Epigenetics and Mechanobiology in Heart Development and Congenital Heart Disease


Abstract
:1. Introduction
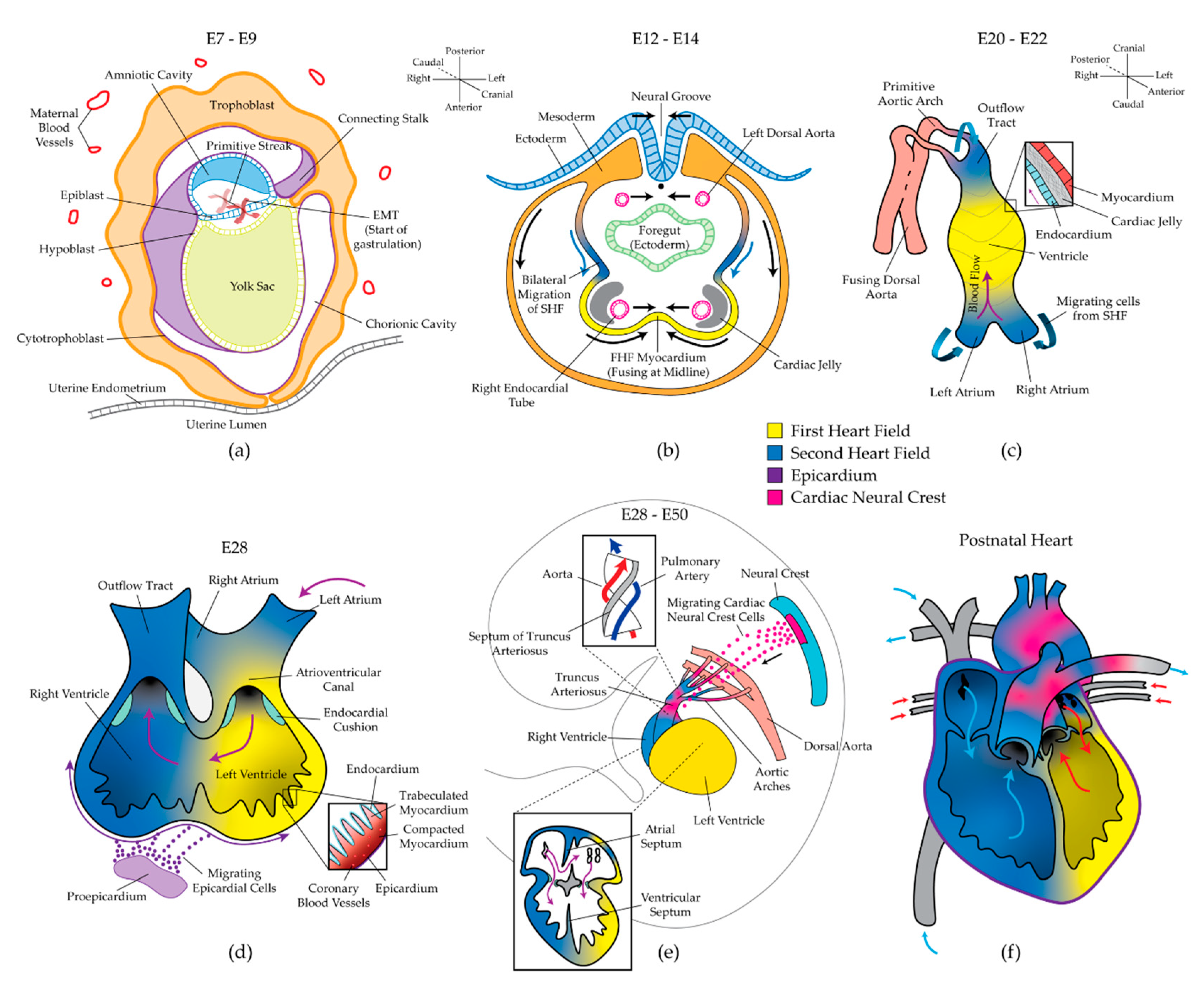
2. Stages of Heart Development and Manifestations of Congenital Heart Disease
2.1. Gastrulation and the Two Heart Fields
2.2. Formation of the Linear Heart Tube
2.3. Cardiac Looping and Chamber Specification
2.4. Septation and Formation of the Valves
2.5. Cardiac Maturation
3. Epigenetics and Congenital Heart Disease
3.1. DNA Methylation
3.2. Histone Modification
3.2.1. Histone Deacetylases in CHD
3.2.2. Polycomb Group Proteins in CHD
3.2.3. Trithorax Group Proteins in CHD
3.3. Non-Coding RNA
miRNA Profiles in CHD Patient Biopsies
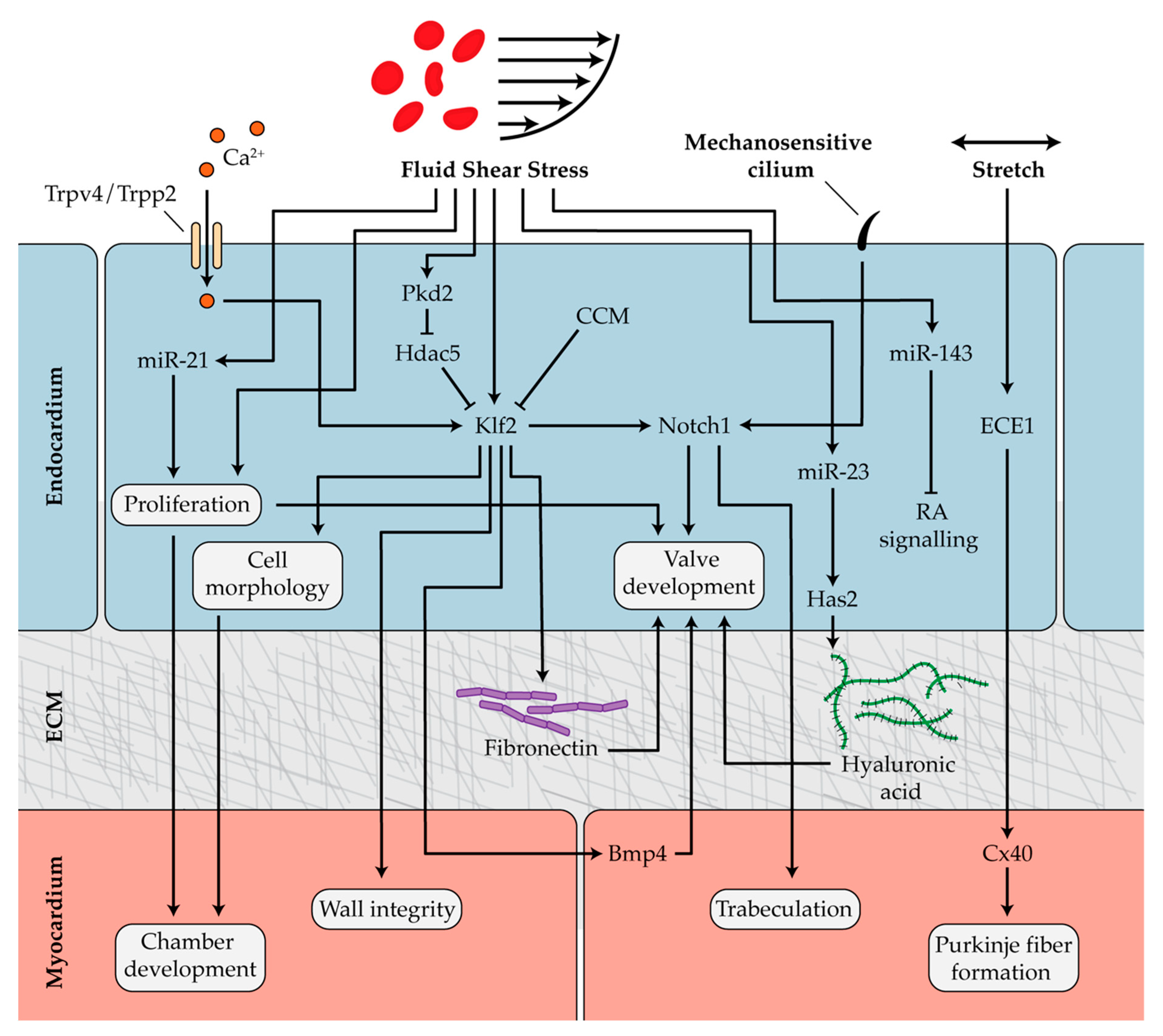
4. Mechanobiology and Cardiac Development
4.1. Mechanosensitive Pathways in Chamber Development and Trabeculation
4.2. Hemodynamics in Endocardial Cushion and Valve Formation
4.3. Mechanotransduction During Formation of the Outflow Tract, Conduction System, and Epicardium
4.4. Mechanical Regulation of Heart Epigenetics
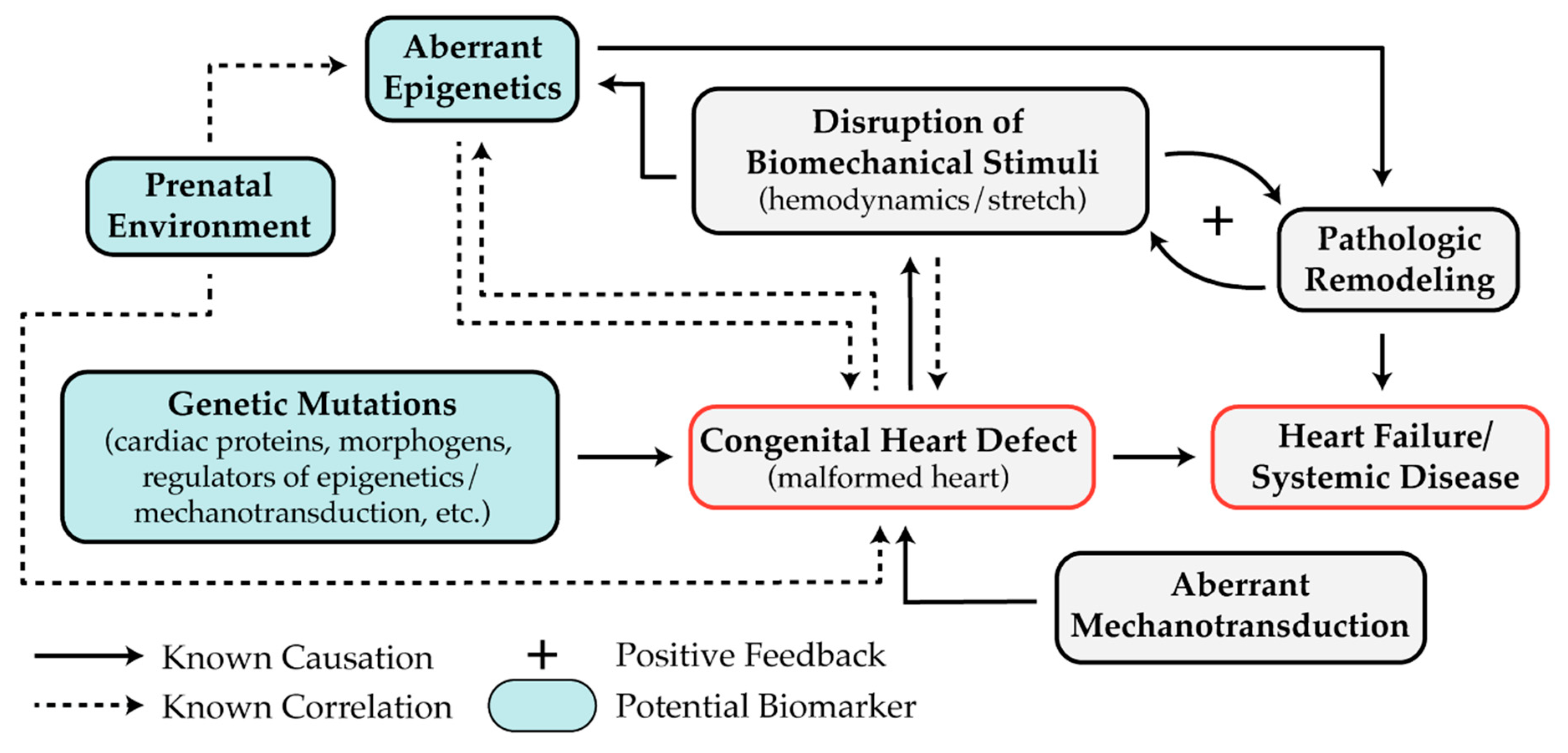
5. Recent Advances
5.1. Epigenetics as Biomarkers for CHD
5.2. Diabetes
6. Discussion
Funding
Conflicts of Interest
References
- Hoffman, J. The global burden of congenital heart disease. Cardiovasc. J. Afr. 2013, 24, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Brennan, J.; Lu, C.C.; Norris, D.P.; Rodriguez, T.A.; Beddington, R.S.; Robertson, E.J. Nodal signalling in the epiblast patterns the early mouse embryo. Nature 2001, 411, 965–969. [Google Scholar] [CrossRef] [PubMed]
- Naito, A.T.; Shiojima, I.; Akazawa, H.; Hidaka, K.; Morisaki, T.; Kikuchi, A.; Komuro, I. Developmental stage-specific biphasic roles of Wnt/beta-catenin signaling in cardiomyogenesis and hematopoiesis. Proc. Natl. Acad. Sci. USA 2006, 103, 19812–19817. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.; Qian, L.; Cheng, P.; Nigam, V.; Arnold, J.; Srivastava, D. A regulatory pathway involving Notch1/beta-catenin/Isl1 determines cardiac progenitor cell fate. Nat. Cell Biol. 2009, 11, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Klaus, A.; Saga, Y.; Taketo, M.M.; Tzahor, E.; Birchmeier, W. Distinct roles of Wnt/beta-catenin and Bmp signaling during early cardiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 18531–18536. [Google Scholar] [CrossRef] [PubMed]
- Prall, O.W.; Menon, M.K.; Solloway, M.J.; Watanabe, Y.; Zaffran, S.; Bajolle, F.; Biben, C.; McBride, J.J.; Robertson, B.R.; Chaulet, H.; et al. An Nkx2-5/Bmp2/Smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell 2007, 128, 947–959. [Google Scholar] [CrossRef]
- Lian, X.; Hsiao, C.; Wilson, G.; Zhu, K.; Hazeltine, L.B.; Azarin, S.M.; Raval, K.K.; Zhang, J.; Kamp, T.J.; Palecek, S.P. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1848–E1857. [Google Scholar] [CrossRef]
- Srivastava, D. Genetic regulation of cardiogenesis and congenital heart disease. Annu. Rev. Pathol. 2006, 1, 199–213. [Google Scholar] [CrossRef]
- Paige, S.L.; Plonowska, K.; Xu, A.; Wu, S.M. Molecular regulation of cardiomyocyte differentiation. Circ. Res. 2015, 116, 341–353. [Google Scholar] [CrossRef]
- Esposito, G.; Butler, T.L.; Blue, G.M.; Cole, A.D.; Sholler, G.F.; Kirk, E.P.; Grossfeld, P.; Perryman, B.M.; Harvey, R.P.; Winlaw, D.S. Somatic mutations in NKX2-5, GATA4, and HAND1 are not a common cause of tetralogy of Fallot or hypoplastic left heart. Am. J. Med. Genet. A 2011, 155A, 2416–2421. [Google Scholar] [CrossRef]
- Yuan, S.; Zaidi, S.; Brueckner, M. Congenital heart disease: emerging themes linking genetics and development. Curr. Opin. Genet. Dev. 2013, 23, 352–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecoli, C.; Pulignani, S.; Foffa, I.; Andreassi, M.G. Congenital heart disease: The crossroads of genetics, epigenetics and environment. Curr. Genomics 2014, 15, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Gittenberger-de Groot, A.C.; Calkoen, E.E.; Poelmann, R.E.; Bartelings, M.M.; Jongbloed, M.R. Morphogenesis and molecular considerations on congenital cardiac septal defects. Ann. Med. 2014, 46, 640–652. [Google Scholar] [CrossRef]
- Wang, X.; Li, P.; Chen, S.; Xi, L.; Guo, Y.; Guo, A.; Sun, K. Influence of genes and the environment in familial congenital heart defects. Mol. Med. Rep. 2014, 9, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Lalani, S.R.; Belmont, J.W. Genetic basis of congenital cardiovascular malformations. Eur. J. Med. Genet. 2014, 57, 402–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, T.A.; Troelsen Kde, L.; Larsen, L.A. Of mice and men: molecular genetics of congenital heart disease. Cell Mol. Life Sci. 2014, 71, 1327–1352. [Google Scholar] [CrossRef] [PubMed]
- Bahado-Singh, R.; Vishweswaraiah, S.; Mishra, N.K.; Guda, C.; Radhakrishna, U. Placental DNA methylation changes for the detection of tetralogy of Fallot. Ultrasound Obstet. Gynecol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Klena, N.T.; Gabriel, G.C.; Liu, X.; Kim, A.J.; Lemke, K.; Chen, Y.; Chatterjee, B.; Devine, W.; Damerla, R.R.; et al. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature 2015, 521, 520–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinberg, A.P. The Key Role of Epigenetics in Human Disease Prevention and Mitigation. N. Engl. J. Med. 2018, 378, 1323–1334. [Google Scholar] [CrossRef]
- Hove, J.R.; Koster, R.W.; Forouhar, A.S.; Acevedo-Bolton, G.; Fraser, S.E.; Gharib, M. Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature 2003, 421, 172–177. [Google Scholar] [CrossRef]
- Tobita, K.; Keller, B.B. Right and left ventricular wall deformation patterns in normal and left heart hypoplasia chick embryos. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H959–H969. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, B.G. Signaling and transcriptional networks in heart development and regeneration. Cold Spring Harb. Perspect. Biol. 2013, 5, a008292. [Google Scholar] [CrossRef] [PubMed]
