
Fluorinated Merophosphinine and Phosphinine Dyes: Synthesis and Evaluation of UV-Visible Light Absorption Properties



Abstract
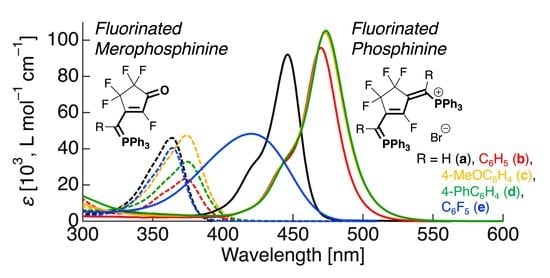
:
1. Introduction
2. Materials and Methods
2.1. General Methods
2.2. Typical Synthetic Procedure for Obtaining 2,4,4,5,5-pentafluoro-3-{1-phenyl-1-(triphenyl-λ5-phosphoniylidene)methyl}-2-cyclopenten-1-one (3b)
2.2.1. 2,4,4,5,5-Pentafluoro-3-{1-phenyl-1-(triphenyl-λ5-phosphoniylidene)methyl}-2-cyclopenten-1-one (3b)
2.2.2. 2,4,4,5,5-Pentafluoro-3-{1-(4-methoxyphenyl)-1-(triphenyl-λ5-phosphoniylidene)methyl}-2-cyclopenten-1-one (3c)
2.2.3. 2,4,4,5,5-Pentafluoro-3-{1-(4-biphenyl)-1-(triphenyl-λ5-phosphoniylidene)methyl}-2-cyclopenten-1-one (3d)
2.2.4. 2,4,4,5,5-Pentafluoro-3-{1-(2,3,4,5,6-pentafluorophenyl)-1-(triphenyl-λ5-phosphoniylidene)methyl}-2-cyclopenten-1-one (3e)
2.3. Typical Synthetic Procedure of [2,4,4,5,5-pentafluoro-3-{1-phenyl-1-(triphenyl-λ5-phosphonilidene)methyl}-2-cyclopentenylidenemethyl]triphenylphosphonium Bromide (4bA)
2.3.1. [2,4,4,5,5-Pentafluoro-3-{1-phenyl-1-(triphenyl-λ5-phosphonilidene)methyl}-2-cyclopentenylidenemethyl]triphenylphosphonium Bromide (4bA)
2.3.2. [2,4,4,5,5-Pentafluoro-3-{1-(4-methoxyphenyl)-1-(triphenyl-λ5-phosphonilidene)methyl}-2-cyclopentenylidenemethyl]triphenylphosphonium Bromide (4cA)
2.3.3. [2,4,4,5,5-Pentafluoro-3-{1-(4-biphenyl)-1-(triphenyl-λ5-phosphonilidene)methyl}-2-cyclopentenylidenemethyl]triphenylphosphonium Bromide (4dA)
