Role of Gut Dysbiosis in Liver Diseases: What Have We Learned So Far?

Abstract
:1. Gut-Liver Axis related to Gut Mycrobiomes
2. Endotoxin and Other Microbial Products
3. Short Chain Fatty Acids
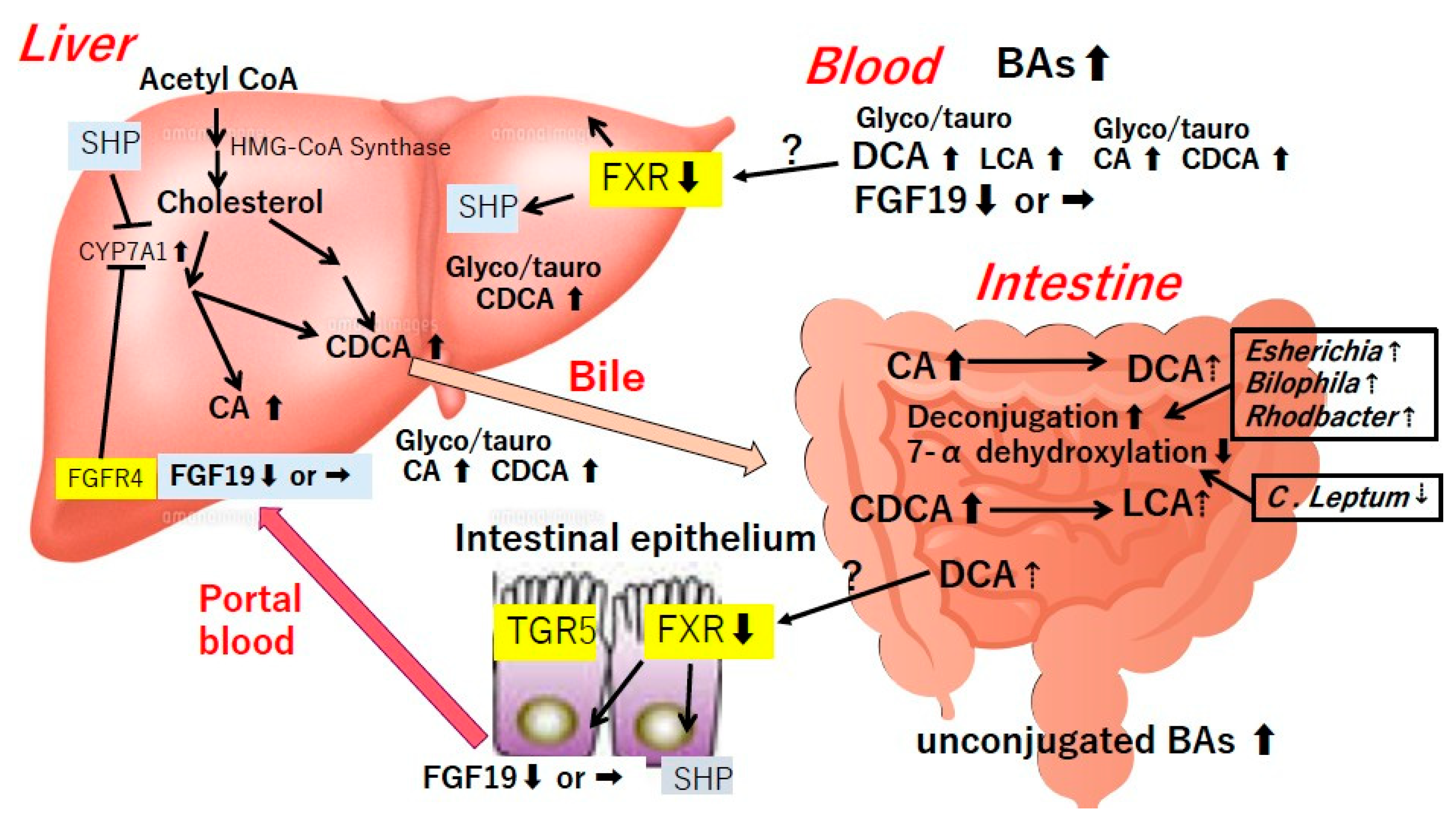
4. Bile Acids
5. Gut Dysbiosis in Liver Diseases
6. Alcoholic Liver Diseases
6.1. Intestinal Hyperpermeability and Bacterial Translocation
6.2. Endotoxemia and Its Consequences
6.3. Dysbiosis and Metabolic Changes
7. Nonalcoholic Steatohepatitis (NASH)
7.1. Obesity, NASH and Dysbiosis
7.2. Dysbiosis and Progression of NAFLD
7.3. Metabolic Changes Related to Gut Microbiota
7.4. Intestinal Permeability, Endotoxemia and Bacterial Translocation
7.5. NASH as a Derangement of Gut-Liver Axis
8. Liver Cirrhosis
8.1. Endotoxemia, Intestinal Permeability and Bacterial Translocation
8.2. SIBO and Gut Dysbiosis
8.3. Fecal and Mucosal Dysbiosis in Cirrhotic Complications
8.4. Metabolic Changes Related to Gut Microbiota
8.5. Gut Microbiome before and after Liver Transplantation
9. Primary Sclerosing Cholangitis (PSC)
10. Primary Biliary Cholangitis (PBC)
11. Hepatocellular Carcinoma (HCC)
12. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Fukui, H. Gut-liver axis in liver cirrhosis: How to manage leaky gut and endotoxemia. World J. Hepatol. 2015, 7, 425–442. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.S.; Shah, V.H. The role of gut-liver axis in the pathogenesis of liver cirrhosis and portal hypertension. Clin. Mol. Hepatol. 2012, 18, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Friedman, S.L. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenes. Tissue Repair 2010, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Schnabl, B. Role of innate immunity and the microbiota in liver fibrosis: Crosstalk between the liver and gut. J. Physiol. 2012, 590, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Topping, D.L.; Clifton, P.M. Short-chain fatty acids and human colonic function: Roles of resistant starch and nonstarch polysaccharides. Physiol. Rev. 2001, 81, 1031–1064. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.D.; Pyrsopoulos, N.; Rustgi, V.K. Microbiota and the liver. Liver Transpl. 2018, 24, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Schwiertz, A.; Taras, D.; Schafer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, L.E.; Koetsier, M.A.; van Deventer, S.J.; van Tol, E.A.F. Short chain fatty acids stimulate epithelial mucin 2 expression through differential effects on prostaglandin E(1) and E(2) production by intestinal myofibroblasts. Gut 2003, 52, 1442–1447. [Google Scholar] [CrossRef] [PubMed]
- Ohira, H.; Tsutsui, W.; Fujioka, Y. Are short chain fatty acids in gut microbiota defensive players for inflammation and atherosclerosis? J. Atheroscler. Thromb. 2017, 24, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid. Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Alves, J.M.; Hylemon, P.B.; Bajaj, J.S. Cirrhosis, bile acids and gut microbiota: Unraveling a complex relationship. Gut Microbes 2013, 4, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- Keitel, V.; Haussinger, D. Perspective: TGR5 (Gpbar-1) in liver physiology and disease. Clin. Res. Hepatol. Gastroenterol. 2012, 36, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Li, F.; Gonzalez, F.J. FXR signaling in the enterohepatic system. Mol. Cell Endocrinol. 2013, 368, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Pijls, K.E.; Jonkers, D.M.; Elamin, E.E.; Elamin, E.E.; Masclee, A.d.A.M.; Koek, G.H. Intestinal epithelial barrier function in liver cirrhosis: An extensive review of the literature. Liver Int. 2013, 33, 1457–1469. [Google Scholar] [CrossRef] [PubMed]
- Stanimirov, B.; Stankov, K.; Mikov, M. Bile acid signaling through farnesoid X and TGR5 receptors in hepatobiliary and intestinal diseases. Hepatobiliary Pancreat. Dis. Int. 2015, 14, 18–33. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Gut microbiota, cirrhosis, and alcohol regulate bile acid metabolism in the gut. Dig. Dis. 2015, 33, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, P.; Seebauer, C.T.; Schnabl, B. Alcoholic liver disease: The gut microbiome and liver cross talk. Alcohol Clin. Exp. Res. 2015, 39, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Bode, J.C.; Bode, C.; Heidelbach, R.; Dürr, H.K.; Martini, G.A. Jejunal microflora in patients with chronic alcohol abuse. Hepatogastroenterology 1984, 31, 30–34. [Google Scholar] [PubMed]
- Morencos, F.C.; de las Heras Castano, G.; Martin Ramos, L.; López Arias, M.J.; Ledesma, F.; Romero, F.P. Small bowel bacterial overgrowth in patients with alcoholic cirrhosis. Dig. Dis. Sci. 1995, 40, 1252–1256. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, P.; Chen, P.; Wang, H.J.; Wang, L.; McCole, D.F.; Brandl, K.; Stärkel, P.; Belzer, C.; Hellerbrand, C.; Tsukamoto, H.; et al. Deficiency of intestinal mucin-2 ameliorates experimental alcoholic liver disease in mice. Hepatology 2013, 58, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Vassallo, G.; Mirijello, A.; Ferrulli, A.; Antonelli, M.; Landolfi, R.; Gasbarrini, A.; Addolorato, G. Review article: Alcohol and gut microbiota-the possible role of gut microbiota modulation in the treatment of alcoholic liver disease. Aliment. Pharmacol. Ther. 2015, 41, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Brauner, B.; Bode, J.C.; Bode, C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: Reevaluation with an improved chromogenic assay. J. Hepatol. 1991, 12, 162–169. [Google Scholar] [CrossRef]
- Yan, A.W.; Fouts, D.E.; Brandl, J.; Stärkel, P.; Torralba, M.; Schott, E.; Tsukamot, H.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 2011, 53, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Bull-Otterson, L.; Feng, W.; Kirpich, I.; Wang, Y.; Qin, X.; Liu, Y.; Gobejishvili, L.; Joshi-Barve, S.; Ayvaz, T.; Petrosino, J.; et al. Metagenomic analyses of alcohol induced pathogenic alterations in the intestinal microbiome and the effect of Lactobacillus rhamnosus GG treatment. PLoS ONE 2013, 8, e53028. [Google Scholar] [CrossRef] [PubMed]
- Canesso, M.C.C.; Lacerda, N.L.; Ferreira, C.M.; Gonçalves, J.L.; Almeida, D.; Gamba, C.; Cassali, G.; Pedroso, S.H.; Moreira, C.; Martins, F.S.; et al. Comparing the effects of acute alcohol consumption in germ-free and conventional mice: The role of the gut microbiota. BMC Microbiol. 2014, 14, 240. [Google Scholar]
- Malaguarnera, G.; Giordano, M.; Nunnari, G.; Gaetano, B.; Michele, M. Gut microbiota in alcoholic liver disease: Pathogenetic role and therapeutic perspectives. World J. Gastroenterol. 2014, 20, 16639–16648. [Google Scholar] [CrossRef] [PubMed]
- Kirpich, I.A.; Solovieva, N.V.; Leikhter, S.N.; Shidakovaa, N.A.; Lebedevac, O.V.; Sidorovd, P.I.; Bazhukovac, T.A.; Solovievd, A.G.; Barvee, S.S.; McClain, C.J.; et al. Probiotics restore bowel flora and improve liver enzymes in human alcohol-induced liver injury: A pilot study. Alcohol 2008, 42, 675–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutlu, E.A.; Gillevet, P.M.; Rangwala, H.; Sikaroodi, M.; Naqvi, A.; Engen, P.A.; Kwasny, M.; Lau, C.K.; Keshavarzian, A. Colonic microbiome is altered in alcoholism. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G966–G978. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, S.; Matamoros, S.; Cani, P.D.; Neyrinck, A.M.; Jamar, F.; Stärkel, P.; Windey, K.; Tremaroli, V.; Bäckhed, F.; Verbeke, K.; et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc. Natl. Acad. Sci. USA 2014, 111, E4485–E4493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.-J.; Blugeon, S.; Bridonneau, C.; Furet, J.-P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, G.; Zhong, W.; Zheng, X.; Li, Q.; Qiu, Y.; Li, H.; Chen, H.; Zhou., Z.; Jia, W. Chronic ethanol consumption alters mammalian gastrointestinal content metabolites. J. Proteome Res. 2013, 12, 3297–3306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubinkina, V.B.; Tyakht, A.V.; Odintsova, V.Y.; Yarygin, K.S.; Kovarsky, B.A.; Pavlenko, A.V.; Ischenko, D.S.; Popenko, A.S.; Alexeev, D.G.; Taraskina, A.Y.; et al. Links of gut microbiota composition with alcohol dependence syndrome and alcoholic liver disease. Microbiome 2017, 5, 141. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, F.; Lu, H.; Wang, B.; Chen, Y.; Lei, D.; Wang, Y.; Zhu, B.; Li, L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 2011, 54, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Kakiyama, G.; Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Heuman, D.M.; Kang, D.G.; Takei, H.; Nittono, H.; Ridlon, J.M.; Fuchs, M.; et al. Colonic inflammation and secondary bile acids in alcoholic cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G929–G937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, J.S.; Heuman, D.M.; Hylemon, P.B.; Sanyal, A.J.; White, M.B.; Monteith, P.; Noble, N.A.; Unser, A.B.; Daita, K.; Fisher, A.R.; et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J. Hepatol. 2014, 60, 940–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, J.S.; Kakiyama, G.; Zhao, D.; Takei, H.; Fagan, A.; Hylemon, P.; Zhou, H.; Pandak, W.M.; Nittono, H.; Fiehn, O.; et al. Continued alcohol misuse in human cirrhosis is associated with an impaired gut-liver axis. Alcohol Clin. Exp. Res. 2017, 41, 1857–1865. [Google Scholar] [CrossRef] [PubMed]
