| Erybraedin C and bitucarpin A | Hemocytometer cell count | HT29 and LoVo human colon adenocarcinoma | LoVo, 26 h and HT29, 29 h | Erybraedin C and bitucarpin A induced antitumor activity against two human adenocarcinoma cell line (HT29 and LoVo) proficient and deficient in MMR (mismatch repair), p53 and Bcl-2 | [66] |
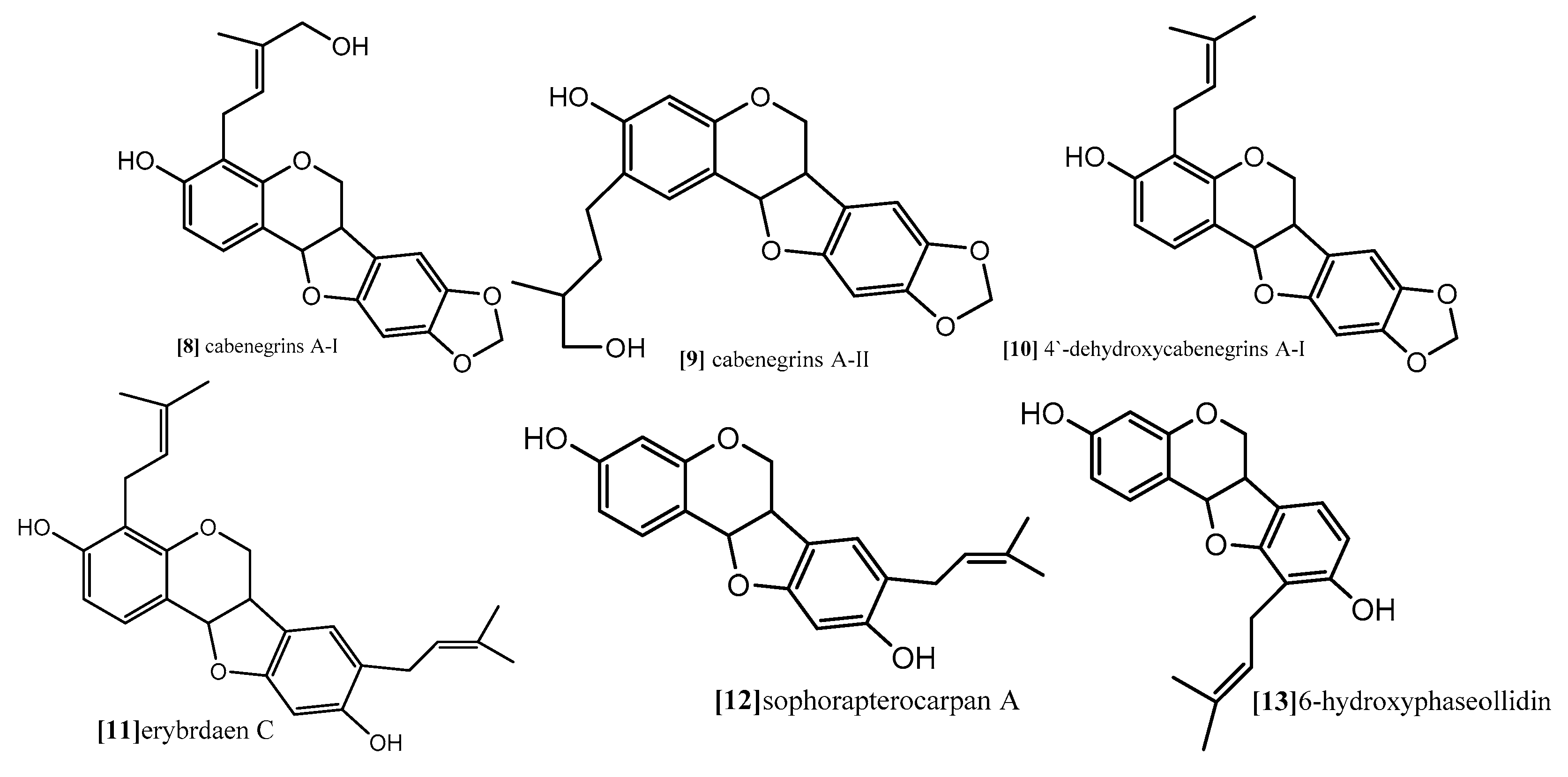
| Sophorapterocarpan A, 6α-hydroxyphaseollidin | resazurin reduction assay Flow cytometry for cell cycle analysis Analysis of mitochondrial membrane potential (MMP) | panel of nine sensitive and resistance cell lines, drug-sensitive (CCRF-CEM) and multidrug-resistant P-glycoprotein (P-gp) over expressing (CEM/ADR5000) leukemia, the (MDA-MB-231-pcDNA3) breast cancer and its resistant subline (MDA-MB- 231-BCRP)(breast cancer resistance protein clone 23), the (HCT116) (p53+/+) colon cancer cells and its knockout clones (HCT116) (p53−/−), the (U87MG) glioblastoma cells and its resistant subline epidermal growth factor receptor (U87MG. ΔEGFR) and human hepatocellular carcinoma cells (HepG2) and the normal hepatocytes (AML12). | 48 and 72 h | pterocarpansSophorapterocarpan (compound 12;
Figure 9) and 6α-hydroxyphaseollidin(compound 13; Figure 9) exhibited strong cytotoxic effects against all tested cancer cell lines with IC50 values 3.6 to 6.4 µM and 3.7 to 14.8 µM, respectively. In the same study the cell cycle analysis of the two pterocarpans compounds 12 and 13 against leukemia (CCRF-CEM) cells demonstrated that both of them induced cell cycle arrest in the Go/G1 phase. Compound 13 was able to increase the activity of caspases which are responsible for cutting the cellular proteins at specific Aspartate residues to regulate the process of apoptosis, compound 13 activated the initiator caspase 3/7 and effectors caspases 8 and 9 two-fold in concentration range (0.5–2 fold IC50) In another method, compound 12 induces apoptosis through breakdown of mitochondrial membrane potential (MMP) the disruption of which is a common event in the process of apoptosis by (17%–92.9%) in concentration range form (1/4-fold IC50 to 2-fold IC50). | [67] |