2.3.4. [2,4,4,5,5-Pentafluoro-3-{1-(2,3,4,5,6-pentafluorophenyl)-1-(triphenyl-λ5-phosphonilidene)methyl}-2-cyclopentenylidenemethyl]triphenylphosphonium Bromide (4eA)
2.3.5. [2,4,4,5,5-Pentafluoro-3-{(triphenyl-λ5-phosphonilidene)methyl}-2-cyclopentenylidenemethyl]triphenylphosphonium Iodide (4aB)
2.3.6. [2,4,4,5,5-Pentafluoro-3-{(triphenyl-λ5-phosphonilidene)methyl}-2-cyclopentenylidenemethyl]triphenylphosphonium Tetrafluoroborate (4aC)
2.3.7. [2,4,4,5,5-Pentafluoro-3-{(triphenyl-λ5-phosphonilidene)methyl}-2-cyclopentenylidenemethyl]triphenylphosphonium Trifluoromethanesulfonate (4aD)
2.3.8. [2,4,4,5,5-Pentafluoro-3-{(triphenyl-λ5-phosphonilidene)methyl}-2-cyclopentenylidenemethyl]triphenylphosphonium Perchlorate (4aE)
2.4. Quantum Chemical Calculation
2.5. Photophysical Properties
2.6. Thermogravimetric Analyses
3. Results and Discussion
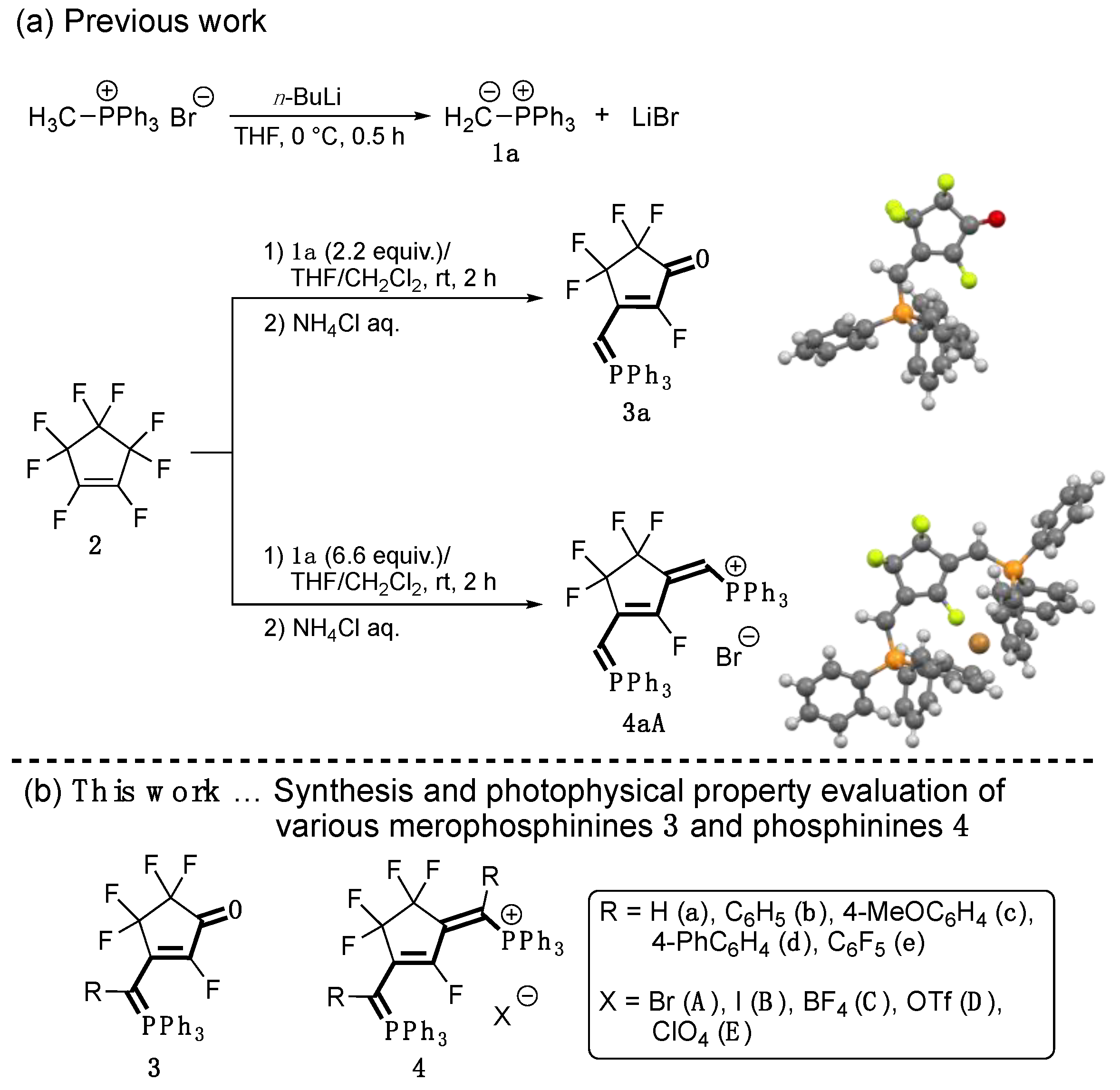
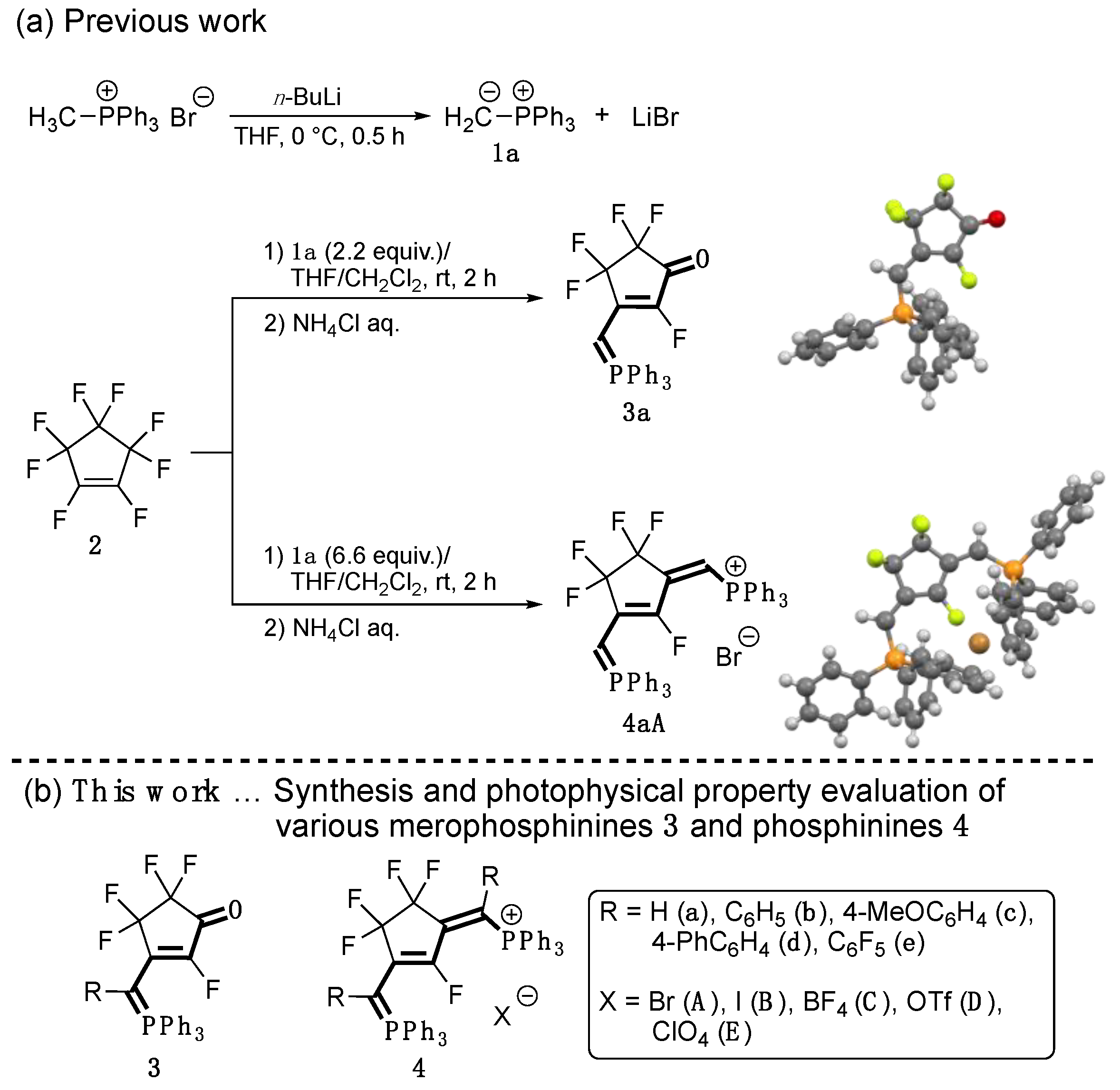
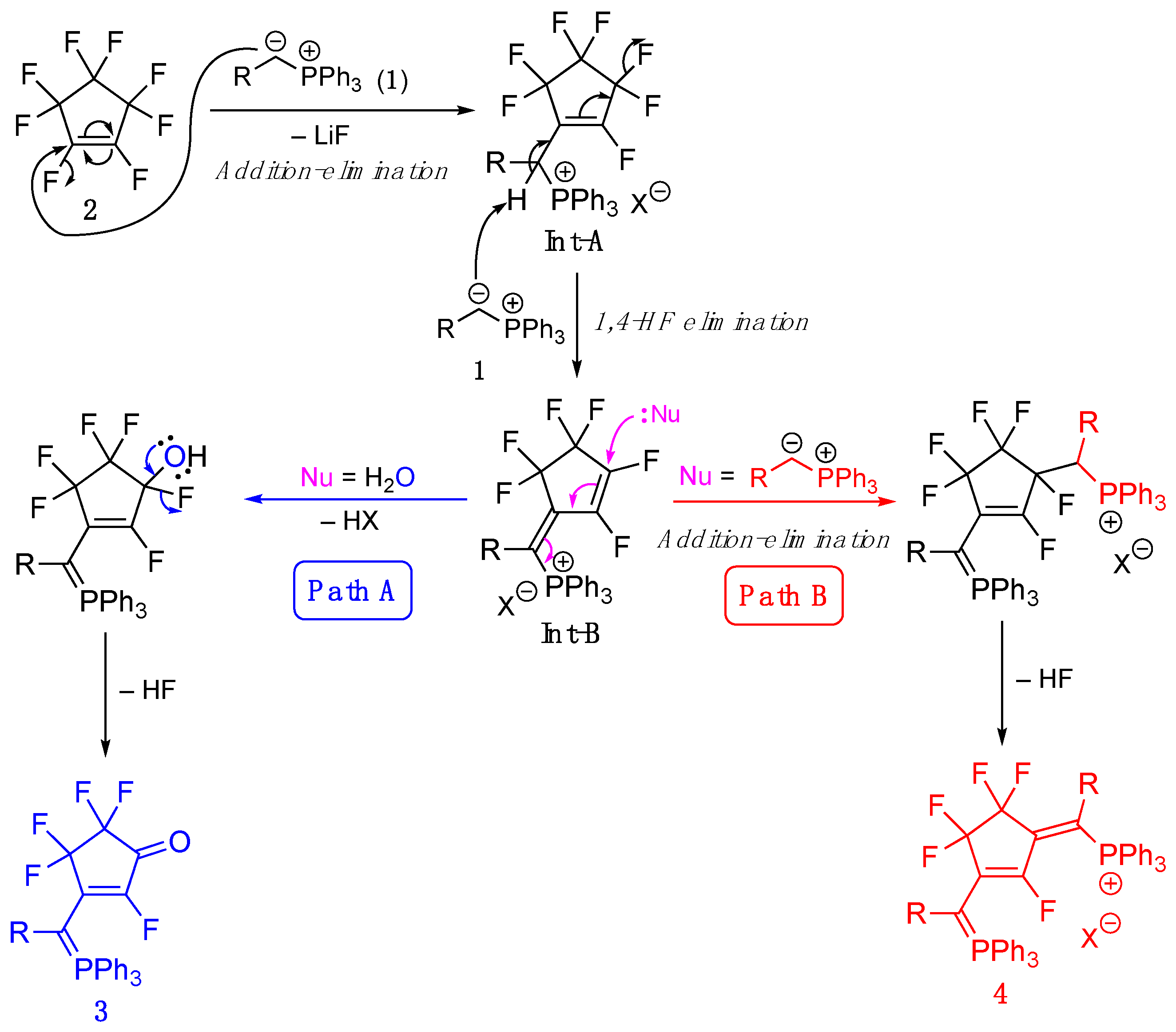
3.1. Synthesis
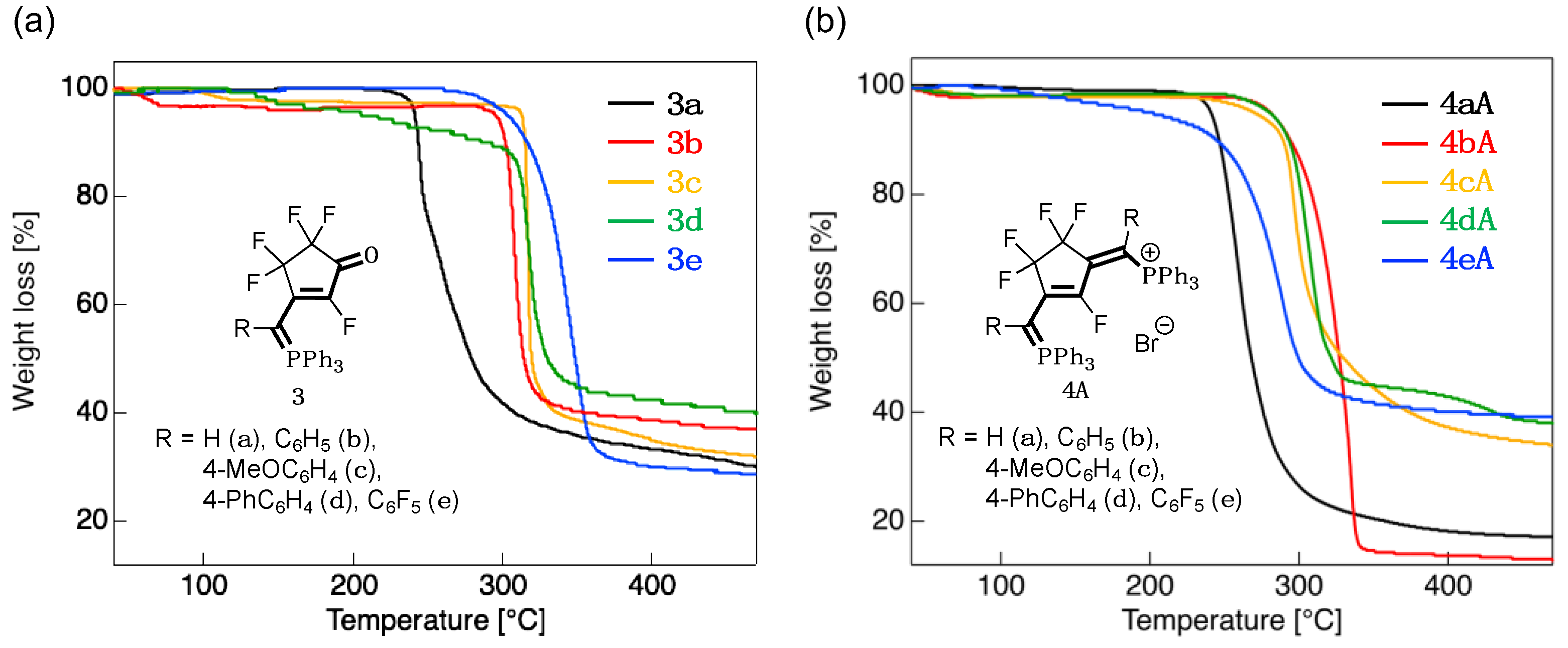
3.2. Thermal Stability
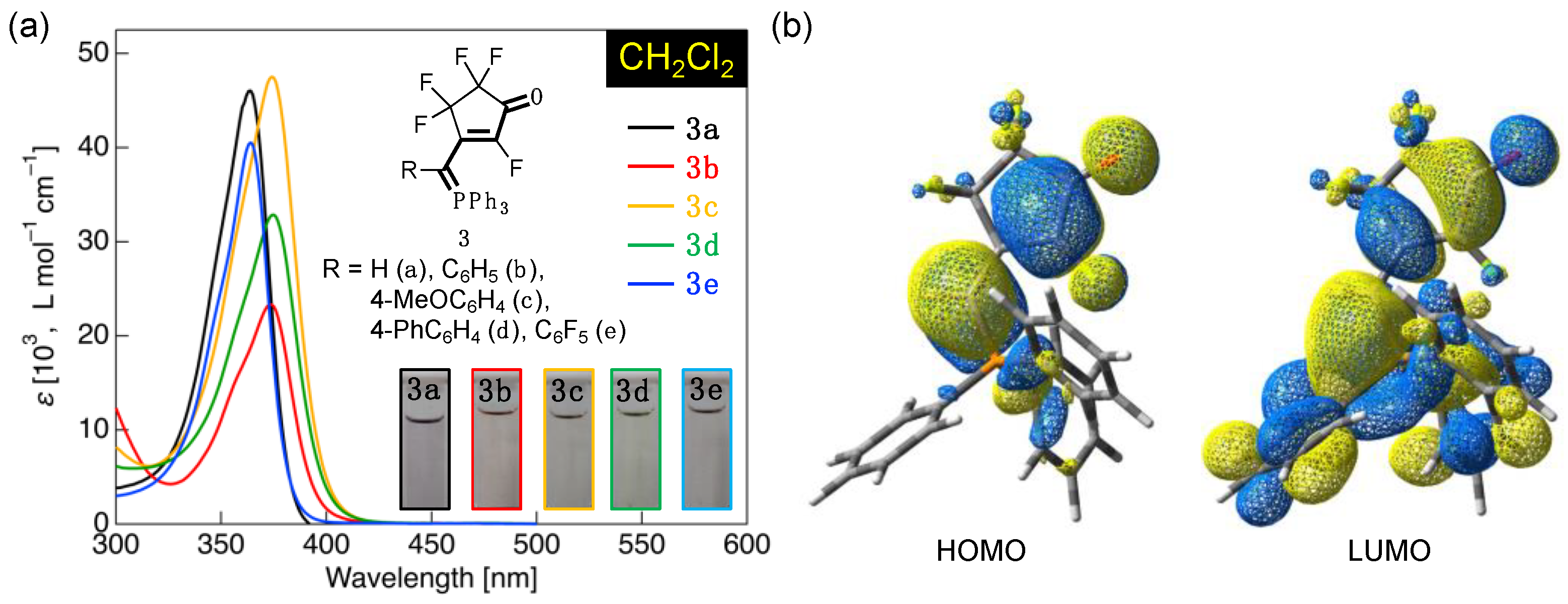
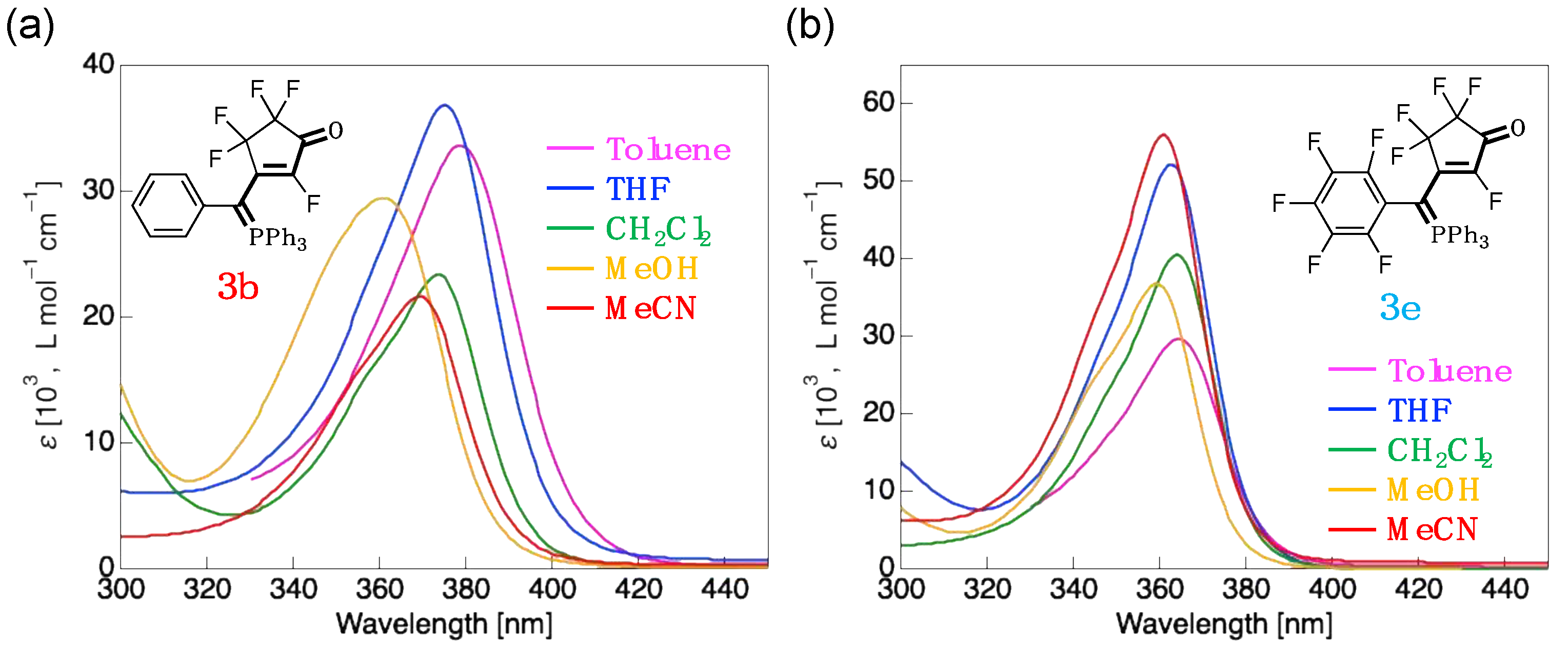
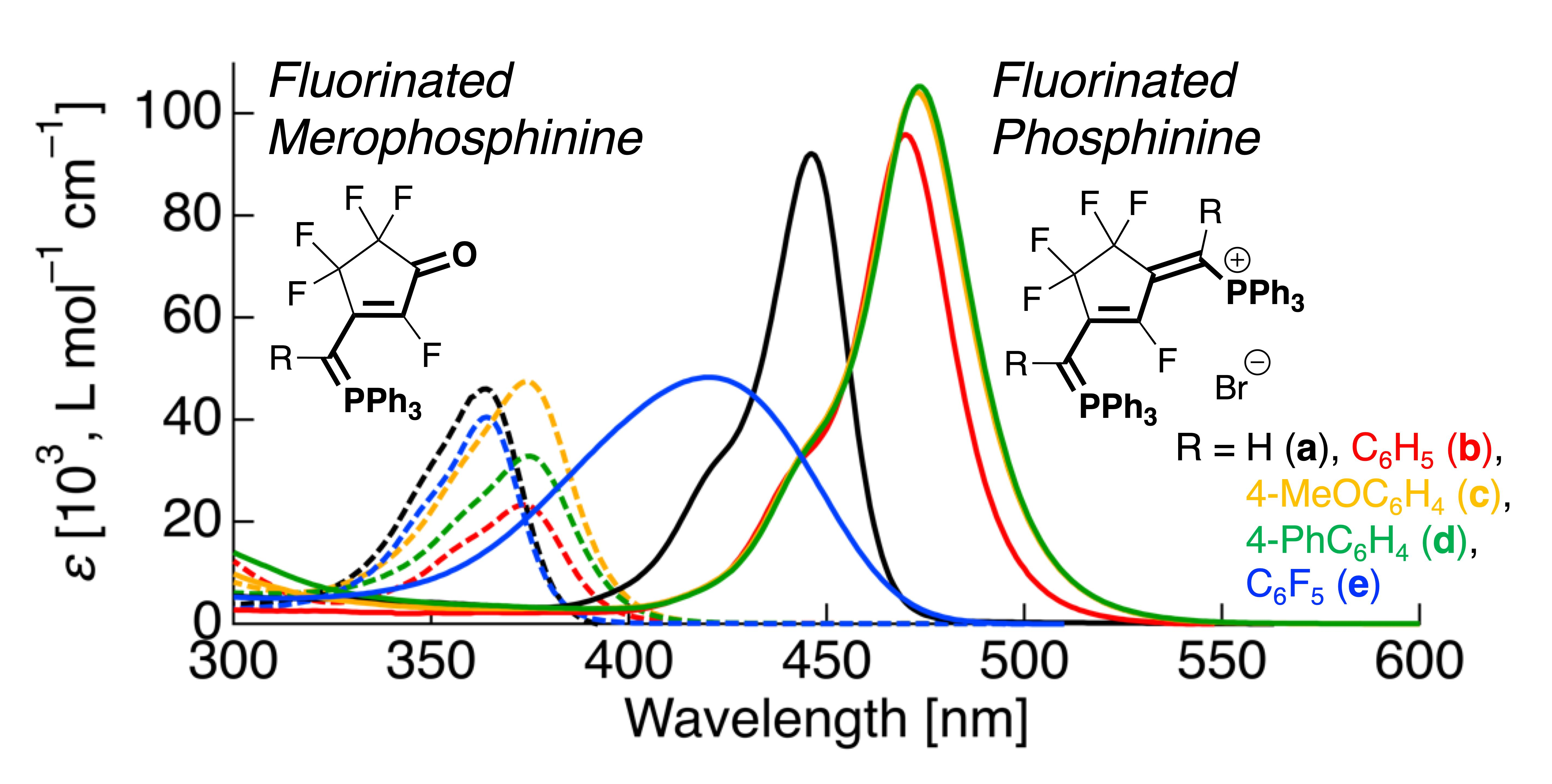
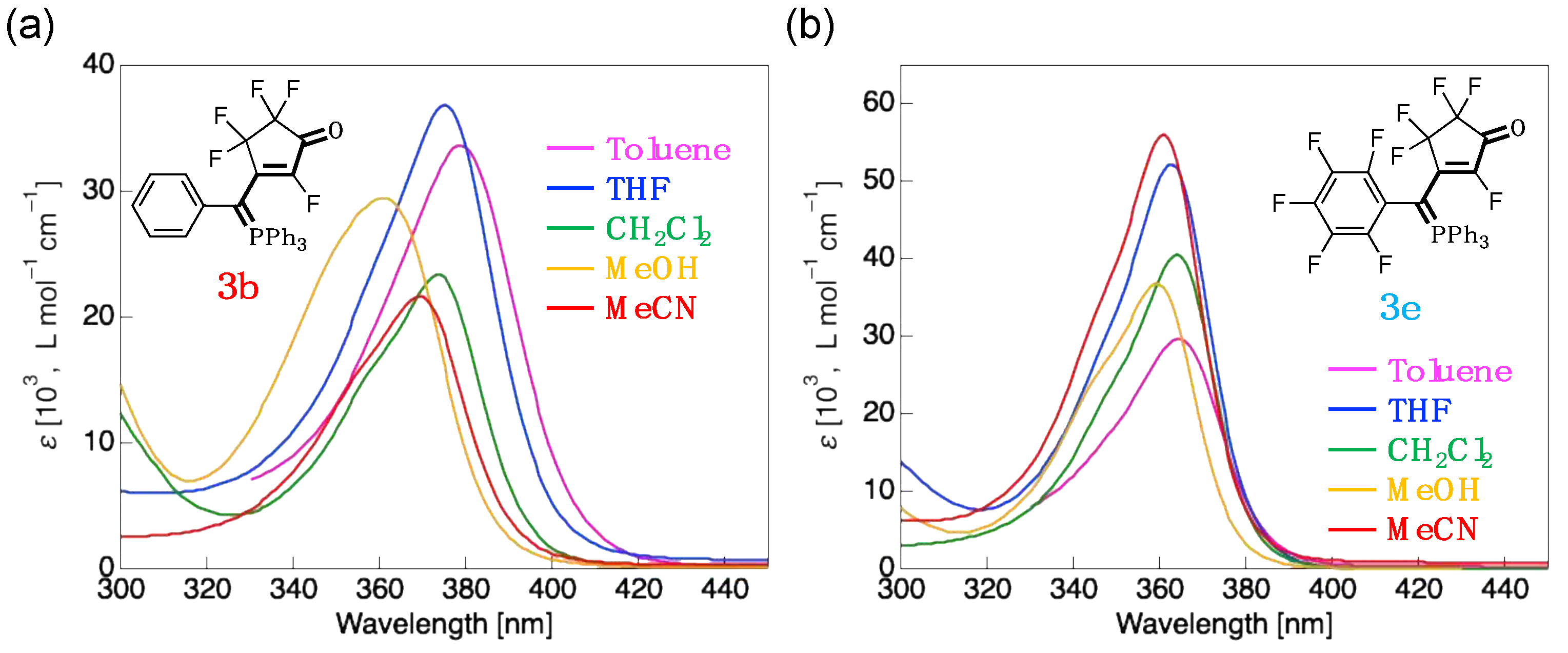
3.3. UV-Vis Absorption Properties of Fluorinated Merophosphinines
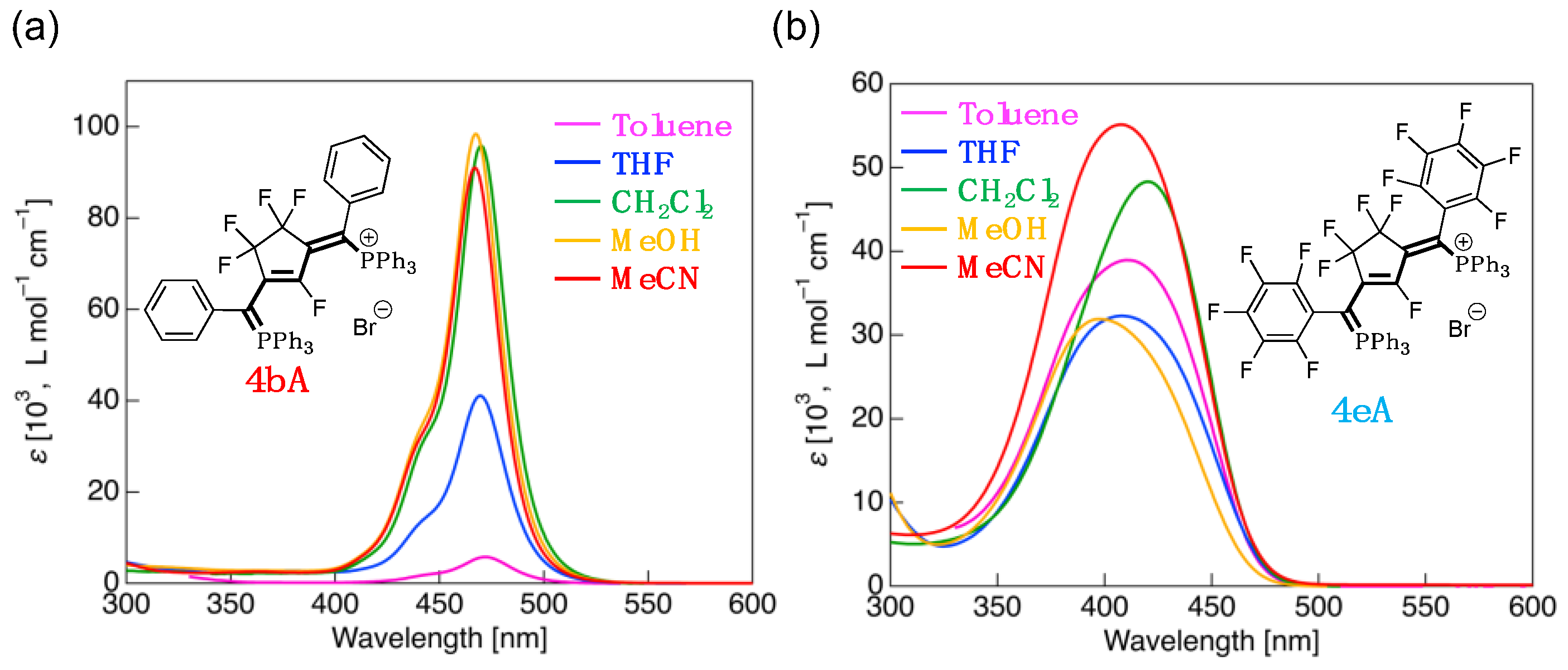
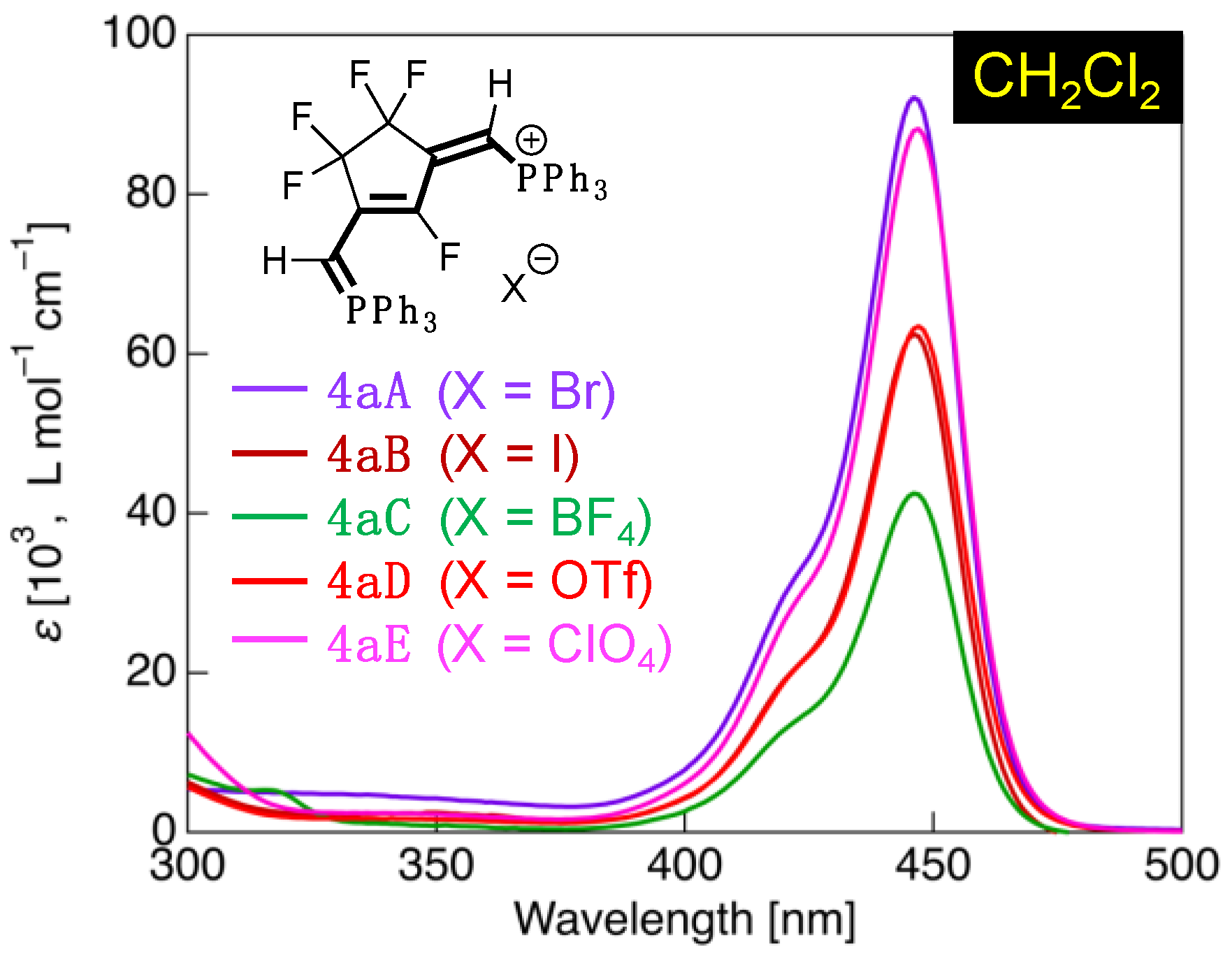
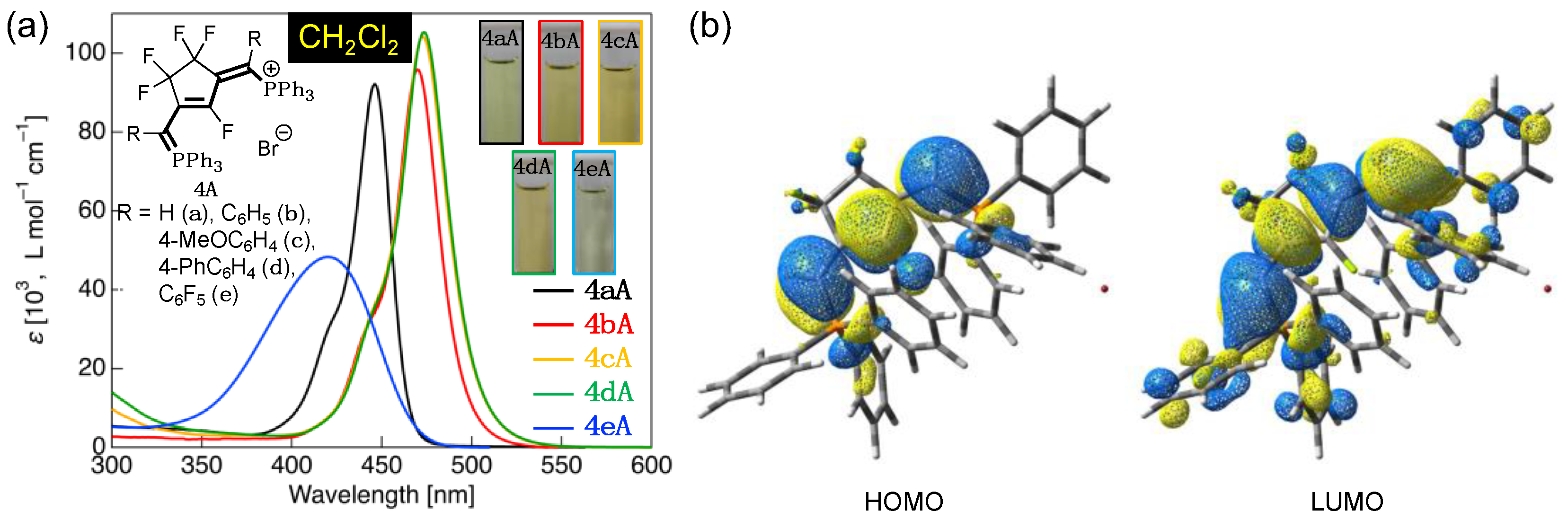
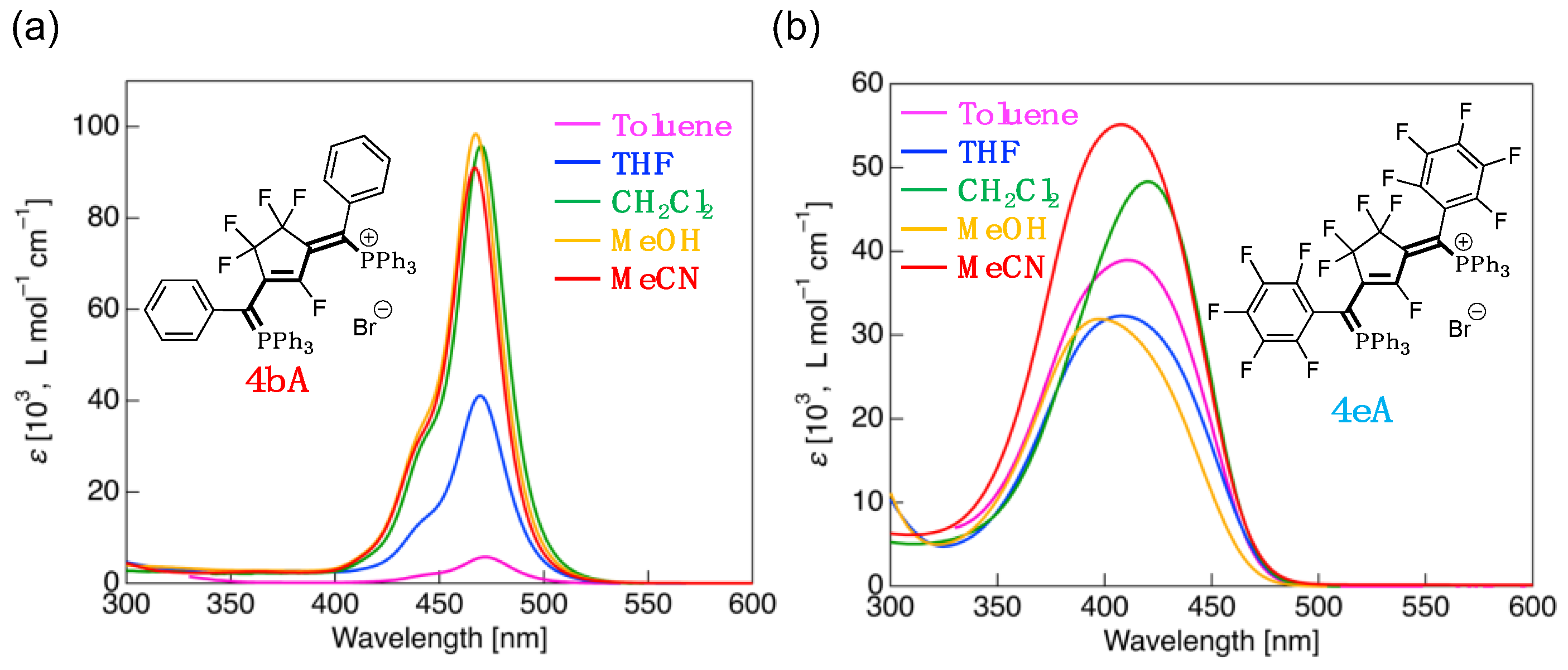
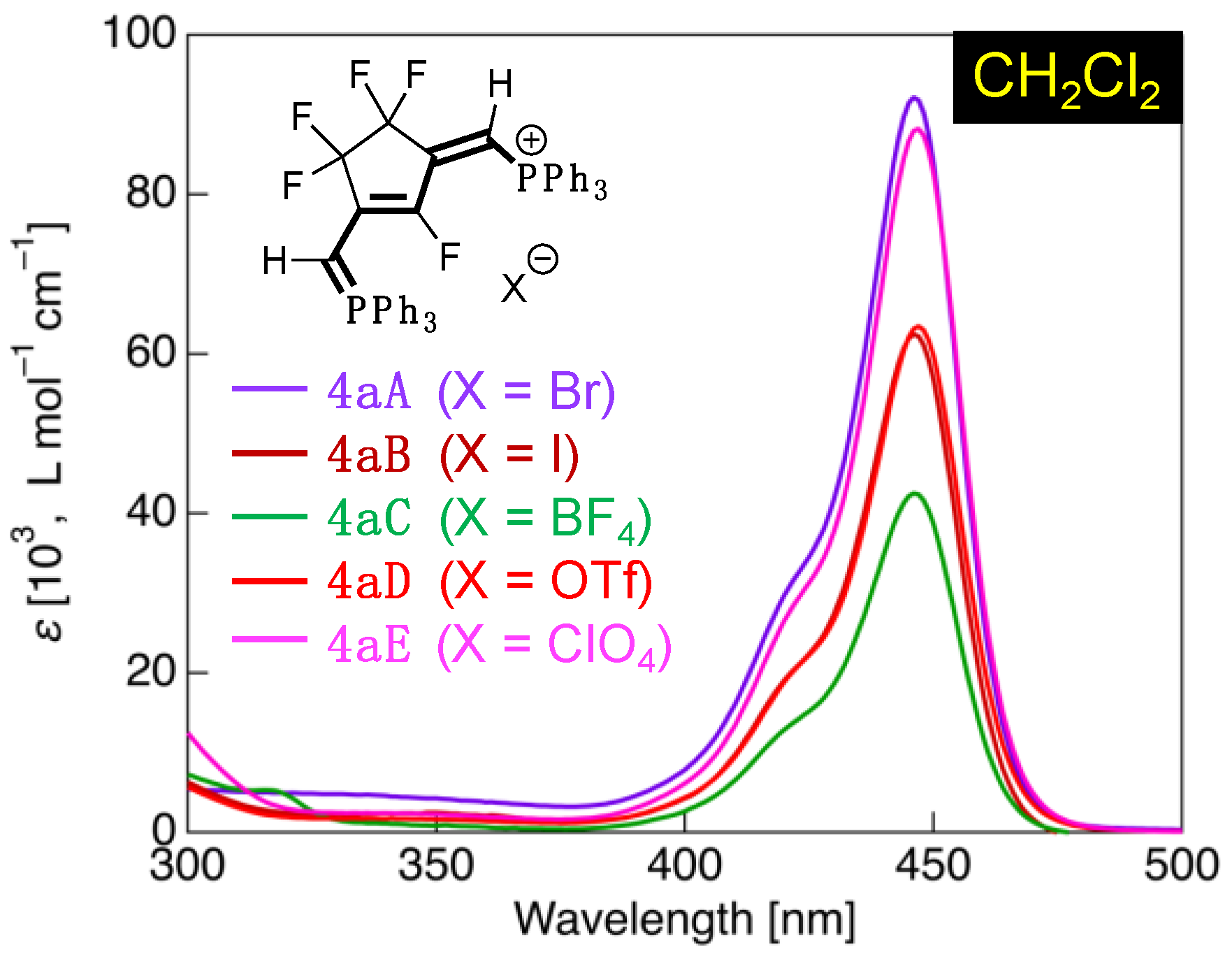
3.4. UV-Vis Absorption Properties of Fluorinated Phosphinines