- Santini, M.P.; Forte, E.; Harvey, R.P.; Kovacic, J.C. Developmental origin and lineage plasticity of endogenous cardiac stem cells. Development 2016, 143, 1242–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, D. Making or breaking the heart: from lineage determination to morphogenesis. Cell 2006, 126, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Lescroart, F.; Chabab, S.; Lin, X.; Rulands, S.; Paulissen, C.; Rodolosse, A.; Auer, H.; Achouri, Y.; Dubois, C.; Bondue, A.; et al. Early lineage restriction in temporally distinct populations of Mesp1 progenitors during mammalian heart development. Nat. Cell Biol. 2014, 16, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Meilhac, S.M.; Esner, M.; Kelly, R.G.; Nicolas, J.F.; Buckingham, M.E. The clonal origin of myocardial cells in different regions of the embryonic mouse heart. Dev. Cell 2004, 6, 685–698. [Google Scholar] [CrossRef]
- van Wijk, B.; van den Berg, G.; Abu-Issa, R.; Barnett, P.; van der Velden, S.; Schmidt, M.; Ruijter, J.M.; Kirby, M.L.; Moorman, A.F.; van den Hoff, M.J. Epicardium and myocardium separate from a common precursor pool by crosstalk between bone morphogenetic protein- and fibroblast growth factor-signaling pathways. Circ. Res. 2009, 105, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Crucean, A.; Alqahtani, A.; Barron, D.J.; Brawn, W.J.; Richardson, R.V.; O’Sullivan, J.; Anderson, R.H.; Henderson, D.J.; Chaudhry, B. Re-evaluation of hypoplastic left heart syndrome from a developmental and morphological perspective. Orphanet J. Rare Dis. 2017, 12, 138. [Google Scholar] [CrossRef]
- Cai, C.L.; Liang, X.; Shi, Y.; Chu, P.H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef]
- Liang, X.; Wang, G.; Lin, L.; Lowe, J.; Zhang, Q.; Bu, L.; Chen, Y.; Chen, J.; Sun, Y.; Evans, S.M. HCN4 dynamically marks the first heart field and conduction system precursors. Circ. Res. 2013, 113, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; Black, B.L. Development of the endocardium. Pediatr. Cardiol. 2010, 31, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Zhang, Z.; Lui, W.; Chen, X.; Wang, Y.; Chamberlain, A.A.; Moreno-Rodriguez, R.A.; Markwald, R.R.; O’Rourke, B.P.; Sharp, D.J.; et al. Endocardial cells form the coronary arteries by angiogenesis through myocardial-endocardial VEGF signaling. Cell 2012, 151, 1083–1096. [Google Scholar] [CrossRef] [PubMed]
- Luxan, G.; D′Amato, G.; MacGrogan, D.; de la Pompa, J.L. Endocardial Notch Signaling in Cardiac Development and Disease. Circ. Res. 2016, 118, e1–e18. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.; Abdelilah-Seyfried, S. The force within: endocardial development, mechanotransduction and signalling during cardiac morphogenesis. Development 2016, 143, 373–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.J.; Lin, C.Y.; Chen, C.H.; Zhou, B.; Chang, C.P. Partitioning the heart: mechanisms of cardiac septation and valve development. Development 2012, 139, 3277–3299. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wu, B.; Chamberlain, A.A.; Lui, W.; Koirala, P.; Susztak, K.; Klein, D.; Taylor, V.; Zhou, B. Endocardial to myocardial notch-wnt-bmp axis regulates early heart valve development. PLoS ONE 2013, 8, e60244. [Google Scholar] [CrossRef] [PubMed]
- Manner, J.; Yelbuz, T.M. Functional Morphology of the Cardiac Jelly in the Tubular Heart of Vertebrate Embryos. J. Cardiovasc. Dev. Dis. 2019, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, M.; Wirrig, E.; Phelps, A.; Wessels, A. Extracellular matrix and heart development. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Camenisch, T.D.; Biesterfeldt, J.; Brehm-Gibson, T.; Bradley, J.; McDonald, J.A. Regulation of cardiac cushion development by hyaluronan. Exp. Clin. Cardiol. 2001, 6, 4–10. [Google Scholar]
- Manner, J.; Wessel, A.; Yelbuz, T.M. How does the tubular embryonic heart work? Looking for the physical mechanism generating unidirectional blood flow in the valveless embryonic heart tube. Dev. Dyn. 2010, 239, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Meilhac, S.M.; Kelly, R.G.; Rocancourt, D.; Eloy-Trinquet, S.; Nicolas, J.F.; Buckingham, M.E. A retrospective clonal analysis of the myocardium reveals two phases of clonal growth in the developing mouse heart. Development 2003, 130, 3877–3889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desgrange, A.; Le Garrec, J.F.; Meilhac, S.M. Left-right asymmetry in heart development and disease: forming the right loop. Development 2018, 145, dev162776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoes, F.C.; Riley, P.R. The ontogeny, activation and function of the epicardium during heart development and regeneration. Development 2018, 145, dev155994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smits, A.M.; Dronkers, E.; Goumans, M.J. The epicardium as a source of multipotent adult cardiac progenitor cells: Their origin, role and fate. Pharmacol. Res. 2018, 127, 129–140. [Google Scholar] [CrossRef]
- Jiang, X.; Rowitch, D.H.; Soriano, P.; McMahon, A.P.; Sucov, H.M. Fate of the mammalian cardiac neural crest. Development 2000, 127, 1607–1616. [Google Scholar] [PubMed]
- Foglia, M.J.; Poss, K.D. Building and re-building the heart by cardiomyocyte proliferation. Development 2016, 143, 729–740. [Google Scholar] [CrossRef] [Green Version]
- Sedmera, D.; Pexieder, T.; Vuillemin, M.; Thompson, R.P.; Anderson, R.H. Developmental patterning of the myocardium. Anat. Rec. 2000, 258, 319–337. [Google Scholar] [CrossRef]
- Risebro, C.A.; Riley, P.R. Formation of the ventricles. Sci. World J. 2006, 6, 1862–1880. [Google Scholar] [CrossRef]