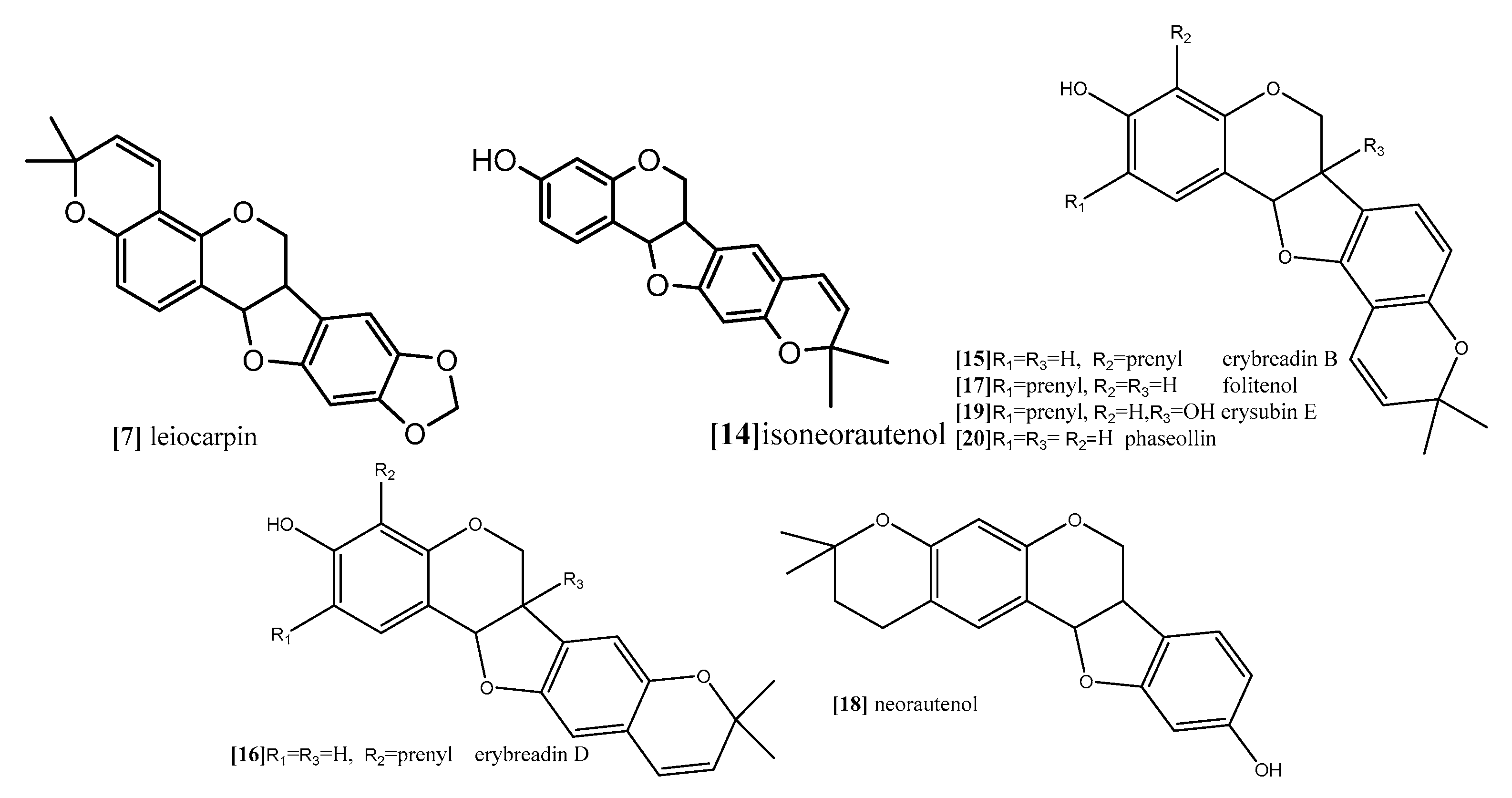
| Isoneorautenol | Resazurin reduction assay Flow cytometry for cell cycle analysis Analysis of mitochondrial membrane potential (MMP) | panel of nine sensitive and resistance cell lines, drug-sensitive (CCRF-CEM) and multidrug-resistant P-glycoprotein (P-gp) over expressing (CEM/ADR5000) leukemia, the (MDA-MB-231-pcDNA3) breast cancer and its resistant subline (MDA-MB- 231-BCRP) (breast cancer resistance protein clone 23), the (HCT116) (p53+/+) colon cancer cells and its knockout clones (HCT116) (p53−/−), the (U87MG) glioblastoma cells and its resistant subline epidermal growth factor receptor (U87MG. ΔEGFR) and human hepatocellular carcinoma cells (HepG2) and the normal hepatocytes (AML12). | 48 and 72 h | Isoneorautenol (compound 14;
Figure 10) showed cytotoxicity against the nine tested cell lines with IC50 values below 22 µM. Compound 14 induced cell cycle arrest between Go/G1 phase and S phase in time-dependent manner. It has been proven from the results that the tested pterocarpans induces apoptosis in leukemia (CCRF-CEM) cells via different mode of action. It also activated caspases 3/7 more than caspases 8 and 9 at a concentration of 2-fold IC50. It displayed dose-dependent disruption of MMP (89% at concentration of 2-fold IC50) as well. | [68] |
| Erybraedin B, erybraedin D, folitenol, neorautenol and erysubin E | The cell viability was assessed using a 4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate (WST-1) based cytotoxicity assay Inhibitory effects on protein tyrosine phosphatase-1B (PTP1B) | MCF7, tamoxifen-resistant MCF7 (MCF7/TAMR), adriamycin-resistant MCF7 (MCF7/ADR) and MDA-MB-231 breast cancer cell lines. | Erybraedin B (compound 