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mustroph, H.; Stollenwerk, M.; Bressau, V. Current developments in optical data storage with organic dyes. Angew. Chem. Int. Ed. 2006, 45, 2016–2035. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Chen, J. Arylamine organic dyes for dye-sensitized solar cells. Chem. Soc. Rev. 2013, 42, 3453–3488. [Google Scholar] [CrossRef]
- Cai, Y.; Si, W.; Huang, W.; Chen, P.; Shao, J.; Dong, X. Organic dye based nanoparticles for cancer phototheranostics. Small 2018, 14, e1704247. [Google Scholar] [CrossRef] [PubMed]
- Kulinich, A.V.; Ishchenko, A.A.; Bulavko, G.V.; Davidenko, N.A. Effect of structure on the photovoltaic properties of merocyanine dyes in polymer films. Theor. Exp. Chem. 2018, 54, 178–185. [Google Scholar] [CrossRef]
- Reichardt, C. Solvatochromic dyes as solvent polarity indicators. Chem. Rev. 1994, 94, 2319–2358. [Google Scholar] [CrossRef]
- Brooker, L.G.S.; Keyes, G.H.; Sprague, R.H.; van Dyke, R.H.; van Lare, E.; van Zandt, G.; White, F.L.; Cressman, H.W.J.; Dent, S.G., Jr. Color and constitution. X. 1 Absorption of the merocyanines 2. J. Am. Chem. Soc. 1951, 73, 5332–5350. [Google Scholar] [CrossRef]
- Matikonda, S.S.; Helmerich, D.A.; Meub, M.; Beliu, G.; Kollmannsberger, P.; Greer, A.; Sauer, M.; Schnermann, M.J. Defining the basis of cyanine phototruncation enables a new approach to single-molecule localization microscopy. ACS Cent. Sci. 2021, 7, 1144–1155. [Google Scholar] [CrossRef]
- Meguellati, K.; Spichty, M.; Ladame, S. Reversible synthesis and characterization of dynamic imino analogues of trimethine and pentamethine cyanine dyes. Org. Lett. 2009, 11, 1123–1126. [Google Scholar] [CrossRef]