- Sedmera, D.; Thompson, R.P. Myocyte proliferation in the developing heart. Dev. Dyn. 2011, 240, 1322–1334. [Google Scholar] [CrossRef] [Green Version]
- Samsa, L.A.; Yang, B.; Liu, J. Embryonic cardiac chamber maturation: Trabeculation, conduction, and cardiomyocyte proliferation. Am. J. Med. Genet. C Semin Med. Genet. 2013, 163C, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Minot, C.S. On a Hitherto Unrecognized Form of Blood Circulation without Capillaries in the Organs of Vertebrates. J. Boston Soc. Med. Sci. 1900, 4, 133–134. [Google Scholar] [PubMed]
- Rentschler, S.; Vaidya, D.M.; Tamaddon, H.; Degenhardt, K.; Sassoon, D.; Morley, G.E.; Jalife, J.; Fishman, G.I. Visualization and functional characterization of the developing murine cardiac conduction system. Development 2001, 128, 1785–1792. [Google Scholar] [PubMed]
- Leach, J.P.; Martin, J.F. Cardiomyocyte Proliferation for Therapeutic Regeneration. Curr. Cardiol. Rep. 2018, 20, 63. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.F.; Serpooshan, V.; Morris, V.B.; Sayed, N.; Pardon, G.; Abilez, O.J.; Nakayama, K.H.; Pruitt, B.L.; Wu, S.M.; Yoon, Y.S.; et al. Big bottlenecks in cardiovascular tissue engineering. Commun. Biol. 2018, 1, 199. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A. Left ventricular noncompaction: a new form of heart failure. Heart Fail. Clin. 2010, 6, 453–469. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.K.; Bader, D.M. Signals from both sides: Control of cardiac development by the endocardium and epicardium. Semin. Cell Dev. Biol. 2007, 18, 84–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuckmann, I.; Evans, S.; Lassar, A.B. Erythropoietin and retinoic acid, secreted from the epicardium, are required for cardiac myocyte proliferation. Dev. Biol. 2003, 255, 334–349. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Chang, T.C.; Kang, J.O.; Choudhary, B.; Makita, T.; Tran, C.M.; Burch, J.B.; Eid, H.; Sucov, H.M. Epicardial induction of fetal cardiomyocyte proliferation via a retinoic acid-inducible trophic factor. Dev. Biol. 2002, 250, 198–207. [Google Scholar] [CrossRef]
- Li, P.; Cavallero, S.; Gu, Y.; Chen, T.H.; Hughes, J.; Hassan, A.B.; Bruning, J.C.; Pashmforoush, M.; Sucov, H.M. IGF signaling directs ventricular cardiomyocyte proliferation during embryonic heart development. Development 2011, 138, 1795–1805. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Gu, Y.; Li, P.; Johnson, B.L.; Sucov, H.M.; Thomas, P.S. PDGF-A as an epicardial mitogen during heart development. Dev. Dyn. 2008, 237, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Asai, R.; Kurihara, Y.; Fujisawa, K.; Sato, T.; Kawamura, Y.; Kokubo, H.; Tonami, K.; Nishiyama, K.; Uchijima, Y.; Miyagawa-Tomita, S.; et al. Endothelin receptor type A expression defines a distinct cardiac subdomain within the heart field and is later implicated in chamber myocardium formation. Development 2010, 137, 3823–3833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collesi, C.; Zentilin, L.; Sinagra, G.; Giacca, M. Notch1 signaling stimulates proliferation of immature cardiomyocytes. J. Cell Biol. 2008, 183, 117–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A.; et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 2014, 157, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Bassat, E.; Mutlak, Y.E.; Genzelinakh, A.; Shadrin, I.Y.; Baruch Umansky, K.; Yifa, O.; Kain, D.; Rajchman, D.; Leach, J.; Riabov Bassat, D.; et al. The extracellular matrix protein agrin promotes heart regeneration in mice. Nature 2017, 547, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Ho, B.X.; Pang, J.K.S.; Pek, N.M.Q.; Hor, J.H.; Ng, S.Y.; Soh, B.S. Wnt/beta-catenin-mediated signaling re-activates proliferation of matured cardiomyocytes. Stem Cell Res. Ther. 2018, 9, 338. [Google Scholar] [CrossRef]
- D’Addario, C.; Di Francesco, A.; Pucci, M.; Finazzi Agro, A.; Maccarrone, M. Epigenetic mechanisms and endocannabinoid signalling. FEBS J. 2013, 280, 1905–1917. [Google Scholar] [CrossRef]
- Joosten, S.C.; Smits, K.M.; Aarts, M.J.; Melotte, V.; Koch, A.; Tjan-Heijnen, V.C.; van Engeland, M. Epigenetics in renal cell cancer: mechanisms and clinical applications. Nat. Rev. Urol. 2018, 15, 430–451. [Google Scholar] [CrossRef]
- Jones, P.A. The DNA methylation paradox. Trends Genet. 1999, 15, 34–37. [Google Scholar] [CrossRef]
- Takai, D.; Jones, P.A. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. USA 2002, 99, 3740–3745. [Google Scholar] [CrossRef] [Green Version]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurdyukov, S.; Bullock, M. DNA Methylation Analysis: Choosing the Right Method. Biology (Basel) 2016, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, A.A.; Lin, M.; Lister, R.L.; Maslov, A.A.; Wang, Y.; Suzuki, M.; Wu, B.; Greally, J.M.; Zheng, D.; Zhou, B. DNA methylation is developmentally regulated for genes essential for cardiogenesis. J. Am. Heart Assoc. 2014, 3, e000976. [Google Scholar] [CrossRef] [PubMed]
- Yin, A.; Feng, M.; Cheng, Z.; Zhang, Q.; Li, H.; Xu, J.; Zhang, H.; Li, Y.; Qian, L. Altered DNA Methylation of Long Noncoding RNA uc.167 Inhibits Cell Differentiation in Heart Development. Biomed. Res. Int. 2018, 2018, 4658024. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishna, U.; Albayrak, S.; Alpay-Savasan, Z.; Zeb, A.; Turkoglu, O.; Sobolewski, P.; Bahado-Singh, R.O. Genome-Wide DNA Methylation Analysis and Epigenetic Variations Associated with Congenital Aortic Valve Stenosis (AVS). PLoS ONE 2016, 11, e0154010. [Google Scholar] [CrossRef]
- Grunert, M.; Dorn, C.; Cui, H.; Dunkel, I.; Schulz, K.; Schoenhals, S.; Sun, W.; Berger, F.; Chen, W.; Sperling, S.R. Comparative DNA methylation and gene expression analysis identifies novel genes for structural congenital heart diseases. Cardiovasc. Res. 2016, 112, 464–477. [Google Scholar] [CrossRef]