- Hendrikx, T.; Schnabl, B. Antimicrobial proteins: Intestinal guards to protect against liver disease. J. Gastroenterol. 2019, 54, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandl, K.; Plitas, G.; Schnabl, B.; DeMatteo, R.P.; Pamer, E.G. MyD88-mediated signals induce the bactericidal lectin RegIII gamma and protect mice against intestinal Listeria monocytogenes infection. J. Exp. Med. 2007, 204, 1891–1900. [Google Scholar] [CrossRef] [PubMed]
- Brandl, K.; Plitas, G.; Mihu, C.N.; Ubeda, C.; Jia, T.; Fleisher, M.; Schnabl, B.; DeMatteo, R.P.; Pamer, E.G. Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature 2008, 455, 804–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dessein, R.; Gironella, M.; Vignal, C.; Peyrin-Biroulet, L.; Sokol, H.; Secher, T.; Lacas-Gervais, S.; Gratadoux, J.-J.; Lafont, F.; Dagorn, J.-C.; et al. Toll-like receptor 2 is critical for induction of Reg3 beta expression and intestinal clearance of Yersinia pseudotuberculosis. Gut 2009, 58, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fouts, D.E.; Starkel, P.; Hartmann, P.; Chen, P.; Llorente, C.; DePew, J.; Moncera, K.; Ho, S.B.; Brenner, D.A.; et al. Intestinal REG3 lectins protect against alcoholic steatohepatitis by reducing mucosa-associated microbiota and preventing bacterial translocation. Cell Host Microbe 2016, 19, 227–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Kirpich, I.; Liu, Y.; Ma, Z.; Barve, S.; McClain, C.J.; Feng, W. Lactobacillus rhamnosus GG treatment potentiates intestinal hypoxia-inducible factor, promotes intestinal integrity and ameliorates alcohol-induced liver injury. Am. J. Pathol. 2011, 179, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Llopis, M.; Cassard, A.M.; Wrzosek, L.; Boschat, L.; Bruneau, A.; Ferrere, G.; Puchois, V.; Martin, J.C.; Lepage, P.; Royet, T.L.; et al. Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut 2016, 65, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Sarin, S.K.; Pande, A.; Schnabl, B. Microbiome as a therapeutic target in alcohol-related liver disease. J. Hepatol. 2019, 70, 260–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.M.; Inamine, T.; Hochrath, K.; Chen, P.; Wang, L.; Llorente, C.; Bluemel, S.; Hartmann, P.; Xu, J.; Koyama, Y.; et al. Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Invest. 2017, 127, 2829–2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjarnason, I.; Peters, T.J.; Wise, R.J. The leaky gut of alcoholism: Possible route of entry for toxic compounds. Lancet 1984, 1, 179–182. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Holmes, E.W.; Patel, M.; Iber, F.; Fields, J.Z.; Pethkar, S. Leaky gut in alcoholic cirrhosis: A possible mechanism for alcohol-induced liver damage. Am. J. Gastroenterol. 1999, 94, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Parlesak, A.; Schafer, C.; Schutz, T.; Bode, J.C.; Bode, C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J. Hepatol. 2000, 32, 742–747. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Fields, J.Z.; Vaeth, J.; Holmes, W. The differing effects of acute and chronic alcohol on gastric and intestinal permeability. Am. J. Gastroenterol. 1994, 89, 2205–2211. [Google Scholar] [PubMed]
- Elamin, E.; Masclee, A.; Troost, F.; Pieters, H.-J.; Keszthelyi, D.; Aleksa, K.; Dekker, J.; Jonkers, D. Ethanol impairs intestinal barrier function in humans through mitogen activated protein kinase signaling: A combined in vivo and in vitro approach. PLoS ONE 2014, 9, e107421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalaitzakis, E. Gastrointestinal dysfunction in liver cirrhosis. World J. Gastroenterol. 2014, 20, 14686–14695. [Google Scholar] [CrossRef] [PubMed]
- Elamin, E.E.; Masclee, A.A.; Dekker, J.; Jonkers, D.M. Ethanol metabolism and its effects on the intestinal epithelial barrier. Nutr. Rev. 2013, 71, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, E.; Keshavarzian, A.; Engen, P.; Forsyth, C.B.; Sikaroodi, M.; Gillevet, P. Intestinal dysbiosis: A possible mechanism of alcohol-induced endotoxemia and alcoholic steatohepatitis in rats. Alcohol Clin. Exp. Res. 2009, 33, 1836–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Starkel, P.; Turner, J.R.; Ho, S.B.; Schnabl, B. Dysbiosis-induced intestinal inflammation activates tumor necrosis factor receptor I and mediates alcoholic liver disease in mice. Hepatology 2015, 61, 883–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodhouse, C.A.; Patel, V.C.; Singanayagam, A.; Shawcross, D.L. Review article: The gut microbiome as a therapeutic target in the pathogenesis and treatment of chronic liver disease. Aliment. Pharmacol. Ther. 2018, 47, 192–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, G. Gut-liver axis in alcoholic liver disease. Gastroenterology 2015, 148, 30–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, M.; Uemura, M.; Nakatani, Y.; Tsujita, S.; Hoppo, K.; Tamagawa, T.; Kitano, H.; Kikukawa, M.; Ann, T.; Ishii, Y.; et al. Plasma endotoxin and serum cytokine levels in patients with alcoholic hepatitis: Relation to severity of liver disturbance. Alcohol Clin. Exp. Res. 2000, 24, 48S–54S. [Google Scholar] [CrossRef] [PubMed]
- Brandl, K.; Hartmann, P.; Jih, L.J.; Pizzo, D.P.; Argemi, J.; Ventura-Cots, M.; Coulter, S.; Liddle, C.; Ling, L.; Rossi, S.J.; et al. Dysregulation of serum bile acids and FGF19 in alcoholic hepatitis. J. Hepatol. 2018, 69, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, P.; Hochrath, K.; Horvath, A.; Chen, P.; Seebauer, C.T.; Llorente, C.; Wang, L.; Alnouti, Y.; Fouts, D.E.; Stärkel, P.; et al. Modulation of the intestinal bile acid/farnesoid X receptor/fibroblast growth factor 15 axis improves alcoholic liver disease in mice. Hepatology 2018, 67, 2150–2166. [Google Scholar] [CrossRef] [PubMed]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Aron-Wisnewsky, J.; Gaborit, B.; Dutour, A.; Clement, K. Gut microbiota and non-alcoholic fatty liver disease: New insights. Clin. Microbiol. Infect. 2013, 19, 338–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilan, Y. Leaky gut and the liver: A role for bacterial translocation in nonalcoholic steatohepatitis. World J. Gastroenterol. 2012, 18, 2609–2618. [Google Scholar] [CrossRef] [PubMed]
- Wigg, A.J.; Roberts-Thomson, I.C.; Dymock, R.B.; McCarthy, P.J.; Grose, R.H.; Cummins, A.G. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 2001, 48, 206–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajjad, A.; Mottershead, M.; Syn, W.K.; Jones, R.; Smith, S.; Nwokolo, C.U. Ciprofloxacin suppresses bacterial overgrowth, increases fasting insulin but does not correct low acylated ghrelin concentration in non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2005, 22, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Shanab, A.A.; Scully, P.; Crosbie, O.; Buckley, M.; O’Mahony, L.; Shanahan, F.; Gazareen, S.; Murphy, E.; Quigley, E.M.M. Small intestinal bacterial overgrowth in nonalcoholic steatohepatitis: Association with toll-like receptor 4 expression and plasma levels of interleukin 8. Dig. Dis. Sci. 2011, 56, 1524–1534. [Google Scholar] [CrossRef] [PubMed]
- Volynets, V.; Kuper, M.A.; Strahl, S.; Maier, I.B.; Spruss, A.; Wagnerberger, S.; Königsrainer, A.; Bischoff, S.C.; Bergheim, I. Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease (NAFLD). Dig. Dis. Sci. 2012, 57, 1932–1941. [Google Scholar] [CrossRef] [PubMed]
- Ferolla, S.M.; Armiliato, G.N.; Couto, C.A.; Ferrari, T.C.A. The role of intestinal bacteria overgrowth in obesity-related nonalcoholic fatty liver disease. Nutrients 2014, 6, 5583–5599. [Google Scholar] [CrossRef] [PubMed]
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Fung, S.K.; Fischer, S.E.; McGilvray, I.D.; Allard, J.P. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 2013, 58, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnabl, B.; Brenner, D.A. Interactions between the intestinal microbiome and liver diseases. Gastroenterology 2014, 146, 1513–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.F.; Murphy, E.F.; Nilaweera, K.; Ross, P.R.; Shanahan, F.; O’Toole, P.W.; Cotter, P.D. The gut microbiota and its relationship to diet and obesity: New insights. Gut Microbes 2012, 3, 186–202. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Backhed, F.; Fulton, L.; Gordon, J.I. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrandt, M.A.; Hoffmann, C.; Sherrill-Mix, S.A.; Keilbaugh, S.A.; Hamady, M.; Chen, Y.-Y.; Knight, R.; Ahima, R.S.; Bushman, F.; Wu, G.D. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 2009, 137, 1716–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panasevich, M.R.; Peppler, W.T.; Oerther, D.B.; Wright, D.C.; Rector, R.S. Microbiome and NAFLD: Potential influence of aerobic fitness and lifestyle modification. Physiol. Genom. 2017, 49, 385–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armougom, F.; Henry, M.; Vialettes, B.; Raccah, D.; Raoult, D. Monitoring bacterial community of human gut microbiota reveals an increase in Lactobacillus in obese patients and Methanogens in anorexic patients. PLoS ONE 2009, 4, e7125. [Google Scholar] [CrossRef] [PubMed]