- Gopika, G.S.; Prasad, P.M.H.; Lekshmi, A.G.; Lekshmypriya, S.; Sreesaila, S.; Arunima, C.; Kumar, M.S.; Anil, A.; Sreekumar, A.; Pillai, Z.S. Chemistry of cyanine dyes—A review. Mater. Today Proc. 2021, 46, 3102–3108. [Google Scholar] [CrossRef]
- Shindy, H.A. Fundamentals in the chemistry of cyanine dyes: A review. Dye. Pigment. 2017, 145, 505–513. [Google Scholar] [CrossRef]
- Tatikolov, A.S. Polymethine dyes as spectral-fluorescent probes for biomacromolecules. J. Photochem. Photobiol. C Photochem. Rev. 2012, 13, 55–90. [Google Scholar] [CrossRef]
- Weimei, L.; Zhenghua, Z.; Zhuguang, Y.; Mengzhen, H.; Bingkui, W. Novel laser dyes: Some bridged pentamethine phosphinines. Dye. Pigment. 1990, 14, 211–216. [Google Scholar] [CrossRef]
- Depoorter, H.; Nys, J.; van Dormael, A. New classes of phosphorus-containing dyes. Tetrahedron Lett. 1961, 2, 199–205. [Google Scholar] [CrossRef]
- Depoorter, H.; Nys, J.; van Dormael, A. Phosphorus containing polymethine dyes I. Methods of preparation and chemical properties. Bull. Soc. Chim. Belg. 1964, 73, 921–943. [Google Scholar] [CrossRef]
- Yamada, S.; Ishii, E.; Konno, T.; Ishihara, T. Preparation of perfluorocyclopentenylmetal species and their cross-coupling reaction with electrophiles—Remarkable accesses to versatile perfluorocyclopentene derivatives. Tetrahedron 2008, 64, 4215–4223. [Google Scholar] [CrossRef]