- Sheng, W.; Qian, Y.; Wang, H.; Ma, X.; Zhang, P.; Diao, L.; An, Q.; Chen, L.; Ma, D.; Huang, G. DNA methylation status of NKX2-5, GATA4 and HAND1 in patients with tetralogy of fallot. BMC Med. Genomics 2013, 6, 46. [Google Scholar] [CrossRef]
- Lyu, G.; Zhang, C.; Ling, T.; Liu, R.; Zong, L.; Guan, Y.; Huang, X.; Sun, L.; Zhang, L.; Li, C.; et al. Genome and epigenome analysis of monozygotic twins discordant for congenital heart disease. BMC Genomics 2018, 19, 428. [Google Scholar] [CrossRef]
- Nimura, K.; Ura, K.; Shiratori, H.; Ikawa, M.; Okabe, M.; Schwartz, R.J.; Kaneda, Y. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature 2009, 460, 287–291. [Google Scholar] [CrossRef]
- Mysliwiec, M.R.; Carlson, C.D.; Tietjen, J.; Hung, H.; Ansari, A.Z.; Lee, Y. Jarid2 (Jumonji, AT rich interactive domain 2) regulates NOTCH1 expression via histone modification in the developing heart. J. Biol. Chem. 2012, 287, 1235–1241. [Google Scholar] [CrossRef]
- Trivedi, C.M.; Zhu, W.; Wang, Q.; Jia, C.; Kee, H.J.; Li, L.; Hannenhalli, S.; Epstein, J.A. Hopx and Hdac2 interact to modulate Gata4 acetylation and embryonic cardiac myocyte proliferation. Dev. Cell 2010, 19, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, S.L.; Janardhan, H.P.; Smee, K.M.; Bachman, M.; Sun, Z.; Lazar, M.A.; Trivedi, C.M. Histone deacetylase 3 modulates Tbx5 activity to regulate early cardiogenesis. Hum. Mol. Genet. 2014, 23, 3801–3809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ow, J.R.; Palanichamy Kala, M.; Rao, V.K.; Choi, M.H.; Bharathy, N.; Taneja, R. G9a inhibits MEF2C activity to control sarcomere assembly. Sci. Rep. 2016, 6, 34163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Just, S.; Berger, I.M.; Meder, B.; Backs, J.; Keller, A.; Marquart, S.; Frese, K.; Patzel, E.; Rauch, G.J.; Tubingen Screen, C.; et al. Protein kinase D2 controls cardiac valve formation in zebrafish by regulating histone deacetylase 5 activity. Circulation 2011, 124, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Sims, R.J.; Weihe, E.K.; Zhu, L.; O’Malley, S.; Harriss, J.V.; Gottlieb, P.D. m-Bop, a repressor protein essential for cardiogenesis, interacts with skNAC, a heart- and muscle-specific transcription factor. J. Biol. Chem. 2002, 277, 26524–26529. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Rotllant, J.; Li, H.; De Deyne, P.; Du, S.J. SmyD1, a histone methyltransferase, is required for myofibril organization and muscle contraction in zebrafish embryos. Proc. Natl. Acad. Sci. USA 2006, 103, 2713–2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, T.L.; Ma, Y.; Park, C.Y.; Harriss, J.; Pierce, S.A.; Dekker, J.D.; Valenzuela, N.; Srivastava, D.; Schwartz, R.J.; Stewart, M.D.; et al. Smyd1 facilitates heart development by antagonizing oxidative and ER stress responses. PLoS ONE 2015, 10, e0121765. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ma, Y.; Kim, E.Y.; Yu, W.; Schwartz, R.J.; Qian, L.; Wang, J. Conditional ablation of Ezh2 in murine hearts reveals its essential roles in endocardial cushion formation, cardiomyocyte proliferation and survival. PLoS ONE 2012, 7, e31005. [Google Scholar] [CrossRef]
- He, H.H.; Meyer, C.A.; Shin, H.; Bailey, S.T.; Wei, G.; Wang, Q.; Zhang, Y.; Xu, K.; Ni, M.; Lupien, M.; et al. Nucleosome dynamics define transcriptional enhancers. Nat. Genet. 2010, 42, 343–347. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, J.; Yoshida, M.; Tarui, S.; Hirata, M.; Nagai, Y.; Kasahara, S.; Naruse, K.; Ito, H.; Sano, S.; Oh, H. Directed differentiation of patient-specific induced pluripotent stem cells identifies the transcriptional repression and epigenetic modification of NKX2-5, HAND1, and NOTCH1 in hypoplastic left heart syndrome. PLoS ONE 2014, 9, e102796. [Google Scholar] [CrossRef]
- Ohtani, K.; Zhao, C.; Dobreva, G.; Manavski, Y.; Kluge, B.; Braun, T.; Rieger, M.A.; Zeiher, A.M.; Dimmeler, S. Jmjd3 controls mesodermal and cardiovascular differentiation of embryonic stem cells. Circ. Res. 2013, 113, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, J.W.; Lee, S.K. UTX, a histone H3-lysine 27 demethylase, acts as a critical switch to activate the cardiac developmental program. Dev. Cell 2012, 22, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Liu, L.; Ding, X.; Zhou, P.; Yuan, X.; Zhou, Z.; Hu, P.; Zhou, H.; Li, Q.; Zhang, S.; et al. Mll2 controls cardiac lineage differentiation of mouse embryonic stem cells by promoting H3K4me3 deposition at cardiac-specific genes. Stem Cell Rev. 2014, 10, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.B.; Bigham, A.W.; Buckingham, K.J.; Hannibal, M.C.; McMillin, M.J.; Gildersleeve, H.I.; Beck, A.E.; Tabor, H.K.; Cooper, G.M.; Mefford, H.C.; et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010, 42, 790–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lickert, H.; Takeuchi, J.K.; Von Both, I.; Walls, J.R.; McAuliffe, F.; Adamson, S.L.; Henkelman, R.M.; Wrana, J.L.; Rossant, J.; Bruneau, B.G. Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature 2004, 432, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, J.K.; Lou, X.; Alexander, J.M.; Sugizaki, H.; Delgado-Olguin, P.; Holloway, A.K.; Mori, A.D.; Wylie, J.N.; Munson, C.; Zhu, Y.; et al. Chromatin remodelling complex dosage modulates transcription factor function in heart development. Nat. Commun. 2011, 2, 187. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Fulcoli, F.G.; Ferrentino, R.; Martucciello, S.; Illingworth, E.A.; Baldini, A. Transcriptional control in cardiac progenitors: Tbx1 interacts with the BAF chromatin remodeling complex and regulates Wnt5a. PLoS Genet. 2012, 8, e1002571. [Google Scholar] [CrossRef]