- Santacruz, A.; Collado, M.C.; Garcia-Valdes, L.; Segura, M.T.; Martín-Lagos, J.A.; Anjos, T.; Martí-Romero, M.; Lopez, R.M.; Florido, J.; Campoy, C.; et al. Gut microbiota composition is associated with body weight, weight gain and biochemical parameters in pregnant women. Br. J. Nutr. 2010, 104, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Baker, S.S.; Liu, W.; Alkhouri, R.; Baker, R.D.; Xie, J.; Ji, G.; Zhu, L. Endotoxemia unrequired in the pathogenesis of pediatric nonalcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2014, 29, 1292–1298. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; DiBaise, J.K.; Zuccolo, A.; Kudrna, D.; Braidotti, M.; Yu, Y.; Parameswaran, P.; Crowell, M.D.; Wing, R.; Rittmann, B.E.; et al. Human gut microbiota in obesity and after gastric bypass. Proc. Natl. Acad. Sci. USA 2009, 106, 2365–2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Million, M.; Maraninchi, M.; Henry, M.; Armougom, F.; Richet, H.; Carrieri, P.; Valero, R.; Raccah, D.; Vialettes, B.; Raoult, D. Obesity-associated gut microbiota is enriched in Lactobacillus reuteri and depleted in Bifidobacterium animalis and Methanobrevibacter smithii. Int. J. Obes. 2012, 36, 817–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, V.W.; Tse, C.H.; Lam, T.T.; Wong, G.L.-H.; Chim, A.M.-L.; Chu, W.C.-W.; Yeung, D.K.-W.; Law, P.T.-W.; Kwan, H.-S.; Yu, J.; et al. Molecular characterization of the fecal microbiota in patients with nonalcoholic steatohepatitis—A longitudinal study. PLoS ONE 2013, 8, e62885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, H.E.; Teterina, A.; Comelli, E.M.; Taibi, A.; Arendt, B.M.; Fischer, S.E.; Lou, W.; Allard, J.P. Nonalcoholic fatty liver disease is associated with dysbiosis independent of body mass index and insulin resistance. Sci. Rep. 2018, 8, 1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raman, M.; Ahmed, I.; Gillevet, P.M.; Probert, C.S.; Ratcliffe, R.S.; Smith, S.; Greenwood, R.; Sikaroodi, M.; Lam, V.; Crotty, P.; et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2013, 11, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Zheng, R.D.; Sun, X.Q.; Ding, W.-J.; Wang, X.-Y.; Fan, J.-G. Gut microbiota dysbiosis in patients with non-alcoholic fatty liver disease. Hepatobiliary Pancreat. Dis. Int. 2017, 16, 375–381. [Google Scholar] [CrossRef]
- Jiang, W.; Wu, N.; Wang, X.; Chi, Y.; Zhang, Y.; Qiu, X.; Hu, Y.; Li, J.; Liu, Y. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci. Rep. 2015, 5, 8096. [Google Scholar] [CrossRef] [PubMed]
- Del Chierico, F.; Nobili, V.; Vernocchi, P.; Russo, A.; De Stefanis, C.; Gnani, D.; Furlanello, C.; Zandona, A.; Paci, P.; Capuani, G.; et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology 2017, 65, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Sobhonslidsuk, A.; Chanprasertyothin, S.; Pongrujikorn, T.; Kaewduang, P.; Promson, K.; Petraksa, S.; Ongphiphadhanakul, B. The Association of gut microbiota with nonalcoholic steatohepatitis in thais. Biomed. Res. Int. 2018, 2018, 9340316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Long, W.; Zhang, C.; Liu, S.; Zhao, L.; Hamaker, B.R. Fiber-utilizing capacity varies in Prevotella-versus Bacteroides-dominated gut microbiota. Sci. Rep. 2017, 7, 2594. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.M. The immune response to Prevotella bacteria in chronic inflammatory disease. Immunology 2017, 151, 363–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özkul, C.; Yalinay, M.; Karakan, T.; Yilmaz, G. Determination of certain bacterial groups in gut microbiota and endotoxin levels in patients with nonalcoholic steatohepatitis. Turk. J. Gastroenterol. 2017, 28, 361–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammes, T.O.; Leke, R.; Escobar, T.D.C.; Beduschi Fracasso, L.; Schons Meyer, F.; Éverton Andrades, M.; Reverbel da Silveira, T. Lactobacillus rhamnosusGG reduces hepatic fibrosis in a model of chronic liver disease in rats. Nutr. Hosp. 2017, 34, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai1, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab. 2017, 25, 1054–1062. [Google Scholar] [CrossRef] [PubMed]
- Saltzman, E.T.; Palacios, T.; Thomsen, M.; Vitetta, L. Intestinal microbiome shifts, dysbiosis, inflammation, and non-alcoholic fatty liver disease. Front. Microbiol. 2018, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Belenguer, A.; Holtrop, G.; Johnstone, A.M.; Flint, H.J.; Lobley, G.E. Reduced dietary intake of carbohydrates by obese subjects results in decreased concentrations of butyrate and butyrate-producing bacteria in feces. Appl. Environ. Microbiol. 2007, 73, 1073–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, S.H.; Lobley, G.E.; Holtrop, G.; Ince, J.; Johnstone, A.M.; Flint, H.J. Human colonic microbiota associated with diet, obesity and weight loss. Int. J. Obes. 2008, 32, 1720–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Baker, R.D.; Baker, S.S. Gut microbiome and nonalcoholic fatty liver diseases. Pediatr. Res. 2015, 77, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Ulven, T. Short-chain free fatty acid receptors FFA2/GPR43 and FFA3/GPR41 as new potential therapeutic targets. Front. Endocrinol. 2012, 3, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjursell, M.; Admyre, T.; Goransson, M.; Marley, A.E.; Smith, D.M.; Oscarsson, J.; Bohlooly-Y, M. Improved glucose control and reduced body fat mass in free fatty acid receptor 2-deficient mice fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E211–E220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferslew, B.C.; Xie, G.; Johnston, C.K.; Su, M.; Stewart, P.W.; Jia, W.; Brouwer, K.L.R.; Barritt IV, A.S. Altered bile acid metabolome in patients with nonalcoholic steatohepatitis. Dig. Dis. Sci. 2015, 60, 3318–3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouzaki, M.; Wang, A.Y.; Bandsma, R.; Comelli, E.M.; Arendt, B.M.; Zhang, L.; Fung, S.; Fischer, S.E.; McGilvray, I.G.; Allard, G.P. Bile acids and dysbiosis in non-alcoholic fatty liver disease. PLoS ONE 2016, 11, e0151829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, P.; Zhang, Y.; Klaassen, C.D. Dose-response of five bile acids on serum and liver bile Acid concentrations and hepatotoxicty in mice. Toxicol. Sci. 2011, 123, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alisi, A.; Ceccarelli, S.; Panera, N.; Prono, F.; Petrini, S.; De Stefanis, C.; Pezzullo, M.; Tozzi, A.; Villani, A.; Bedogni, G.; et al. Association between serum atypical fibroblast growth factors 21 and 19 and pediatric nonalcoholic fatty liver disease. PLoS ONE 2013, 8, e67160. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, D.; Marschall, H.U.; Lammert, F. Response of fibroblast growth factor 19 and bile acid synthesis after a body weight-adjusted oral fat tolerance test in overweight and obese NAFLD patients: A non-randomized controlled pilot trial. BMC Gastroenterol. 2018, 18, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreuder, T.C.; Marsman, H.A.; Lenicek, M.; van Werven, J.R.; Nederveen, A.J.; Jansen, P.L.M.; Schaap, F.G. The hepatic response to FGF19 is impaired in patients with nonalcoholic fatty liver disease and insulin resistance. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G440–G445. [Google Scholar] [CrossRef] [PubMed]
- Jiao, N.; Baker, S.S.; Chapa-Rodriguez, A.; Liu, W.; Nugent, C.A.; Tsompana, M.; Mastrandrea, L.; Buck, M.L.; Baker, R.J.; Genco, R.J.; et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 2018, 67, 1881–1891. [Google Scholar] [CrossRef] [PubMed]
- Haub, S.; Kanuri, G.; Volynets, V.; Brune, T.; Bischoff, S.C.; Bergheim, I. Serotonin reuptake transporter (SERT) plays a critical role in the onset of fructose-induced hepatic steatosis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G335–G344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.D.; Chen, W.D.; Wang, M.; Yu, D.; Forman, B.M.; Huang, W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 2008, 48, 1632–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Wang, J.; Liu, Q.; Harnish, D.C. Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J. Hepatol. 2009, 51, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, S.; Mencarelli, A.; Palladino, G.; Fiorucci, S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J. Lipid. Res. 2010, 51, 771–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahan, R.H.; Wang, X.X.; Cheng, L.L.; Krisko, T.; Smith, M.; Kasmi, K.E.; Pruzanski, M.; Adorini, L.; Golden-Mason, L.; Levi, M.; et al. Bile acid receptor activation modulates hepatic monocyte activity and improves nonalcoholic fatty liver disease. J. Biol. Chem. 2013, 288, 11761–11770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.D.; Chen, W.D.; Yu, D.; Forman, B.M.; Huang, W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-kappaB) in mice. Hepatology 2011, 54, 1421–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.-U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Banerjee, A.; Jang, S.; Yoo, S.-H.; Yun, J.-W.; Gonzalez, F.J.; Keshavarzian, A.; Song, B.-J. CYP2E1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis. Free Radic. Biol. Med. 2013, 65, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.H.; Carey, E.J.; Lindor, K.D. Recent advances in the development of farnesoid X receptor agonists. Ann. Transl. Med. 2015, 3, 5. [Google Scholar] [PubMed]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Liu, X.; Peng, X.; Xue, R.; Ji, L.; Shen, X.; Chen, S.; Gu, J.; Zhang, S. Bile acids override steatosis in farnesoid X receptor deficient mice in a model of non-alcoholic steatohepatitis. Biochem. Biophys. Res. Commun. 