- Yamada, S.; Noma, M.; Hondo, K.; Konno, T.; Ishihara, T. Preparation and addition-elimination reactions of benzyl α,β,β-trifluoracrylate. A new stereoselective approach to (Z)-β-substituted α,β-difluoracrylates. J. Org. Chem. 2008, 73, 522–528. [Google Scholar] [CrossRef]
- Yamada, S.; Takahashi, T.; Konno, T.; Ishihara, T. A novel fluorine-metal exchange reaction of pentafluorocrotonate with organocuprate. Generation of β-metallated tetrafluorocrotonate and its cross-coupling reaction. Chem. Commun. 2007, 21, 3679–3681. [Google Scholar] [CrossRef]
- Yamada, S.; Ishii, E.; Konno, T.; Ishihara, T. Reaction of perfluorocyclopentene with various carbon nucleophiles—Heteroaromatic lithium reagents, enolate and phosphonium ylide. Org. Biomol. Chem. 2007, 5, 1442–1449. [Google Scholar] [CrossRef]
- Yamada, S.; Noma, M.; Konno, T.; Ishihara, T.; Yamanaka, H. Novel synthesis of (Z)-difluoroacrylates via a highly stereoselective addition-elimination reaction. Org. Lett. 2006, 8, 843–845. [Google Scholar] [CrossRef]
- Yamada, S.; Konno, T.; Ishihara, T.; Yamanaka, H. Reaction of octafluorocyclopentene with various carbon nucleophiles. J. Fluor. Chem. 2005, 126, 125–133. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Hohenstein, E.G.; Chill, S.T.; Sherrill, C.D. Assessment of the performance of the M05-2X and M06-2X exchange-correlation functionals for noncovalent interactions in biomolecules. J. Chem. Theory Comput. 2008, 4, 1996–2000. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jensen, J.H. Improving the efficiency and convergence of geometry optimization with the polarizable continuum model: New energy gradients and molecular surface tessellation. J. Comput. Chem. 2004, 25, 1449–1462. [Google Scholar] [CrossRef] [PubMed]