- Lange, M.; Kaynak, B.; Forster, U.B.; Tonjes, M.; Fischer, J.J.; Grimm, C.; Schlesinger, J.; Just, S.; Dunkel, I.; Krueger, T.; et al. Regulation of muscle development by DPF3, a novel histone acetylation and methylation reader of the BAF chromatin remodeling complex. Genes Dev. 2008, 22, 2370–2384. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Samal, E.; Srivastava, D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 2005, 436, 214–220. [Google Scholar] [CrossRef]
- Wu, Y.; Ma, X.J.; Wang, H.J.; Li, W.C.; Chen, L.; Ma, D.; Huang, G.Y. Expression of Cx43-related microRNAs in patients with tetralogy of Fallot. World J. Pediatr. 2014, 10, 138–144. [Google Scholar] [CrossRef]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Olson, E.N. MEF2: A central regulator of diverse developmental programs. Development 2007, 134, 4131–4140. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Young, A.G.; Winslow, M.M.; Lintault, L.; Meissner, A.; Erkeland, S.J.; Newman, J.; Bronson, R.T.; Crowley, D.; Stone, J.R.; et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008, 132, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Liu, Z.; Zhou, L. Roles of miR-17-92 Cluster in Cardiovascular Development and Common Diseases. Biomed. Res. Int. 2017, 2017, 9102909. [Google Scholar] [CrossRef] [PubMed]
- Banjo, T.; Grajcarek, J.; Yoshino, D.; Osada, H.; Miyasaka, K.Y.; Kida, Y.S.; Ueki, Y.; Nagayama, K.; Kawakami, K.; Matsumoto, T.; et al. Haemodynamically dependent valvulogenesis of zebrafish heart is mediated by flow-dependent expression of miR-21. Nat. Commun. 2013, 4, 1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagendijk, A.K.; Goumans, M.J.; Burkhard, S.B.; Bakkers, J. MicroRNA-23 restricts cardiac valve formation by inhibiting Has2 and extracellular hyaluronic acid production. Circ. Res. 2011, 109, 649–657. [Google Scholar] [CrossRef]
- Stauffer, B.L.; Russell, G.; Nunley, K.; Miyamoto, S.D.; Sucharov, C.C. miRNA expression in pediatric failing human heart. J. Mol. Cell. Cardiol. 2013, 57, 43–46. [Google Scholar] [CrossRef] [Green Version]
- Sucharov, C.C.; Sucharov, J.; Karimpour-Fard, A.; Nunley, K.; Stauffer, B.L.; Miyamoto, S.D. Micro-RNA expression in hypoplastic left heart syndrome. J. Cardiac Fail. 2015, 21, 83–88. [Google Scholar] [CrossRef]
- Duisters, R.F.; Tijsen, A.J.; Schroen, B.; Leenders, J.J.; Lentink, V.; van der Made, I.; Herias, V.; van Leeuwen, R.E.; Schellings, M.W.; Barenbrug, P.; et al. miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ. Res. 2009, 104, 170–178. [Google Scholar] [CrossRef]
- Li, D.; Ji, L.; Liu, L.; Liu, Y.; Hou, H.; Yu, K.; Sun, Q.; Zhao, Z. Characterization of circulating microRNA expression in patients with a ventricular septal defect. PLoS ONE 2014, 9, e106318. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chang, J.J.; Xu, F.; Ma, X.J.; Wu, Y.; Li, W.C.; Wang, H.J.; Huang, G.Y.; Ma, D. MicroRNA deregulation in right ventricular outflow tract myocardium in nonsyndromic tetralogy of fallot. Can. J. Cardiol. 2013, 29, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- Bittel, D.C.; Kibiryeva, N.; Marshall, J.A.; O’Brien, J.E. MicroRNA-421 Dysregulation is Associated with Tetralogy of Fallot. Cells 2014, 3, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Xu, X.; Deng, F.; Feng, J.; Zhang, H.; Liu, Y.; Zhang, Y.; Pan, L.; Liu, Y.; Zhang, D.; et al. miRNA-940 reduction contributes to human Tetralogy of Fallot development. J. Cell Mol. Med. 2014, 18, 1830–1839. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, K.Y.; Kida, Y.S.; Banjo, T.; Ueki, Y.; Nagayama, K.; Matsumoto, T.; Sato, M.; Ogura, T. Heartbeat regulates cardiogenesis by suppressing retinoic acid signaling via expression of miR-143. Mech. Dev. 2011, 128, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Gilsbach, R.; Preissl, S.; Gruning, B.A.; Schnick, T.; Burger, L.; Benes, V.; Wurch, A.; Bonisch, U.; Gunther, S.; Backofen, R.; et al. Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat. Commun. 2014, 5, 5288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Lin, S.; Garcia, B.A.; Zhao, Y. Quantitative proteomic analysis of histone modifications. Chem. Rev. 2015, 115, 2376–2418. [Google Scholar] [CrossRef]
- Mysliwiec, M.R.; Bresnick, E.H.; Lee, Y. Endothelial Jarid2/Jumonji is required for normal cardiac development and proper Notch1 expression. J. Biol. Chem. 2011, 286, 17193–17204. [Google Scholar] [CrossRef]
- Blakeslee, W.W.; Demos-Davies, K.M.; Lemon, D.D.; Lutter, K.M.; Cavasin, M.A.; Payne, S.; Nunley, K.; Long, C.S.; McKinsey, T.A.; Miyamoto, S.D. Histone deacetylase adaptation in single ventricle heart disease and a young animal model of right ventricular hypertrophy. Pediatr. Res. 2017, 82, 642–649. [Google Scholar] [CrossRef] [Green Version]
- McKinsey, T.A. Therapeutic potential for HDAC inhibitors in the heart. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 303–319. [Google Scholar] [CrossRef]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Kolanowska, M.; Kubiak, A.; Jazdzewski, K.; Wojcicka, A. MicroRNA Analysis Using Next-Generation Sequencing. Methods Mol. Biol. (Clifton, N.J.) 2018, 1823, 87–101. [Google Scholar]
- Wang, J.; Liu, S.; Heallen, T.; Martin, J.F. The Hippo pathway in the heart: pivotal roles in development, disease, and regeneration. Nat. Rev. Cardiol. 2018, 15, 672–684. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.E., Jr.; Kibiryeva, N.; Zhou, X.G.; Marshall, J.A.; Lofland, G.K.; Artman, M.; Chen, J.; Bittel, D.C. Noncoding RNA expression in myocardium from infants with tetralogy of Fallot. Circ. Cardiovasc. Genet. 2012, 5, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Midgett, M.; Rugonyi, S. Congenital heart malformations induced by hemodynamic altering surgical interventions. Front. Physiol. 2014, 5, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auman, H.J.; Coleman, H.; Riley, H.E.; Olale, F.; Tsai, H.J.; Yelon, D. Functional modulation of cardiac form through regionally confined cell shape changes. PLoS Biol. 2007, 5, e53. [Google Scholar] [CrossRef]