2014, 448, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Dumas, M.E.; Barton, R.H.; Toye, A.; Cloarec, O.; Blancher, C.; Rothwell, A.; Fearnside, J.; Tatoud, R.; Blanc, V.; Lindon, J.C.; et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12511–12516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.C.; Li, Z.; Liu, R.; Yang, X.P.; Pan, M.; Lagrost, L.; Fisher, E.A.; Williams, K.J. Phospholipid transfer protein deficiency impairs apolipoprotein-B secretion from hepatocytes by stimulating a proteolytic pathway through a relative deficiency of vitamin E and an increase in intracellular oxidants. J. Biol. Chem. 2005, 280, 18336–18340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgio, V.; Miele, L.; Principessa, L.; Ferretti, F.; Villa, M.P.; Negro, V.; Grieco, A.; Alisi, A.; Nobili, V. Intestinal permeability is increased in children with non-alcoholic fatty liver disease, and correlates with liver disease severity. Digestive and Liver Disease 2014, 46, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Guercio Nuzio, S.; Di Stasi, M.; Pierri, L.; Troisi, J.; Poeta, M.; Bisogno, A.; Belmonte, F.; Tripodi, M.; Di Salvio, D.; Massa, G. Multiple gut–liver axis abnormalities in children with obesity with and without hepatic involvement. Pediatric obesity 2017, 12, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Manco, M.; Devito, R.; Piemonte, F.; Nobili, V. Endotoxin and plasminogen activator inhibitor-1 serum levels associated with nonalcoholic steatohepatitis in children. J. Pediatric Gastroenterol. Nutr. 2010, 50, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Farhadi, A.; Gundlapalli, S.; Shaikh, M.; Frantzides, C.; Harrell, L.; Kwasny, M.M.; Keshavarzian, A. Susceptibility to gut leakiness: A possible mechanism for endotoxaemia in non-alcoholic steatohepatitis. Liver International 2008, 28, 1026–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapil, S.; Duseja, A.; Sharma, B.K.; Singla, B.; Chakraborti, A.; Das, A.; Ray, P.; Dhiman, R.K.; Chawla, Y. Small intestinal bacterial overgrowth and toll-like receptor signaling in patients with non-alcoholic fatty liver disease. J. gastroenterol. Hepatol. 2016, 31, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Sharifnia, T.; Antoun, J.; Verriere, T.G.; Suarez, G.; Wattacheril, J.; Wilson, K.T.; Peek, R.M., Jr.; Abumrad, N.N.; Flynn, C.R. Hepatic TLR4 signaling in obese NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G270–G278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiziltas, S.; Ata, P.; Colak, Y.; Mesçi, B.; Senates, E.; Enc, F.; Ulasoglu, C.; Tuncer, I.; Oguz, A. TLR4 gene polymorphism in patients with nonalcoholic fatty liver disease in comparison to healthy controls. Metab. Syndr. Relat. Disord. 2014, 12, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Brun, P.; Castagliuolo, I.; Leo, V.D.; Buda, A.; Pinzani, M.; Palù, G.; Martines, D. Increased intestinal permeability in obese mice: New evidence in the pathogenesis of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G518–G525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amar, J.; Chabo, C.; Waget, A.; Klopp, P.; Vachoux, C.; Bermúdez-Humarán, L.G.; Smirnova, N.; Bergé, M.; Sulpice, T.; Lahtinen, S. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: Molecular mechanisms and probiotic treatment. EMBO molecular medicine 2011, 3, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.S.; Baker, R.D.; Liu, W.; Nowak, N.J.; Zhu, L. Role of alcohol metabolism in non-alcoholic steatohepatitis. PloS ONE 2010, 5, e9570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010, 139, 323–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Minicis, S.; Rychlicki, C.; Agostinelli, L.; Saccomanno, S.; Candelaresi, C.; Trozzi, L.; Mingarelli, E.; Facinelli, B.; Magi, G.; Palmieri, C. Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology 2014, 59, 1738–1749. [Google Scholar] [CrossRef] [PubMed]
- Kawaratani, H.; Tsujimoto, T.; Kitazawa, T.; Kitade, M.; Yoshiji, H.; Uemura, M.; Fukui, H. Innate immune reactivity of the liver in rats fed a choline-deficient L-amino-acid-defined diet. World J. Gastroenterol. 2008, 14, 6655–6661. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, T.; Kawaratani, H.; Kitazawa, T.; Hirai, T.; Ohishi, H.; Kitade, M.; Yoshiji, H.; Uemura, M.; Fukui, H. Decreased phagocytic activity of Kupffer cells in a rat nonalcoholic steatohepatitis model. World J. Gastroenterol. 2008, 14, 6036–6043. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, T.; Kawaratani, H.; Kitazawa, T.; Uemura, M.; Fukui, H. Innate immune reactivity of the ileum-liver axis in nonalcoholic steatohepatitis. Dig. Dis. Sci. 2012, 57, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Dunzendorfer, S.; Lee, H.-K.; Soldau, K.; Tobias, P.S. TLR4 is the signaling but not the lipopolysaccharide uptake receptor. J. Immunol. 2004, 173, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Douhara, A.; Moriya, K.; Yoshiji, H.; Noguchi, R.; Namisaki, T.; Kitade, M.; Kaji, K.; Aihara, Y.; Nishimura, N.; Takeda, K. Reduction of endotoxin attenuates liver fibrosis through suppression of hepatic stellate cell activation and remission of intestinal permeability in a rat non-alcoholic steatohepatitis model. Mol. Med. Rep. 2015, 11, 1693–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukumo, D.M.; Carvalho-Filho, M.A.; Carvalheira, J.B.; Prada, P.O.; Hirabara, S.M.; Schenka, A.A.; Araújo, E.P.; Vassallo, J.; Curi, R.; Velloso, L.A. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes 2007, 56, 1986–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csak, T.; Velayudham, A.; Hritz, I.; Petrasek, J.; Levin, I.; Lippai, D.; Catalano, D.; Mandrekar, P.; Dolganiuc, A.; Kurt-Jones, E. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G433–G441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, C.L.; Elfers, C.T.; Figlewicz, D.P.; Melhorn, S.J.; Morton, G.J.; Hoofnagle, A.; Yeh, M.M.; Nelson, J.E.; Kowdley, K.V. Vitamin D deficiency in obese rats exacerbates nonalcoholic fatty liver disease and increases hepatic resistin and Toll-like receptor activation. Hepatology 2012, 55, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Ohnishi, H. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 7381–7391. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Yang, L.; van Rooijen, N.; Brenner, D.A.; Ohnishi, H.; Seki, E. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology 2013, 57, 577–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinzani, M.; Rosselli, M.; Zuckermann, M. Liver cirrhosis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Tandon, P.; Garcia-Tsao, G. Bacterial infections, sepsis, and multiorgan failure in cirrhosis. Semin. Liver Dis. 2008, 28, 26–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvaniti, V.; D’Amico, G.; Fede, G.; Manousou, P.; Tsochatzis, E.; Pleguezuelo, M.; Burroughs, A.K. Infections in patients with cirrhosis increase mortality four-fold and should be used in determining prognosis. Gastroenterology 2010, 139, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Campillo, B.; Richardet, J.-P.; Kheo, T.; Dupeyron, C. Nosocomial spontaneous bacterial peritonitis and bacteremia in cirrhotic patients: Impact of isolate type on prognosis and characteristics of infection. Clin. Infect. Dis. 2002, 35, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, J.S.; Heuman, D.M.; Hylemon, P.B.; Sanyal, A.J.; White, M.B.; Monteith, P.; Noble, N.A.; Unser, A.B.; Daita, K.; Fisher, A.R.; et al. The cirrhosis dysbiosis ratio defines changes in the gut microbiome associated with cirrhosis and its complications. J. Hepatol. 2014, 60, 940–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prytz, H.; Holst-Christensen, J.; Korner, B.; Liehr, H. Portal venous and systemic endotoxaemia in patients without liver disease and systemic endotoxaemia in patients with cirrhosis. Scand. J. Gastroenterol. 1976, 11, 857–863. [Google Scholar] [PubMed]
- Fukui, H.; Tsujita, S.; Matsumoto, M.; Kitano, H.; Hoppou, K.; Morimura, M.; Takaya, A.; Okamoto, S.; Tsujii, T.; Bode, C. Endotoxemia in chronic hepatitis and cirrhosis: Epiphenomenon or of pathological relevence? In Gut and the Liver; Blum, H., Bode, C., Bode, J.C., Sartor, R.B., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1998; pp. 251–262. [Google Scholar]
- Lin, R.-S.; Lee, F.-Y.; Lee, S.-D.; Tsai, Y.-T.; Lin, H.C.; Rei-Hwa, L.; Wan-Ching, H.; Cheng-Chun, H.; Sun-Sang, W.; Kwang-Juei, L. Endotoxemia in patients with chronic liver diseases: Relationship to severity of liver diseases, presence of esophageal varices, and hyperdynamic circulation. J. Hepatol. 1995, 22, 165–172. [Google Scholar] [CrossRef]
- Lumsden, A.B.; Henderson, J.M.; Kutner, M.H. Endotoxin levels measured by a chromogenic assay in portal, hepatic and peripheral venous blood in patients with cirrhosis. Hepatology 1988, 8, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Tachiyama, G.; Sakon, M.; Kambayashi, J.; Iijima, S.; Tsujinaka, T.; Mori, T. Endogenous endotoxemia in patients with liver cirrhosis-a quantitative analysis of endotoxin in portal and peripheral blood. Jpn. J. Surg. 1988, 18, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H. Endotoxin and other microbial translocation markers in the blood: A clue to understand leaky gut syndrome. Cell Mol. Med. 2016, 2, 3. [Google Scholar] [CrossRef]