15; Figure 10) is the most active compound with IC50 values (5.6 to 11.8 µM), it exhibited 2-fold activity of tamoxifen the reference drug against the drug resistance cell lines, tamoxifen-resistant MCF7 (MCF7/TAMR) and adriamycin-resistant MCF7 (MCF7/ADR) cell lines with IC50 (6.2 ± 0.2 and 5.6 ± 0.7 µM), respectively. It has been proven that the presence of 2,2 dimethypyran substitution in ring D of the pterocarpan molecule is the most important feature of the cytotoxic activity against breast cancer cell lines where the presence of this moiety in erybraedin B (compound 15; Figure 10), erybraedin D(compound 16; Figure 10), folitenol (compound 17; Figure 10) demonstrated potent cytotoxicity more than compound which lack this moiety. Moreover, it was concluded from this study that there is strong correlation between the cytotoxicity against the breast cancer cell lines and the inhibition of protein tyrosine phosphatase 1B (PTP1B) activity where pterocarpans that showed strong cytotoxicity exhibited potent inhibition to PTP1B activity. Prenylation of the pterocarpan molecules appear to be important pharmacophoric feature for both inhibition of PTP 1B and cytotoxic effect on breast cancer cell lines, while the absence of this moiety is accompanied with diminished activity in both assay systems. Prenylation at position C-4 in the tested pterocarpans seemed to be an important requirement for the potent inhibition of PTP1B activity, where compounds 15, 16 and 11 share the same feature and showed IC50 (4.2, 6.4, 7.3 µM) respectively. On the other hand, pterocarpans with prenylation at C-2 showed lower inhibitory effect, whereas neorautenol (compound 18; Figure 10), folitenol (compound 17; Figure 10) and erysubin E (compound 19; Figure 10) showed IC50 values of 7.6, 7.8, 8.8 µM, respectively. The interesting point here is hydroxylation of C-6 of compound 18 diminishes its cytotoxicity but keeps its inhibition property of PTP1B. It could be concluded that the inhibition of PTP1B is supposed to be related to breast carcinogenesis inhibition, and the selective inhibition of PTP1B is emerging as a new strategy for the treatment of breast cancer | [69] | |