- Stainier, D.Y.; Weinstein, B.M.; Detrich, H.W., 3rd; Zon, L.I.; Fishman, M.C. Cloche, an early acting zebrafish gene, is required by both the endothelial and hematopoietic lineages. Development 1995, 121, 3141–3150. [Google Scholar]
- Holtzman, N.G.; Schoenebeck, J.J.; Tsai, H.J.; Yelon, D. Endocardium is necessary for cardiomyocyte movement during heart tube assembly. Development 2007, 134, 2379–2386. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, A.C.; Lombardo, V.A.; Veerkamp, J.; Priller, F.; Abdelilah-Seyfried, S. Blood flow and Bmp signaling control endocardial chamber morphogenesis. Dev. Cell 2014, 30, 367–377. [Google Scholar] [CrossRef]
- Rasouli, S.J.; El-Brolosy, M.; Tsedeke, A.T.; Bensimon-Brito, A.; Ghanbari, P.; Maischein, H.M.; Kuenne, C.; Stainier, D.Y. The flow responsive transcription factor Klf2 is required for myocardial wall integrity by modulating Fgf signaling. Elife 2018, 7, e38889. [Google Scholar] [CrossRef] [PubMed]
- Peshkovsky, C.; Totong, R.; Yelon, D. Dependence of cardiac trabeculation on neuregulin signaling and blood flow in zebrafish. Dev. Dyn. 2011, 240, 446–456. [Google Scholar] [CrossRef]
- Slough, J.; Cooney, L.; Brueckner, M. Monocilia in the embryonic mouse heart suggest a direct role for cilia in cardiac morphogenesis. Dev. Dyn. 2008, 237, 2304–2314. [Google Scholar] [CrossRef] [PubMed]
- Samsa, L.A.; Givens, C.; Tzima, E.; Stainier, D.Y.; Qian, L.; Liu, J. Cardiac contraction activates endocardial Notch signaling to modulate chamber maturation in zebrafish. Development 2015, 142, 4080–4091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messerschmidt, V.; Bailey, Z.; Baek, K.I.; Bryant, R.; Li, R.; Hsiai, T.K.; Lee, J. Light-sheet Fluorescence Microscopy to Capture 4-Dimensional Images of the Effects of Modulating Shear Stress on the Developing Zebrafish Heart. J. Vis. Exp. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bartman, T.; Walsh, E.C.; Wen, K.K.; McKane, M.; Ren, J.; Alexander, J.; Rubenstein, P.A.; Stainier, D.Y. Early myocardial function affects endocardial cushion development in zebrafish. PLoS Biol. 2004, 2, E129. [Google Scholar] [CrossRef]
- Vermot, J.; Forouhar, A.S.; Liebling, M.; Wu, D.; Plummer, D.; Gharib, M.; Fraser, S.E. Reversing blood flows act through klf2a to ensure normal valvulogenesis in the developing heart. PLoS Biol. 2009, 7, e1000246. [Google Scholar] [CrossRef]
- Franco, C.A.; Jones, M.L.; Bernabeu, M.O.; Geudens, I.; Mathivet, T.; Rosa, A.; Lopes, F.M.; Lima, A.P.; Ragab, A.; Collins, R.T.; et al. Dynamic endothelial cell rearrangements drive developmental vessel regression. PLoS Biol. 2015, 13, e1002125. [Google Scholar]
- Boselli, F.; Steed, E.; Freund, J.B.; Vermot, J. Anisotropic shear stress patterns predict the orientation of convergent tissue movements in the embryonic heart. Development 2017, 144, 4322–4327. [Google Scholar] [CrossRef] [Green Version]
- Steed, E.; Faggianelli, N.; Roth, S.; Ramspacher, C.; Concordet, J.P.; Vermot, J. klf2a couples mechanotransduction and zebrafish valve morphogenesis through fibronectin synthesis. Nat. Commun. 2016, 7, 11646. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Rawnsley, D.R.; Goddard, L.M.; Pan, W.; Cao, X.J.; Jakus, Z.; Zheng, H.; Yang, J.; Arthur, J.S.; Whitehead, K.J.; et al. The cerebral cavernous malformation pathway controls cardiac development via regulation of endocardial MEKK3 signaling and KLF expression. Dev. Cell 2015, 32, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Tang, A.T.; Wong, W.Y.; Bamezai, S.; Goddard, L.M.; Shenkar, R.; Zhou, S.; Yang, J.; Wright, A.C.; Foley, M.; et al. Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signalling. Nature 2016, 532, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Renz, M.; Otten, C.; Faurobert, E.; Rudolph, F.; Zhu, Y.; Boulday, G.; Duchene, J.; Mickoleit, M.; Dietrich, A.C.; Ramspacher, C.; et al. Regulation of beta1 integrin-Klf2-mediated angiogenesis by CCM proteins. Dev. Cell 2015, 32, 181–190. [Google Scholar] [CrossRef]
- Maddaluno, L.; Rudini, N.; Cuttano, R.; Bravi, L.; Giampietro, C.; Corada, M.; Ferrarini, L.; Orsenigo, F.; Papa, E.; Boulday, G.; et al. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 2013, 498, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Donat, S.; Lourenco, M.; Paolini, A.; Otten, C.; Renz, M.; Abdelilah-Seyfried, S. Heg1 and Ccm1/2 proteins control endocardial mechanosensitivity during zebrafish valvulogenesis. Elife 2018, 7, e28939. [Google Scholar] [CrossRef] [PubMed]
- Heckel, E.; Boselli, F.; Roth, S.; Krudewig, A.; Belting, H.G.; Charvin, G.; Vermot, J. Oscillatory Flow Modulates Mechanosensitive klf2a Expression through trpv4 and trpp2 during Heart Valve Development. Curr. Biol. CB 2015, 25, 1354–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.J.; Vedula, V.; Baek, K.I.; Chen, C.; Chen, J.; Chou, M.I.; Lam, J.; Subhedar, S.; Wang, J.; Ding, Y.; et al. Contractile and hemodynamic forces coordinate Notch1b-mediated outflow tract valve formation. JCI Insight 2019, 5, e124460. [Google Scholar] [CrossRef]
- Menon, V.; Eberth, J.F.; Goodwin, R.L.; Potts, J.D. Altered Hemodynamics in the Embryonic Heart Affects Outflow Valve Development. J. Cardiovasc. Dev. Dis. 2015, 2, 108–124. [Google Scholar] [CrossRef]