- Bellot, P.; Frances, R.; Such, J. Pathological bacterial translocation in cirrhosis: Pathophysiology, diagnosis and clinical implications. Liver Int. 2013, 33, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H. Gut microbiome-based therapeutics in liver cirrhosis: Basic consideration for the next step. J. Clin. Transl. Hepatol. 2017, 5, 249–260. [Google Scholar] [PubMed] [Green Version]
- Campillo, B.; Pernet, P.; Bories, P.; Richardet, J.; Devanlay, M.; Aussel, C. Intestinal permeability in liver cirrhosis: Relationship with severe septic complications. Eur. J. Gastroenterol. Hepatol. 1999, 11, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Pascual, S.; Such, J.; Esteban, A.; Zapater, P.; Casellas, J.A.; Aparicio, J.R.; Girona, E.; Gutierrez, A.; Carnices, F.; Palazon, J.M. Intestinal permeability is increased in patients with advanced cirrhosis. Hepatogastroenterology 2003, 50, 1482–1486. [Google Scholar] [PubMed]
- Scarpellini, E.; Valenza, V.; Gabrielli, M.; Lauritano, E.C.; Perotti, G.; Merra, G.; Dal Lago, A.; Ojetti, V.; Ainora, M.E.; Santoro, M. Intestinal permeability in cirrhotic patients with and without spontaneous bacterial peritonitis: Is the ring closed? Am. J. Gastroenterol. 2010, 105, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Fink, D.; Romanowski, K.; Valuckaite, V.; Babrowski, T.; Kim, M.; Matthews, J.B.; Liu, D.; Zaborina, O.; Alverdy, J.C. Pseudomonas aeruginosa potentiates the lethal effect of intestinal ischemia-reperfusion injury: The role of in vivo virulence activation. J. Trauma 2011, 71, 1575–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pijls, K.E.; Koek, G.H.; Elamin, E.E.; de Vries, H.; Masclee, A.A.; Jonkers, D.M. Large intestine permeability is increased in patients with compensated liver cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G147–G153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francés, R.; González-Navajas, J.M.; Zapater, P.; Muñoz, C.; Caño, R.; Pascual, S.; Márquez, D.; Santana, F.; Pérez-Mateo, M.; Such, J. Bacterial DNA induces the complement system activation in serum and ascitic fluid from patients with advanced cirrhosis. J. Clin. Immunol. 2007, 27, 438–444. [Google Scholar] [CrossRef] [PubMed]
- González-Navajas, J.M.; Bellot, P.; Francés, R.; Zapater, P.; Muñoz, C.; García-Pagán, J.C.; Pascual, S.; Pérez-Mateo, M.; Bosch, J.; Such, J. Presence of bacterial-DNA in cirrhosis identifies a subgroup of patients with marked inflammatory response not related to endotoxin. J. Hepatol. 2008, 48, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.M.; Schwacha, H.; Steinbrückner, B.; Brinkmann, F.E.; Ditzen, A.K.; Aponte, J.J.; Pelz, K.; Berger, D.; Kist, M.; Blum, H.E. Small intestinal bacterial overgrowth in human cirrhosis is associated with systemic endotoxemia. Am. J. Gastroenterol. 2002, 97, 2364–2370. [Google Scholar] [CrossRef] [PubMed]
- Morencos, F.C.; Castano, G.D.L.H.; Ramos, L.M.; Arias, M.J.L.; Ledesma, F.; Romero, F.P. Small bowel bacterial overgrowth in patients with alcoholic cirrhosis. Dig. Dis. Sci. 1996, 41, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Chang, C.S.; Chen, G.H. Small-intestinal bacterial overgrowth in patients with liver cirrhosis, diagnosed with glucose H2 or CH4 breath tests. Scand. J. Gastroenterol. 1998, 33, 867–871. [Google Scholar] [PubMed]
- Chang, C.S.; Chen, G.H.; Lien, H.C.; Yeh, H.Z. Small intestine dysmotility and bacterial overgrowth in cirrhotic patients with spontaneous bacterial peritonitis. Hepatology 1998, 28, 1187–1190. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Dhiman, R.K.; Kumari, S.; Rana, S.; Agarwal, R.; Duseja, A.; Chawla, Y. Role of small intestinal bacterial overgrowth and delayed gastrointestinal transit time in cirrhotic patients with minimal hepatic encephalopathy. J. Hepatol. 2010, 53, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Sadik, R.; Abrahamsson, H.; Björnsson, E.; Gunnarsdottir, A.; Stotzer, P.O. Etiology of portal hypertension may influence gastrointestinal transit. Scand. J. Gastroenterol. 2003, 38, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Pardo, A.; Bartolí, R.; Lorenzo-Zúñiga, V.; Planas, R.; Viñado, B.; Riba, J.; Cabré, E.; Santos, J.; Luque, T.; Ausina, V. Effect of cisapride on intestinal bacterial overgrowth and bacterial translocation in cirrhosis. Hepatology 2000, 31, 858–863. [Google Scholar] [CrossRef] [PubMed]
- Van Thiel, D.H.; Fagiuoli, S.; Wright, H.I.; Chien, M.C.; Gavaler, J.S. Gastrointestinal transit in cirrhotic patients: Effect of hepatic encephalopathy and its treatment. Hepatology 1994, 19, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Jun, D.W.; Kim, K.T.; Lee, O.Y.; Chae, J.D.; Son, B.K.; Kim, S.H.; Jo, Y.J.; Park, Y.S. Association between small intestinal bacterial overgrowth and peripheral bacterial DNA in cirrhotic patients. Dig. Dis. Sci. 2010, 55, 1465–1471. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Duan, Z.P.; Ha, D.K.; Bengmark, S.; Kurtovic, J.; Riordan, S.M. Synbiotic modulation of gut flora: Effect on minimal hepatic encephalopathy in patients with cirrhosis. Hepatology 2004, 39, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Wu, Z.; Xu, W.; Yang, J.; Chen, Y.; Li, L. Intestinal microbiota was assessed in cirrhotic patients with hepatitis B virus infection. Intestinal microbiota of HBV cirrhotic patients. Microb. Ecol. 2011, 61, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, B.; Fu, Y.; Chen, Y.; Yang, F.; Lu, H.; Chen, Y.; Xu, J.; Li, L. Changes of fecal Bifidobacterium species in adult patients with hepatitis B virus-induced chronic liver disease. Microb. Ecol. 2012, 63, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-W.; Lu, H.-F.; Wu, J.; Zuo, J.; Chen, P.; Sheng, J.-F.; Zheng, S.-S.; Li, L.-J. Assessment of the fecal lactobacilli population in patients with hepatitis B virus-related decompensated cirrhosis and hepatitis B cirrhosis treated with liver transplant. Microb. Ecol. 2012, 63, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Yan, X.; Zou, D.; Yang, Z.; Wang, X.; Liu, W.; Wang, S.; Li, X.; Han, J.; Huang, L. Abnormal fecal microbiota community and functions in patients with hepatitis B liver cirrhosis as revealed by a metagenomic approach. BMC Gastroenterol. 2013, 13, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, J.S.; Ridlon, J.M.; Hylemon, P.B.; Thacker, L.R.; Heuman, D.M.; Smith, S.; Sikaroodi, M.; Gillevet, P.M. Linkage of gut microbiome with cognition in hepatic encephalopathy. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G168–G175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.B.; Wang, P.Y.; Wang, X.; Wan, Y.L.; Liu, Y.C. Butyrate enhances intestinal epithelial barrier function via up-regulation of tight junction protein Claudin-1 transcription. Dig. Dis. Sci. 2012, 57, 3126–3135. [Google Scholar] [CrossRef] [PubMed]
- Usami, M.; Miyoshi, M.; Yamashita, H. Gut microbiota and host metabolism in liver cirrhosis. World J. Gastroenterol. 2015, 21, 11597–11608. [Google Scholar] [CrossRef] [PubMed]
- Corrêa-Oliveira, R.; Fachi, J.L.; Vieira, A.; Sato, F.T.; Vinolo, M.A.R. Regulation of immune cell function by short-chain fatty acids. Clin. Transl. Immunol. 2016, 5, e73. [Google Scholar] [CrossRef] [PubMed]
- Vinolo, M.A.; Rodrigues, H.G.; Nachbar, R.T.; Curi, R. Regulation of inflammation by short chain fatty acids. Nutrients 2011, 3, 858–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, C.; Bajaj, J.S. Gut Microbiota and Complications of Liver Disease. Gastroenterol Clin North Am 2017, 46, 155–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Jin, Y.; Li, J.; Zhao, L.; Li, Z.; Xu, J.; Zhao, F.; Feng, J.; Chen, H.; Fang, C. Small bowel transit and altered gut microbiota in patients with liver cirrhosis. Front. Physiol. 2018, 9, 470. [Google Scholar] [CrossRef] [PubMed]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, J.S.; Liu, E.J.; Kheradman, R.; Fagan, A.; Heuman, D.M.; White, M.; Gavis, E.A.; Hylemon, P.; Sikaroodi, M.; Gillevet, P.M. Fungal dysbiosis in cirrhosis. Gut 2018, 67, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Betrapally, N.S.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; White, M.B.; Unser, A.; Thacker, L.R.; Sanyal, A.J.; Kang, D.J. Salivary microbiota reflects changes in gut microbiota in cirrhosis with hepatic encephalopathy. Hepatology 2015, 62, 1260–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, J.S.; Betrapally, N.S.; Gillevet, P.M. Decompensated cirrhosis and microbiome interpretation. Nature 2015, 525, E1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betrapally, N.S.; Gillevet, P.M.; Bajaj, J.S. Gut microbiome and liver disease. Transl. Res. 2017, 179, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarker, S.A.; Gyr, K. Non-immunological defence mechanisms of the gut. Gut 1992, 33, 987–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, J.S.; Acharya, C.; Fagan, A.; White, M.B.; Gavis, E.; Heuman, D.M.; Hylemon, P.B.; Fuchs, M.; Puri, P.; Schubert, M.L.; et al. Proton pump inhibitor initiation and withdrawal affects gut microbiota and readmission risk in cirrhosis. Am. J. Gastroenterol. 2018, 113, 1177. [Google Scholar] [CrossRef] [PubMed]