| Neorautenol and phaseollin | (HAII4) rat hepatoma cell line | Both compounds possess potent cytotoxicity with EC50 1, 1.5 µM respectively. The analysis of their mechanism of cytotoxicity indicates that both compounds significantly increased the activity of caspase 3/7 enzymes, at concentration of 1, 2 µM respectively, and the amount of fragmented nuclei also increased, which are signs of apoptosis. Also, it was found that both compounds disrupted the cell membrane of (HAII4) cells which also indicates the ability of induction necrosis. Furthermore, it was found that Neorautenol significantly breaks DNA strands while phaseollin showed no activity | [70] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Selim, K.A.; Abdelrasoul, H.; Aboelmagd, M.; Tawila, A.M. The Role of the MAPK Signaling, Topoisomerase and Dietary Bioactives in Controlling Cancer Incidence. Diseases 2017, 5, 13. https://doi.org/10.3390/diseases5020013
Selim KA, Abdelrasoul H, Aboelmagd M, Tawila AM. The Role of the MAPK Signaling, Topoisomerase and Dietary Bioactives in Controlling Cancer Incidence. Diseases. 2017; 5(2):13. https://doi.org/10.3390/diseases5020013
Chicago/Turabian StyleSelim, Khaled A., Hend Abdelrasoul, Mohamed Aboelmagd, and Ahmed M. Tawila. 2017. "The Role of the MAPK Signaling, Topoisomerase and Dietary Bioactives in Controlling Cancer Incidence" Diseases 5, no. 2: 13. https://doi.org/10.3390/diseases5020013
APA StyleSelim, K. A., Abdelrasoul, H., Aboelmagd, M., & Tawila, A. M. (2017). The Role of the MAPK Signaling, Topoisomerase and Dietary Bioactives in Controlling Cancer Incidence. Diseases, 5(2), 13. https://doi.org/10.3390/diseases5020013