- Yin, J.; Kuebler, W.M. Mechanotransduction by TRP channels: general concepts and specific role in the vasculature. Cell Biochem. Biophys. 2010, 56, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hou, B.; Tumova, S.; Muraki, K.; Bruns, A.; Ludlow, M.J.; Sedo, A.; Hyman, A.J.; McKeown, L.; Young, R.S.; et al. Piezo1 integration of vascular architecture with physiological force. Nature 2014, 515, 279–282. [Google Scholar] [CrossRef]
- Takebayashi-Suzuki, K.; Yanagisawa, M.; Gourdie, R.G.; Kanzawa, N.; Mikawa, T. In vivo induction of cardiac Purkinje fiber differentiation by coexpression of preproendothelin-1 and endothelin converting enzyme-1. Development 2000, 127, 3523–3532. [Google Scholar] [PubMed]
- Hall, C.E.; Hurtado, R.; Hewett, K.W.; Shulimovich, M.; Poma, C.P.; Reckova, M.; Justus, C.; Pennisi, D.J.; Tobita, K.; Sedmera, D.; et al. Hemodynamic-dependent patterning of endothelin converting enzyme 1 expression and differentiation of impulse-conducting Purkinje fibers in the embryonic heart. Development 2004, 131, 581–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bressan, M.C.; Louie, J.D.; Mikawa, T. Hemodynamic forces regulate developmental patterning of atrial conduction. PLoS ONE 2014, 9, e115207. [Google Scholar] [CrossRef] [PubMed]
- Peralta, M.; Steed, E.; Harlepp, S.; Gonzalez-Rosa, J.M.; Monduc, F.; Ariza-Cosano, A.; Cortes, A.; Rayon, T.; Gomez-Skarmeta, J.L.; Zapata, A.; et al. Heartbeat-driven pericardiac fluid forces contribute to epicardium morphogenesis. Curr. Biol. CB 2013, 23, 1726–1735. [Google Scholar] [CrossRef] [PubMed]
- Andres-Delgado, L.; Ernst, A.; Galardi-Castilla, M.; Bazaga, D.; Peralta, M.; Munch, J.; Gonzalez-Rosa, J.M.; Marques, I.; Tessadori, F.; de la Pompa, J.L.; et al. Actin dynamics and the Bmp pathway drive apical extrusion of proepicardial cells. Development 2019. [Google Scholar] [CrossRef]
- Camenisch, T.D.; Spicer, A.P.; Brehm-Gibson, T.; Biesterfeldt, J.; Augustine, M.L.; Calabro, A.; Kubalak, S.; Klewer, S.E.; McDonald, J.A. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J. Clin. Investig. 2000, 106, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Huynh, Q.K.; McKinsey, T.A. Protein kinase D directly phosphorylates histone deacetylase 5 via a random sequential kinetic mechanism. Arch. Biochem. Biophys. 2006, 450, 141–148. [Google Scholar] [CrossRef]
- Chen, L.J.; Wei, S.Y.; Chiu, J.J. Mechanical regulation of epigenetics in vascular biology and pathobiology. J. Cell Mol. Med. 2013, 17, 437–448. [Google Scholar] [CrossRef]
- Wu, W.; Xiao, H.; Laguna-Fernandez, A.; Villarreal, G., Jr.; Wang, K.C.; Geary, G.G.; Zhang, Y.; Wang, W.C.; Huang, H.D.; Zhou, J.; et al. Flow-Dependent Regulation of Kruppel-Like Factor 2 Is Mediated by MicroRNA-92a. Circulation 2011, 124, 633–641. [Google Scholar] [CrossRef]
- van Velzen, C.L.; Clur, S.A.; Rijlaarsdam, M.E.; Bax, C.J.; Pajkrt, E.; Heymans, M.W.; Bekker, M.N.; Hruda, J.; de Groot, C.J.; Blom, N.A.; et al. Prenatal detection of congenital heart disease--results of a national screening programme. BJOG 2016, 123, 400–407. [Google Scholar] [CrossRef]
- Eckersley, L.; Sadler, L.; Parry, E.; Finucane, K.; Gentles, T.L. Timing of diagnosis affects mortality in critical congenital heart disease. Arch. Dis. Child. 2016, 101, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.Q.; Zhou, L.; Chen, Y.; Ni, B. Circulating microRNAs as potential biomarkers for diagnosis of congenital heart defects. World J. Emerg. Med. 2016, 7, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.B.; Han, S.P.; Bai, Y.F.; Zhu, C.; Pan, Y.; Guo, X.R. microRNA expression profiling in fetal single ventricle malformation identified by deep sequencing. Int. J. Mol. Med. 2012, 29, 53–60. [Google Scholar] [PubMed]
- Liu, H.; Hu, Y.; Zhuang, B.; Yin, J.; Chen, X.H.; Wang, J.; Li, M.M.; Xu, J.; Wang, X.Y.; Yu, Z.B.; et al. Differential Expression of CircRNAs in Embryonic Heart Tissue Associated with Ventricular Septal Defect. Int. J. Med. Sci. 2018, 15, 703–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef]
- Liu, Z.Q.; Chen, O.; Zheng, M.; Wang, L.; Zhou, Y.; Yin, C.Y.; Liu, J.D.; Qian, L. Re-patterning of H3K27me3, H3K4me3 and DNA methylation during fibroblast conversion into induced cardiomyocytes. Stem Cell Res. 2016, 16, 507–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, H.; Chen, L.; Xue, J.; Huang, T.; Wei, X.; Liu, D.; Ma, W.; Cao, S.; Yuan, Z. Expression profile of maternal circulating microRNAs as non-invasive biomarkers for prenatal diagnosis of congenital heart defects. Biomed. Pharm. 2019, 109, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Bahado-Singh, R.O.; Zaffra, R.; Albayarak, S.; Chelliah, A.; Bolinjkar, R.; Turkoglu, O.; Radhakrishna, U. Epigenetic markers for newborn congenital heart defect (CHD). J. Matern Fetal Neonatal Med. 2016, 29, 1881–1887. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishna, U.; Vishweswaraiah, S.; Veerappa, A.M.; Zafra, R.; Albayrak, S.; Sitharam, P.H.; Saiyed, N.M.; Mishra, N.K.; Guda, C.; Bahado-Singh, R. Newborn blood DNA epigenetic variations and signaling pathway genes associated with Tetralogy of Fallot (TOF). PLoS ONE 2018, 13, e0203893. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishna, U.; Albayrak, S.; Zafra, R.; Baraa, A.; Vishweswaraiah, S.; Veerappa, A.M.; Mahishi, D.; Saiyed, N.; Mishra, N.K.; Guda, C.; et al. Placental epigenetics for evaluation of fetal congenital heart defects: Ventricular Septal Defect (VSD). PLoS ONE 2019, 14, e0200229. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Joseph, K.S.; Lisonkova, S.; Rouleau, J.; Van den Hof, M.; Sauve, R.; Kramer, M.S.; Canadian Perinatal Surveillance System. Association between maternal chronic conditions and congenital heart defects: A population-based cohort study. Circulation 2013, 128, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Helle, E.I.T.; Biegley, P.; Knowles, J.W.; Leader, J.B.; Pendergrass, S.; Yang, W.; Reaven, G.R.; Shaw, G.M.; Ritchie, M.; Priest, J.R. First Trimester Plasma Glucose Values in Women without Diabetes are Associated with Risk for Congenital Heart Disease in Offspring. J. Pediatric. 