- Grønkjær, L.L.; Holmstrup, P.; Schou, S.; Schwartz, K.; Kongstad, J.; Jepsen, P.; Vilstrup, H. Presence and consequence of tooth periapical radiolucency in patients with cirrhosis. Hepat. Med. 2016, 8, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, C.; Sahingur, S.E.; Bajaj, J.S. Microbiota, cirrhosis, and the emerging oral-gut-liver axis. JCI Insight 2017, 2, e94416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhai, H.; Geng, J.; Yu, R.; Ren, H.; Fan, H.; Shi, P. Large-scale survey of gut microbiota associated with MHE Via 16S rRNA-based pyrosequencing. Am. J. Gastroenterol. 2013, 108, 1601–1611. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, V.; Betrapally, N.S.; Hylemon, P.B.; White, M.B.; Gillevet, P.M.; Unser, A.B.; Fagan, A.; Daita, K.; Heuman, D.M.; Zhou, H.; et al. Impaired gut-liver-brain axis in patients with cirrhosis. Sci. Rep. 2016, 6, 26800. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Saraswat, V.A.; Dhiman, R.K. Gut microbiota: Its role in hepatic encephalopathy. J. Clin. Exp. Hepatol. 2015, 5, S29–S36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, W.; Alam, M.; Roy, C.; Pyne, P.; George, A.; Chakraborty, R.; Majumder, S.; Agarwal, A.; Chakraborty, S.; Majumdar, S.; et al. Genome implosion elicits host-confinement in Alcaligenaceae: Evidence from the comparative genomics of Tetrathiobacter kashmirensis, a pathogen in the making. PLoS ONE 2013, 8, e64856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, C.M.; Chen, K.F.; Lin, Y.F.; Ke, H.M.; Huang, H.Y.; Gong, Y.N.; Tsai, W.S.; You, J.F.; Lu, M.J.; Cheng, H.T.; et al. Predicting clinical outcomes of cirrhosis patients with hepatic encephalopathy from the fecal microbiome. Cell Mol. Gastroenterol. Hepatol. 2019, 8, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Such, J.; Francés, R.; Muñoz, C.; Zapater, P.; Casellas, J.A.; Cifuentes, A.; Rodrı́guez-Valera, F.; Pascual, S.; Sola-Vera, J.; Carnicer, F.; et al. Detection and identification of bacterial DNA in patients with cirrhosis and culture-negative, nonneutrocytic ascites. Hepatology 2002, 36, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Koulaouzidis, A.; Bhat, S.; Karagiannidis, A.; Tan, W.C.; Linaker, B.D. Spontaneous bacterial peritonitis. Postgrad. Med. J. 2007, 83, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.; Arendt, B.M.; Bhat, V.; Renner, E.L.; Humar, A.; Allard, J.P. Implication of the intestinal microbiome in complications of cirrhosis. World J. Hepatol. 2016, 8, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Bruns, T.; Reuken, P.A.; Stengel, S.; Gerber, L.; Appenrodt, B.; Schade, J.H.; Lammert, F.; Zeuzem, S.; Stallmach, A. The prognostic significance of bacterial DNA in patients with decompensated cirrhosis and suspected infection. Liver Int. 2016, 36, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Sanduzzi Zamparelli, M.; Rocco, A.; Compare, D.; Nardone, G. The gut microbiota: A new potential driving force in liver cirrhosis and hepatocellular carcinoma. United Eur. Gastroenterol. J. 2017, 5, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Hylemon, P.B.; Ridlon, J.M.; Heuman, D.M.; Daita, K.; White, M.B.; Monteith, P.; Noble, N.A.; Sikaroodi, M.; Gillevet, P.M. Colonic mucosal microbiome differs from stool microbiome in cirrhosis and hepatic encephalopathy and is linked to cognition and inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G675–G685. [Google Scholar] [CrossRef] [PubMed]
- Iebba, V.; Guerrieri, F.; Di Gregorio, V.; Levrero, M.; Gagliardi, A.; Santangelo, F.; Sobolev, A.P.; Circi, S.; Giannelli, V.; Mannina, L.; et al. Combining amplicon sequencing and metabolomics in cirrhotic patients highlights distinctive microbiota features involved in bacterial translocation, systemic inflammation and hepatic encephalopathy. Sci. Rep. 2018, 8, 8210. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ji, F.; Guo, J.; Shi, D.; Fang, D.; Li, L. Dysbiosis of small intestinal microbiota in liver cirrhosis and its association with etiology. Sci. Rep. 2016, 6, 34055. [Google Scholar] [CrossRef] [PubMed]
- Kakiyama, G.; Pandak, W.M.; Gillevet, P.M.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; Takei, H.; Muto, A.; Nittono, H.; Ridlon, J.M.; et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J. Hepatol. 2013, 58, 949–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clements, W.D.; Parks, R.; Erwin, P.; Halliday, M.I.; Barr, J.; Rowlands, B.J. Role of the gut in the pathophysiology of extrahepatic biliary obstruction. Gut 1996, 39, 587–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzo-Zuniga, V.; Bartoli, R.; Planas, R.; Hofmann, A.F.; Viñado, B.; Hagey, L.R.; Hernández, J.M.; Mañé, J.; Alvarez, M.A.; Ausina, V.; et al. Oral bile acids reduce bacterial overgrowth, bacterial translocation, and endotoxemia in cirrhotic rats. Hepatology 2003, 37, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Renga, B.; Mencarelli, A.; Cipriani, S.; D’Amore, C.; Carino, A.; Bruno, A.; Francisci, D.; Zampella, A.; Distrutti, E.; Fiorucci, S. The bile acid sensor FXR is required for immune-regulatory activities of TLR-9 in intestinal inflammation. PLoS ONE 2013, 8, e54472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbeke, L.; Farre, R.; Trebicka, J.; Komuta, M.; Roskams, T.; Klein, S.; Elst, I.V.; Windmolders, P.; Vanuytsel, T.; Nevens, F.; et al. Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology 2014, 59, 2286–2298. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, L.; Farre, R.; Verbinnen, B.; Covens, K.; Vanuytsel, T.; Verhaegen, J.; Komuta, M.; Roskams, T.; Chatterjee, S.; Annaert, P.; et al. The FXR agonist obeticholic acid prevents gut barrier dysfunction and bacterial translocation in cholestatic rats. Am. J. Pathol. 2015, 185, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Qin, N.; Guo, J.; Qian, G.; Fang, D.; Shi, D.; Xu, M.; Yang, F.; He, Z.; Van Nostrand, J.D.; et al. Functional gene arrays-based analysis of fecal microbiomes in patients with liver cirrhosis. BMC Genom. 2014, 15, 753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Jiang, S.; Chen, Y.; Zhao, X.; Li, H.; Lin, W.; Li, B.; Wang, X.; Yuan, J.; Sun, Y. Cirrhosis related functionality characteristic of the fecal microbiota as revealed by a metaproteomic approach. BMC Gastroenterol. 2016, 16, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.W.; Ling, Z.X.; Lu, H.F.; Zuo, J.; Sheng, J.F.; Zheng, S.S.; Li, L.J. Changes of gut bacteria and immune parameters in liver transplant recipients. Hepatobiliary Pancreat. Dis. Int. 2012, 11, 40–50. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Fagan, A.; Sikaroodi, M.; White, M.B.; Sterling, R.K.; Gilles, H.; Heuman, D.; Stravitz, R.T.; Matherly, S.C.; Siddiqui, M.S.; et al. Liver transplant modulates gut microbial dysbiosis and cognitive function in cirrhosis. Liver Transpl. 2017, 23, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.Y.; Yang, Y.S.; Qu, W.; Zhu, Z.J.; Wei, L.; Ye, Z.S.; Zhang, J.R.; Sun, X.Y.; Zeng, Z.G. Gut microbiota of liver transplantation recipients. Sci. Rep. 2017, 7, 3762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, J.S.; Kakiyama, G.; Cox, I.J.; Nittono, H.; Takei, H.; White, M.; Fagan, A.; Gavis, E.A.; Heuman, D.M.; Gilles, H.C.; et al. Alterations in gut microbial function following liver transplant. Liver Transpl. 2018, 24, 752–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; He, J.; Wu, Z.; Xu, W.; Zhang, H.; Ye, P.; Yang, J.; Zhen, S.; Li, L. Assessment of microbiome variation during the perioperative period in liver transplant patients: A retrospective analysis. Microb. Ecol. 2013, 65, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Doycheva, I.; Leise, M.D.; Watt, K.D. The intestinal microbiome and the liver transplant recipient: What we know and what we need to know. Transplantation 2016, 100, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Luo, Z.; Li, Z.; Deng, M.; Liu, H.; Zhu, B.; Ruan, B.; Li, L. Structural shifts of fecal microbial communities in rats with acute rejection after liver transplantation. Microb. Ecol. 2012, 64, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Nagao, M.; Miyamoto, K.; Oka, K.; Takahashi, M.; Yamamoto, M.; Matsumura, Y.; Kaido, T.; Uemoto, S.; Ichiyama, S. Longitudinal analysis of the intestinal microbiota in liver transplantation. Transplant. Direct. 2017, 3, e144. [Google Scholar] [CrossRef] [PubMed]
- Tabibian, J.H.; Ali, A.H.; Lindor, K.D. Primary sclerosing cholangitis, part 1, epidemiology, etiopathogenesis, clinical features, and treatment. Gastroenterol. Hepatol. 2018, 14, 293–304. [Google Scholar]
- Dyson, J.K.; Beuers, U.; Jones, D.E.J.; Lohse, A.W.; Hudson, M. Primary sclerosing cholangitis. Lancet 2018, 391, 2547–2559. [Google Scholar] [CrossRef]