- Kulinich, A.V.; Mikitenko, E.K.; Ishchenko, A.A. Scope of negative solvatochromism and solvatofluorochromism of merocyanines. Phys. Chem. Chem. Phys. 2016, 18, 3444–3453. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.; Choi, M.G.; Jeon, H.R.; Chang, S.-K. Negative solvatochromism of merocyanine dyes: Application as water content probes for organic solvents. Sens. Actuators B Chem. 2011, 157, 14–18. [Google Scholar] [CrossRef]
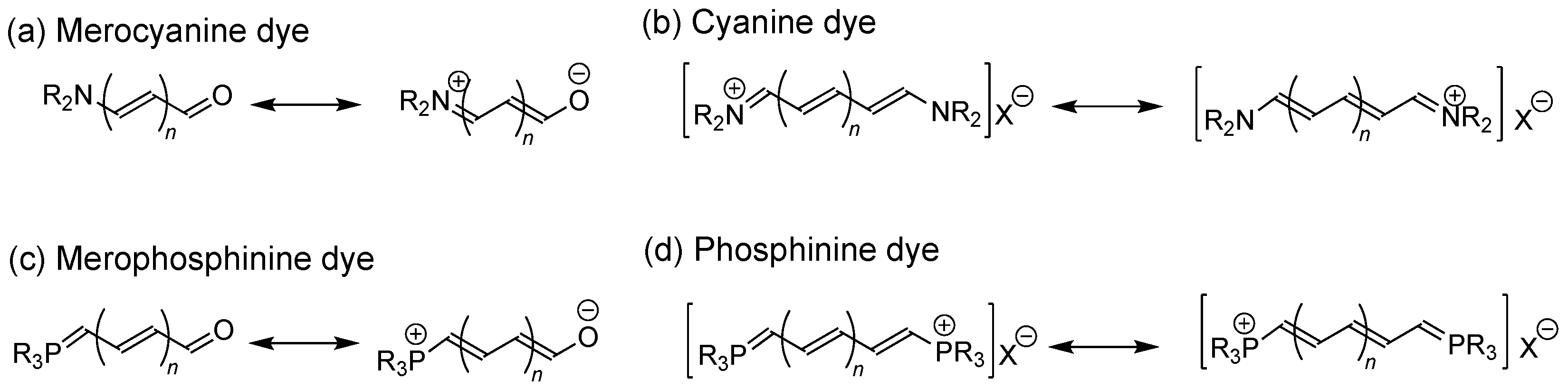



{kind=link}
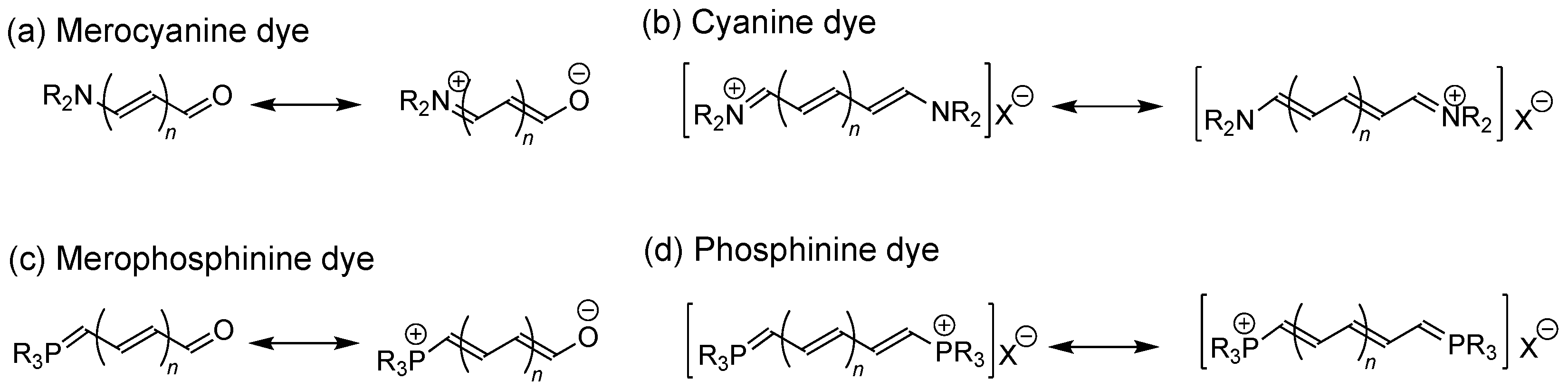{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
| Fluorinated Merophosphinines | |||
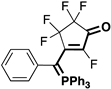 |  |  |  |
| 3b (11%) | 3c (19%) | 3d (2%) | 3e (26%) |
| Fluorinated Phosphinines | |||
 | 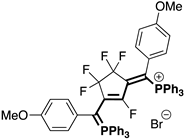 | 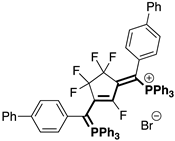 | 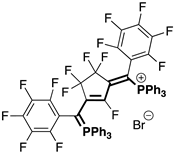 |
| 4bA (23%) | 4cA (44%) | 4dA (51%) | 4eA (10%) |
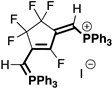 | 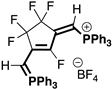 |  | 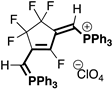 |
| 4aB (54%) | 4aC (32%) | 4aD (89%) | 4aE (20%) |
| Compound | Tdec [°C] 1 | Compound | Tdec [°C] 1 |
|---|---|---|---|
| 3a | 241 | 4aA | 241 |
| 3b | 295 | 4bA | 281 |
| 3c | 313 | 4cA | 270 |
| 3d | 217 | 4dA | 280 |
| 3e | 304 | 4eA | 203 |
| Compound | Solvent | λabs [nm] 1 (ε [L·mol–1·cm–1]) | Theoretical Electronic Transition and the Corresponding Orbital Energies [eV] 2 | ΔEH-L [eV] 3 |