2018, 195, 275–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, D.; Fu, N.; Yang, P. MiR-17 Downregulation by High Glucose Stabilizes Thioredoxin-Interacting Protein and Removes Thioredoxin Inhibition on ASK1 Leading to Apoptosis. Toxicol. Sci. 2016, 150, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Maloyan, A.; Muralimanoharan, S.; Huffman, S.; Cox, L.A.; Nathanielsz, P.W.; Myatt, L.; Nijland, M.J. Identification and comparative analyses of myocardial miRNAs involved in the fetal response to maternal obesity. Physiol. Genomics 2013, 45, 889–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, K.V.; Bhatt, A.; Azamian, M.S.; Sundgren, N.C.; Zapata, G.E.; Hernandez, P.; Fox, K.; Kaiser, J.R.; Belmont, J.W.; Hanchard, N.A. Aberrant DNA methylation as a diagnostic biomarker of diabetic embryopathy. Genet. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Satin, J.; Schroder, E.A. Autoregulation of cardiac l-type calcium channels. Trends Cardiovasc. Med. 2009, 19, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Basu, M.; Zhu, J.Y.; LaHaye, S.; Majumdar, U.; Jiao, K.; Han, Z.; Garg, V. Epigenetic mechanisms underlying maternal diabetes-associated risk of congenital heart disease. JCI Insight 2017, 2, e95085. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Zhao, L.; Cai, W.; Wei, M.; Zhou, X.; Yang, G.; Yuan, L. Maternal exosomes in diabetes contribute to the cardiac development deficiency. Biochem. Biophys. Res. Commun. 2017, 483, 602–608. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modifier | Modification | Target Gene(s) | Disease Phenotype | Ref. |
|---|---|---|---|---|
| DNA Methylation | ||||
| DNMT3B | Hypermethylation | HAS2 | - | [73] |
| - | Hypermethylation | APOA5, PCSK9 | AVS | [75] |
| - | Hypermethylation | SCO2 | TOF, VSD | [76] |
| - | Hypermethylation | NKX2.5 *, HAND1 | TOF | [77] |
| - | Hypermethylation | ZIC3, NR2F2 | DORV | [78] |
| lncRNA uc.167 | Hypermethylation | MEF2C | VSD, increased CM apoptosis | [74] |
| Histone Modifications | ||||
| Whsc1 * | H3K36me3 | ↓ NKX2.5 * target genes | ASD, VSD, Wolf-Hirschhorn Syndrome | [79] |
| JARID2/SETDB1 | H3K9me3 | ↑ NOTCH1 * | DORV, LVNC, VSD | [80] |
| HDAC2 * | Deacetylation | ↓ GATA4 * target genes | Impaired cardiac differentiation and proliferation | [81] |
| HDAC3 * | Deacetylation | ↓ TBX5 * target genes | Impaired cardiac differentiation | [82] |
| G9a/HDAC5 | H3K9me2, HDAC5 association | ↓ MEF2A/KLF2A target genes | Sarcomere disorganization, Valve malformation | [83,84] |
| SMyD1 * | H3K4me3 | ↑ SkNAC target loci | HRHS, CM hypoplasia | [85,86,87] |
| Ezh2/PRC2 | H3K27me3, PRC1 recruitment | TBX2, HEY2 | VSD, ASD, impaired endocardium, trabeculae, compaction, proliferation | [88,89] |
| - | ↑ H3K27me3, ↓ H3K4me2 | NKX2.5 *, HAND1, NOTCH1 *, TBX2, HEY | HLHS | [90] |
| Jmjd3 | H3K27 demethylation | ↑ GATA4 *, T, CTNNB1 | Impaired mesoderm/cardiac differentiation | [91] |
| UTX | H3K27 demethylation | ↑ GATA4 *, NKX2.5 *, TBX5 * | Arrested cardiac development after looping | [92] |
| MLL2 * | H3K4me3 | ↑ NKX2.5 *, TBX5 *, MEF2C | Abolished cardiac differentiation from mesoderm | [93,94] |
| Baf60c/Brg1 | ATP-dependent histone-DNA destabilization | ↑ GATA4, TBX5, NKX2.5 | Impaired cardiac differentiation, OFT, and chamber formation | [95,96] |
| TBX1 * | Enhances Baf60a CRC H3K4me | ↑ WNT5A | DORV, DiGeorge syndrome, HRHS, OFT defects | [97] |
| DPF3 | Recruits BAF CRC to H3 and H4 methylation/acetylation sites | ↑ MEF2A | Impaired looping and contractility | [98] |
| miRNA | ||||
| miR-1 | Suppression | HAND2, GJA1, SRF, MEF2, SOX9, HDAC4 | TOF, VSD | [99,100,101,102,103] |
| miR-17~92 | Suppression | ISL-1, TBX1, GJA1, FOG2 | Cardiac progenitor differentiation, VSD, angiogenesis | [104,105] |
| miR-21 | Flow-dependent knockdown | PDCD4, PTENB, SPRY2 | Valve and endocardial malformation | [106] |
| miR-23 | Flow-dependent knockdown | HAS2 | Valve and endocardial malformation | [107] |
| miR-137b/miR-204 | Suppression | GATA4 *, HAND2 | HLHS, pIDC | [108,109] |
| miR-99/miR-100/miR-145 | Ventricular loading-dependent knockdown | QKI, CDK6, SOX11, BAZ2A, FOG2, GATA6 | HLHS | [109] |
| miR-30/miR-133 | Suppression | CTGF, SRF MEF2A | Impaired proliferation, cardiac fibrosis | [110] |
| miR-433 | Suppression | NOTCH1 *, GATA3 | VSD, RV dysfunction | [111] |
| miR-222 | Suppression | ZFPM-2 | TOF, RV morphogenesis, CM proliferation | [112] |
| miR-421 | Suppression | SOX2 | TOF | [113] |
| miR-940 | Suppression | JARID2 | TOF, impaired cardiac progenitor proliferation/migration | [114] |
| miR-143 | Flow-dependent knockdown | RALDH2, RXRAB | Chamber and OFT development | [115] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jarrell, D.K.; Lennon, M.L.; Jacot, J.G. Epigenetics and Mechanobiology in Heart Development and Congenital Heart Disease. Diseases 2019, 7, 52. https://doi.org/10.3390/diseases7030052
Jarrell DK, Lennon ML, Jacot JG. Epigenetics and Mechanobiology in Heart Development and Congenital Heart Disease. Diseases. 2019; 7(3):52. https://doi.org/10.3390/diseases7030052
Chicago/Turabian StyleJarrell, Dillon K., Mallory L. Lennon, and Jeffrey G. Jacot. 2019. "Epigenetics and Mechanobiology in Heart Development and Congenital Heart Disease" Diseases 7, no. 3: 52. https://doi.org/10.3390/diseases7030052
APA StyleJarrell, D. K., Lennon, M. L., & Jacot, J. G. (2019). Epigenetics and Mechanobiology in Heart Development and Congenital Heart Disease. Diseases, 7(3), 52. https://doi.org/10.3390/diseases7030052