- Xu, B.; Broome, U.; Ericzon, B.G.; Sumitran-Holgersson, S. High frequency of autoantibodies in patients with primary sclerosing cholangitis that bind biliary epithelial cells and induce expression of CD44 and production of interleukin 6. Gut 2002, 51, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terjung, B.; Sohne, J.; Lechtenberg, B.; Gottwein, J.; Muennich, M.; Herzog, V.; Mähler, M.; Sauerbruch, T.; Spengler, U. p-ANCAs in autoimmune liver disorders recognise human beta-tubulin isotype 5 and cross-react with microbial protein FtsZ. Gut 2010, 59, 808–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdier, J.; Luedde, T.; Sellge, G. Biliary mucosal barrier and microbiome. Viszeralmedizin 2015, 31, 156–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karrar, A.; Broome, U.; Sodergren, T.; Jaksch, M.; Bergquist, A.; Björnstedt, M.; Sumitran–Holgersson, S. Biliary epithelial cell antibodies link adaptive and innate immune responses in primary sclerosing cholangitis. Gastroenterology 2007, 132, 1504–1514. [Google Scholar] [CrossRef] [PubMed]
- Rossen, N.G.; Fuentes, S.; Boonstra, K.; D’Haens, G.R.; Heilig, H.G.; Zoetendal, E.G.; de Vos, W.M.; Ponsioen, C.Y. The mucosa-associated microbiota of PSC patients is characterized by low diversity and low abundance of uncultured Clostridiales II. J. Crohns Colitis 2015, 9, 342–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kummen, M.; Holm, K.; Anmarkrud, J.A.; Nygård, S.; Vesterhus, M.; Høivik, M.L.; Trøseid, M.; Marschall, H.U.; Schrumpf, E.; Moum, B.; et al. The gut microbial profile in patients with primary sclerosing cholangitis is distinct from patients with ulcerative colitis without biliary disease and healthy controls. Gut 2017, 66, 611–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabino, J.; Vieira-Silva, S.; Machiels, K.; Joossens, M.; Falony, G.; Ballet, V.; Ferrante, M.; Van Assche, G.; Van der Merwe, S.; Vermeire, S.; et al. Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut 2016, 65, 1681–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughn, B.P.; Kaiser, T.; Staley, C.; Hamilton, M.J.; Reich, J.; Graiziger, C.; Singroy, S.; Kabage, A.J.; Sadowsky, M.J.; Khoruts, A. A pilot study of fecal bile acid and microbiota profiles in inflammatory bowel disease and primary sclerosing cholangitis. Clin. Exp. Gastroenterol. 2019, 12, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalanka-Tuovinen, J.; Salojarvi, J.; Salonen, A.; Immonen, O.; Garsed, K.; Kelly, F.M.; Zaitoun, A.; Palva, A.; Spiller, R.C.; de Vos, W.M. Faecal microbiota composition and host-microbe cross-talk following gastroenteritis and in postinfectious irritable bowel syndrome. Gut 2014, 63, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Rajilic-Stojanovic, M.; de Vos, W.M. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol. Rev. 2014, 38, 996–1047. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Bao, X.; Goel, A.; Colombel, J.F.; Pekow, J.; Jabri, B.; Williams, K.M.; Castillo, A.; Odin, J.A.; Meckel, K.; et al. The features of mucosa-associated microbiota in primary sclerosing cholangitis. Aliment. Pharmacol. Ther. 2016, 43, 790–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, J.; Palmela, C.; Brito, H.; Bao, X.; Goel, A.; Bao, X.; Ruiqi, H.; Moura-Santos, P.; Pereira da Silva, J.; Oliveira, A.; et al. The gut microbiota, bile acids and their correlations in primary sclerosing cholangitis associated with inflammatory bowel disease. United Eur. Gastroenterol. J. 2018, 6, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quraishi, M.N.; Sergeant, M.; Kay, G.; Iqbal, T.; Chan, J.; Constantinidou, C.; Trivedi, P.; Ferguson, J.; Adams, D.H.; Pallen, M.; et al. The gut-adherent microbiota of PSC-IBD is distinct to that of IBD. Gut 2017, 66, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Bajer, L.; Kverka, M.; Kostovcik, M.; Macinga, P.; Dvorak, J.; Stehlikova, Z.; Brezina, J.; Wohl, P.; Spicak, J.; Drastich, P. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J. Gastroenterol. 2017, 23, 4548–4558. [Google Scholar] [CrossRef] [PubMed]
- Kummen, M.; Hov, J.R. Response to ‘Faecal microbiota profiles as diagnostic biomarkers in primary sclerosing cholangitis’. Gut 2017, 66, 755–756. [Google Scholar] [CrossRef] [PubMed]
- Rühlemann, M.C.; Heinsen, F.A.; Zenouzi, R.; Lieb, W.; Franke, A.; Schramm, C. Faecal microbiota profiles as diagnostic biomarkers in primary sclerosing cholangitis. Gut 2017, 66, 753–754. [Google Scholar] [CrossRef] [PubMed]
- Iwasawa, K.; Suda, W.; Tsunoda, T.; Oikawa-Kawamoto, M.; Umetsu, S.; Inui, A.; Fujisawa, T.; Morita, H.; Sogo, T.; Hattori, M. Characterisation of the faecal microbiota in Japanese patients with paediatric-onset primary sclerosing cholangitis. Gut 2017, 66, 1344–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaska, L.; Sledzinski, T.; Chomiczewska, A.; Dettlaff-Pokora, A.; Swierczynski, J. Improved glucose metabolism following bariatric surgery is associated with increased circulating bile acid concentrations and remodeling of the gut microbiome. World J. Gastroenterol. 2016, 22, 8698–8719. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.J.; Hirschfield, G.M.; Gershwin, M.E. Obeticholic acid for the treatment of primary biliary cirrhosis. Expert. Rev. Clin. Pharmacol. 2016, 9, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.C.; Bajaj, J.S. The human gut microbiome in liver diseases. Semin. Liver Dis. 2017, 37, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Sclair, S.N.; Little, E.; Levy, C. Current concepts in primary biliary cirrhosis and primary sclerosing cholangitis. Clin. Transl. Gastroenterol. 2015, 6, e109. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Saito, H.; Ueno, Y.; Uto, H.; Obara, K.; Sakaida, I.; Shibuya, A.; Seike, M.; Nagoshi, S.; Segawa, M.; et al. Evidence-based clinical practice guidelines for liver cirrhosis 2015. J. Gastroenterol. 2016, 51, 629–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, K.; Takahashi, A.; Fujita, M.; Imaizumi, H.; Hayashi, M.; Okai, K.; Ohira, H. Dysbiosis of oral microbiota and its association with salivary immunological biomarkers in autoimmune liver disease. PLoS ONE 2018, 13, e0198757. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.X.; Fang, D.Q.; Shi, D.; Chen, D.Y.; Yan, R.; Zhu, Y.X.; Chen, Y.F.; Shao, L.; Guo, F.F.; Wu, W.R.; et al. Alterations and correlations of the gut microbiome, metabolism and immunity in patients with primary biliary cirrhosis. Environ. Microbiol. 2016, 18, 2272–2286. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Wei, Y.; Li, Y.; Chen, W.; Chen, H.; Wang, Q.; Yang, F.; Miao, Q.; Xiao, X.; Zhang, H.; et al. Gut microbial profile is altered in primary biliary cholangitis and partially restored after UDCA therapy. Gut 2018, 67, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.G.; Feng, Y.; Theve, E.J.; Raczynski, A.R.; Fiala, J.L.; Doernte, A.L.; Williams, M.; McFaline, J.L.; Essigmann, J.M.; Schauer, D.B.; et al. Gut microbes define liver cancer risk in mice exposed to chemical and viral transgenic hepatocarcinogens. Gut 2010, 59, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X.; Yan, H.X.; Liu, Q.; Yang, W.; Wu, H.P.; Dong, W.; Tang, L.; Lin, Y.; He, Y.Q.; Zou, S.S.; et al. Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology 2010, 52, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Yu, L.X.; Yang, W.; Tang, L.; Lin, Y.; Wu, H.; Zhai, B.; Tan, Y.X.; Shan, L.; Liu, Q.; et al. Profound impact of gut homeostasis on chemically-induced pro-tumorigenic inflammation and hepatocarcinogenesis in rats. J. Hepatol. 2012, 57, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Dapito, D.H.; Mencin, A.; Gwak, G.Y.; Pradere, J.P.; Jang, M.K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.X.; Schwabe, R.F. The gut microbiome and liver cancer: Mechanisms and clinical translation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H. Improve gut microbiome: A new horizon of cancer therapy. Hepatobiliary Surg. Nutr. 2017, 6, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Grat, M.; Wronka, K.M.; Krasnodebski, M.; Lewandowski, Z.; Kosińska, I.; Grąt, K.; Stypułkowski, J.; Rejowski, S.; Wasilewicz, M.; Gałęcka, M.; et al. Profile of gut microbiota associated with the presence of hepatocellular cancer in patients with liver cirrhosis. Transplant. Proc. 2016, 48, 1687–1691. [Google Scholar] [CrossRef] [PubMed]
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Paroni Sterbini, F.; Petito, V.; et al. Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in non-alcoholic fatty liver disease. Hepatology 2018, 69, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Li, A.; Jiang, J.; Zhou, L.; Yu, Z.; Lu, H.; Xie, H.; Chen, X.; Shao, L.; Zhang, R.; et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut 2019, 68, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Gupta, H.; Youn, G.S.; Shin, M.J.; Suk, K.T. Role of gut microbiota in hepatocarcinogenesis. Microorganisms 2019, 7, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adolph, T.E.; Grander, C.; Moschen, A.R.; Tilg, H. Liver–Microbiome Axis in Health and Disease. Trends Immunol. 2018, 39, 712–723. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Phylum | Class | Order | Family | Genus (Species) |
|---|---|---|---|---|
| Firmicutes | Bacilli ↑ [33] * | Bacillales | Bacillaceae | Bacillus ↑ [33] * |
| Lactobacilales | Lactobacillaceae | Lactobacillus ↓ [32] (L. salivarius ↑ [37]) | ||
| Streptococcaceae | Streptococcus ↑ [49] (S. infantarius subsp. Infantarius ↓ [37]) | |||
| Lactococcus ↑ [37] (L. lactis subsp Cremoris ↑ [37]) | ||||
| Enterococcaceae | Enterococcus ↓ [32] | |||
| Clostridia ↓ [33] * [34] † | Clostridiales | Clostridiaceae | Clostridium ↓ [34] † [33] *, | |