|---|---|---|---|---|
| 3a | CH2Cl2 | 364 (46,000) | HOMO (–7.02) → LUMO (–1.08) | 5.94 (137) |
| 3b | CH2Cl2 | 373 (23,300) | HOMO (–6.87) → LUMO (–1.09) | 5.78 (133) |
| Toluene | 379 (33,600) | HOMO (–6.71) → LUMO (–1.04) | 5.67 (131) | |
| THF | 375 (36,900) | HOMO (–6.85) → LUMO (–1.08) | 5.77 (133) | |
| MeOH | 361 (29,400) | HOMO (–6.92) → LUMO (–1.11) | 5.81 (134) | |
| MeCN | 369 (21,600) | HOMO (–6.92) → LUMO (–1.11) | 5.81 (134) | |
| 3c | CH2Cl2 | 374 (47,500) | HOMO (–6.82) → LUMO (–1.07) | 5.75 (132) |
| 3d | CH2Cl2 | 375 (32,800) | HOMO (–6.85) → LUMO (–1.11) | 5.74 (132) |
| 3e | CH2Cl2 | 364 (40,500) | HOMO (–7.17) → LUMO (–1.22) | 5.95 (137) |
| Toluene | 364 (29,600) | HOMO (–7.06) → LUMO (–1.19) | 5.87 (135) | |
| THF | 363 (52,200) | HOMO (–7.16) → LUMO (–1.22) | 5.94 (137) | |
| MeOH | 359 (36,700) | HOMO (–7.21) → LUMO (–1.24) | 5.97 (138) | |
| MeCN | 361 (56,000) | HOMO (–7.21) → LUMO (–1.24) | 5.97 (138) |
| Compound | Solvent | λabs [nm] 1 (ε [L mol–1 cm–1]) | Theoretical Transition and Orbital Energies [eV] 2 | ΔEH-L [eV] 3 |
|---|---|---|---|---|
| 4aA | CH2Cl2 | 424sh (33,600), 446 (92,100) | HOMO (–6.74) → LUMO (–1.53) | 5.21 (120) |
| 4bA | CH2Cl2 | 445sh (33,400), 470 (95,900) | HOMO (–6.43) → LUMO (–1.49) | 4.94 (114) |
| Toluene | 444sh (1900), 472 (5800) | HOMO (–6.40) → LUMO (–1.46) | 4.94 (114) | |
| THF | 444sh (14,600), 469 (41,000) | HOMO (–6.43) → LUMO (–1.49) | 4.94 (114) | |
| MeOH | 442sh (34,900), 467 (98,400) | HOMO (–6.44) → LUMO (–1.50) | 4.95 (114) | |
| MeCN | 442sh (33,000), 467 (91,100) | HOMO (–6.44) → LUMO (–1.50) | 4.95 (114) | |
| 4cA | CH2Cl2 | 444sh (33,800), 473 (104,200) | – 4 | – 4 |
| 4dA | CH2Cl2 | 444sh (33,200), 474 (105,300) | – 4 | – 4 |
| 4eA | CH2Cl2 | 420 (48,300) | HOMO (–6.93) → LUMO (–1.80) | 5.13 (118) |
| Toluene | 410 (38,900) | HOMO–3 (–6.95) → LUMO (–1.82) | 5.13 (118) | |
| THF | 408 (32,300) | HOMO (–6.93) → LUMO (–1.80) | 5.13 (118) | |
| MeOH | 397 (31,900) | HOMO (–6.92) → LUMO (–1.79) | 5.13 (118) | |
| MeCN | 408 (55,100) | HOMO (–6.92) → LUMO (–1.79) | 5.13 (118) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamada, S.; Takahashi, Y.; Konno, T. Fluorinated Merophosphinine and Phosphinine Dyes: Synthesis and Evaluation of UV-Visible Light Absorption Properties. Compounds 2023, 3, 153-168. https://doi.org/10.3390/compounds3010013
Yamada S, Takahashi Y, Konno T. Fluorinated Merophosphinine and Phosphinine Dyes: Synthesis and Evaluation of UV-Visible Light Absorption Properties. Compounds. 2023; 3(1):153-168. https://doi.org/10.3390/compounds3010013
Chicago/Turabian StyleYamada, Shigeyuki, Yusuke Takahashi, and Tsutomu Konno. 2023. "Fluorinated Merophosphinine and Phosphinine Dyes: Synthesis and Evaluation of UV-Visible Light Absorption Properties" Compounds 3, no. 1: 153-168. https://doi.org/10.3390/compounds3010013
APA StyleYamada, S., Takahashi, Y., & Konno, T. (2023). Fluorinated Merophosphinine and Phosphinine Dyes: Synthesis and Evaluation of UV-Visible Light Absorption Properties. Compounds, 3(1), 153-168. https://doi.org/10.3390/compounds3010013