| Subdoligranulum ↓ [33] *, | ||||
| Clostridales incertae sedis XIII ↓ [34] † | ||||
| Clostridales incertae sedis XIV ↑ [34] † ↓ [40] | ||||
| Eubacteriaceae | Eubacterium (E. eligenes ↓ [37] | |||
| Ruminococcaceae ↓ [34], [40] † | Ruminococcus ↓ [34] † ↓ [40] (R. lactaris ↓ [37]) | |||
| Faecalibacterium ↓ [34] † (F. prusnitzii ↓ [37], [34] †) | ||||
| Anaerofilum ↓ [34] † | ||||
| Oscillospiraceae | Oscillibacter ↓ [34] † | |||
| Lachnospiraceae ↓ [40] ↑ [34] † | Dorea ↑ [34] † | |||
| Rosaeburia (R. homnis ↓ [37], R. intesatinalis ↓ [37]) | ||||
| Blautia ↑ [34] † | ||||
| Coprococcus ↓ [37] (C. eutactus ↓ [37]) | ||||
| Acidaminococcaceae | Megasphaera ↑ [34] † | |||
| Unclassified ↓ [34] | ||||
| Negativicutes | Selenomonadales | Veillonellaceae ↑ [39] | ||
| Bifidobacteriaceae | Bifidobacterium ↓ [32,34] ↑ [44] | |||
| Verucomicrobia | Verucomicrobiae | Verucomicrobiales | Verucomicrobiaceae | Akkermansia ↓ [37] |
| Bacteroidetes ↓ [33] | Bacteroidia | Bacteroidales | Bacteroidaceae ↓ [33,39,40] | |
| Porphyromonadaceae ↓ [39] | ||||
| Prevotelaceae ↑ [38] ↓ [40] | ||||
| Rikenellaceae | Alistipes (A. putredinis ↓ [37]) | |||
| Proteobacteria ↑ [33] | γ-proteobacteria ↑ [33] * | Enterobacteriales | Enterobacteriaceae ↑ [40] | Klebsiella ↑ [37] (K. pneumonia ↑ [37]) |
| Enterobacter ↑ [44] | ||||
| Oceanospirillales | Halomonadaceae ↑ [40] |
| Phylum | Class | Order | Family | Genus |
|---|---|---|---|---|
| Firmicutes ↑ [90] ↓ [84,88,89] | Bacilli | Lactobacilales | Lactobacillaceae ↑ [89] | Lactobacillus ↑ [89,90,92] |
| Streptococcaceae ↑ [91] | Streptococcus ↑ [92] | |||
| Erysipelotrichales | Erysipelotrichaceae ↑ [91] | Anaerobacter ↑ [92] | ||
| Clostridia ↓ [88] | Clostridiales ↓ [88] | Clostridiaceae | Anaerosporobacter ↓ [88] | |
| Anaerobacter ↑ [92] | ||||
| Clostridium (Cluster XI ↑ [92], C. leptum ↓ [111]) | ||||
| Eubacteriaceae Ruminococcaceae ↓ [84,89,90,92] | Eubacterim (E. rectale ↓ [13]) Ruminococcus ↓ [89] ↑ [93] (R. obeum CA9:39 ↓ [13] R. obeum ↓ [13]) | |||
| Faecalibacterium ↓ [88,89] (F. prausnitzii ↓ [89]) | ||||
| Oscillospira ↓ [93] | ||||
| Oscillospiraceae | Oscillibacter↓ [90,92] | |||
| Lachnospiraceae ↑ [90,91] ↓ [84,89] | Dorea ↑ [90,93] ↓ [89] | |||
| Roseburia ↑ [90] ↓ [89] | ||||
| Robinsoniella ↑ [90] | ||||
| Anaerostipes ↓ [89] | ||||
| Blautia ↑ [91] ↓ [89] | ||||
| Coprococcus ↓ [89] | ||||
| Incertae Sedis Family XI ↑ [92] | Peptoniphilus ↑ [93] | |||
| Anaerococcus ↑ [93] | ||||
| Unclassified | Flavonifracter ↓ [92] | |||
| Negativicutes | Selenomonadales | Veillonellaceae | Allisonella ↑ [88] | |
| Acidaminococcaceae | Phascolarctobacterium ↑ [94] | |||
| Lentisphaerae ↓ [92] | ||||
| Actinobacteria ↓ [84,94] ↑ [93] | Actinobacteria | Actinomycetales | Propionibacteriaceae | Propionibacterium (P. acnes ↑ [93]) |
| Fusobacteria ↑ [91] | ||||
| Verrucomicrobia | Verrucomicrobiae | Verrucomicrobiales | Verrucomicrobiaceae | Akkermansia (A. muciniphila ↓ [100]) |
| Bacteroidetes ↓ [73,93,111] ↑ [84,94] | Bacteroidia | Bacteroidales | Bacteroidaceae ↓ [89] | Bacteroides ↓ [89] |
| Porphyromonadaceae ↑ [88] ↓ [89,90] | Parabacteroides ↑ [88] ↓ [89] | |||
| Odoribacter ↓ [92] | ||||
| Prevotellaceae ↑ [84] | Prevotella ↑ [84,94] ↓ [91,92] | |||
| Rikenellaceae ↓ [89,93] | Alistipes ↓ [89,92] | |||
| Proteobacteria ↑ [84,91] | γ-proteobacteria | Aeromonadales ↑ [88] | Succinivibrionaceae ↑ [88] | |
| Enterobacteriales | Enterobacteriaceae ↑ [84,91,100] | Escherichia ↑ [84,91,92,103,116] | ||
| Shigella ↑ [91] | ||||
| α-proteobacteria | Rhizobiales | Bradyrhizobiaceae | Bradyrhizobium ↑ [93] | |
| Rhodbacterales | Rhodbacteraceae | Rhodbacter ↑ [116] | ||
| δ-proteobacteria | Desulfovibrionale | Desulfovibrionaceae | Biophila ↑ [116] |
| Phylum | Class | Order | Family | Genus (Speices) |
|---|---|---|---|---|
| Firmicutes ↑ [208] # | Bacilli ↑ [38,187] | Bacillales | Stphylococcaceae | Staphylococcus ↑ [183] † |
| Lactobacilales | Lactobacillaceae | Lactobacillus (L. rhamnosus ↓84 L. fermentus ↓ [84], L. ruminis ↓ [37], L. antri ↑ [57], L. crispatus ↑ [57]) | ||
| Streptococcaceae ↑ [38,185,202] | Streptococcus ↑ [37,97] (S. thermophiles ↑ [37], S. infantarius ↑ [37]) | |||
| Enterococcaceae | Enterococcus ↑ [208] # (E. faecalis ↑ [182]) | |||
| Clostridia | Clostridiales | Clostridiaceae | Clostridium ↓ [182] ↑ [97], Clostridium XI ↑ [208] # Clostridium XIV ↓ [208] # [208] (C. methylpentosum ↓ [37], C. saccharolyricun [37] ↓) | |
| Eubacteriaceae | Eubacterium ↓ [97] (E. siraeum ↓ [37]) | |||
| Ruminococcaceae ↓ [37,188,214], [208] # | Subdoligranulum ↓ [211] | |||
| Faecalibacterium ↓ [37,182] (F. prausnitzii ↓ [184]) | ||||
| Ruminococcus (R. sp 18P13 ↓ [37]) | ||||
| Ruminococcacaeae bacterium ↓ [37] | ||||
| Lachnospiraceae ↓ [188,214], [208] # | Dorea ↓ [211] | |||
| Blautia ↓ [214] | ||||
| Butyrivibrio (B. crossotus ↓ [37]) | ||||
| Negativicutes ↑ [185] | Selenomonadales | Veillonellaceae ↑ [38,187,188,204] # | Veillonella ↑ [97], [208] # (V. dispar ↑ [185], V. paruvula [185] ↑) | |
| Acidaminococcaceae | Acidaminococcus ↑ [211] | |||
| Phascolarctobacterium ↓ [37] | ||||
| Actinobacteria ↑ [208] # | Actinobacteriia | Bifidobacteriales | Bifidobacteriaceae | Bifidobacterium ↓ [180,184] ↑ [37] (B. catenulatum ↓ [185]) |
| Fusobacteria [38,97] ↑ | Fusobacteriia | Fusobacteriales | Fusobacteriaceae ↑ [38,188] # | |
| Bacteroidetes ↓ [38,97,185,208] # | Bacteroidia | Bacteroidales | Bacteroidaceae ↓ [38] | Bacteroides ↓ [97] (B. coprocola ↓ [37], B. uniformis ↓ [37]) |
| Prevotellaceae | Prevotella ↑ [97,208] #↓ [37] (P. corpi ↓ [37]) | |||
| Paraprevotella ↓ [37] (P. xylaniphila ↓ [37]) | ||||
| Rikenellaceae ↓ [208] # | Alistipes ↓ [97] (A. shahil ↓ [37]) | |||
| Porphyromonadaceae | Barnecialla ↓ [37] | |||
| Odoribacter ↓ [37] (O. splanchnicus ↓ [37]) | ||||
| Parabacteroides (P. distasonis ↓ [37]) | ||||
| Tannerella ↓ [37] | ||||
| Proteobacteria ↑ [38,97,185,208] # | β-proteobacteria | Burkholderiales | Alcaligenaceae ↑ [188] | |
| Burkholderiaceae | Burkholderia ↑ [214] | |||
| Ralstoniaceae | Ralstonia ↑ [214] | |||
| γ-proteobacteria ↑ [38,187] | Enterobacteriales | Enterobacteriaceae ↑ [38], [188] #, [184,188,217] | Proteus ↑ [214] | |
| Escherichia ↑ (E. coli ↑ [183,187] †) | ||||
| Pasteurellaceae ↑ [38] | ||||
| δ-proteobacteria | Desulfovibrionales | Desulfovibrionaceae | Biophila ↑ (B. wadsworthia ↓ [37]) |
| Phylum | Class | Order | Family | Genus |
|---|---|---|---|---|
| Firmicutes | Bacilli | Lactobacilales | Lactobacillaceae | Lactobacillus ↑ [241] |
| Streptococcaceae | Streptococcus ↑ [249] ↑ # [249] (S. infantis ↑ [249] ↑ # [249] S. alactolyticus ↑ [252] ↑ # [252] S. equi ↑ [249] ↑] # [249], S. parasanguinis [252] ↑ # [252]) | |||
| Enterococcaceae | Enterococcus ↑ [241,249,252] (E. faecium ↑ [252], E.sp.NBRC107345 ↑ [252]) | |||
| Clostridia | Clostridiales | Clostridiaceae | Clostridium ↑ [249] | |
| Clostridiales II ↓ [239] ↓ # [239] | ||||
| Ruminococcaceae | Ruminococcus ↑ # [246,252] (R. gnavus ↓ [249], R. obeum ↑ § [245]) | |||
| Faecalibacterium ↑ # [251] (F. prausnitzii ↓ [249]) | ||||
| Dorea ↓ # [246] | ||||
| Roseburia ↑ # [251] ↓ # [246] ↓ [§267] | ||||
| Blautia ↑ § [245] ↓ # [246] (B. obeum ↑ § [245] ↓ [252], B. faecis ↑ § [245]) | ||||
| Coprococcus ↓ [240,249] (C. catus ↓ [249]) | ||||
| Lachnospiraceae ↑ § [248] | Anaerostipes (A. hardus ↓ [252]) | |||
| Lachnospira ↓ # [246] | ||||
| Negativicutes ↑ [187] | Selenomonadales | Veillonellaceae ↑ [38,187,204] | Veilonella ↑ [240,249,251] ↑ # [240,249] ↓ # [246] | |
| Acidominococcaceae | (V. dispar ↑ [249], ↑ # [249] V. paruvula ↑249], ↑ # [249] V.sp.3_1_44 ↑ [252]) Megasphaera ↑ [240] ↑ § [249] Phascolarctobacterium ↓ [240] | |||
| Actinobacteria | Actinobacteria | Actinomycetales | Micrococcaceae ↑ [249] | Rothia ↑ [249] ↑ # [249] (R. mucilaginosa ↑ [249]) |
| Coriobacteriales | Coriobacteriaceae | Adlercreutzia ↓ [249] (A. equolifaciens ↓ [249] ↓ # [249]) | ||
| Fusobacteria | Fusobacteriia | Fusobacteriales | Fusobacteriaceae | Fusobacterium ↑ [241] ↑ # [246] |
| Bacteroidetes | Bacteroidia | Bacteroidales | Bacteroidaceae | Bacteroides ↓ § [249] |
| Prevotellaceae | Prevotella ↓ § [249] (P. corpi ↓ [249], ↓ # [249]) | |||
| Porphydomonadaceae | Parabacteroides ↓ # [252] (P. distasonis ↓ [252]) | |||
| Barnesiellaceae ↑ [245] | ||||
| Proteobacteria | γ-proteobacteria | Enterobacteriales | Enterobacteriaceae | Escherichia ↑ § [249] |
| Pasteurellales | Pasteurellaceae | Haemophilus ↑ [249] | ||
| Aeromonadales | Succinivibrionaceae | Succinivibrio ↓ [240] | ||
| Desulfovibrionales | Desulfovibrionaceae | Desulfovibrio ↓ [240] |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukui, H. Role of Gut Dysbiosis in Liver Diseases: What Have We Learned So Far? Diseases 2019, 7, 58. https://doi.org/10.3390/diseases7040058
Fukui H. Role of Gut Dysbiosis in Liver Diseases: What Have We Learned So Far? Diseases. 2019; 7(4):58. https://doi.org/10.3390/diseases7040058
Chicago/Turabian StyleFukui, Hiroshi. 2019. "Role of Gut Dysbiosis in Liver Diseases: What Have We Learned So Far?" Diseases 7, no. 4: 58. https://doi.org/10.3390/diseases7040058
APA StyleFukui, H. (2019). Role of Gut Dysbiosis in Liver Diseases: What Have We Learned So Far? Diseases, 7(4), 58. https://doi.org/10.3390/diseases7040058