1. Introduction
The use of peptides as components of sensing systems is particularly attractive, as they are relatively small with straightforward means of synthesis as well as modification, and can provide high-affinity targets for receptor-specific interactions [
1]. In addition, synthetic peptides can serve to mimic antigenic determinant sites within larger proteins of interest, and are thereby capable of binding with antibodies that have specificity toward those protein antigens [
2,
3,
4]. Peptide-based sensors have been used to detect a variety of targets, including proteins, enzymes, and ions. Peptide-based sensing systems will generally exploit the molecular recognition capabilities between the peptide and a given target to serve as the selectivity element, with the coupling of the peptide to a transducer in order to enable signal detection by the user. Such transduction can be as simple as a change in fluorescence afforded by a pendent fluorophore [
5], or more complex transduction, such as having the peptide coupled to a quartz crystal microbalance [
6]. The ability to identify target-selective peptides to serve as recognition moieties has been demonstrated by evolutionary screening approaches [
7], which opens the way for novel peptide-based biosensors [
8,
9]. The rational design of peptides can also be implemented for integration with transduction systems. For example, in the detection of Cu
2+ ions using a fluorescein isothiocyanate-coupled peptide (FITC-Ahx-His-Glu-Phe-Cys-NH
2), researchers have demonstrated that electron transfer occurs resulting in fluorescence quenching after Cu
2+ binds with the histidine and cysteine, and which proceeds with high sensitivity [
10]. Electrochemical sensing has also been demonstrated by designing peptide coatings on gold electrodes that serve as selective substrates for proteases [
11]. In one manifestation, this has been used for the determination of metastasis-linked protease [
12].
Peptides have also been proven to work well as responsive or selective moieties when linked to nanoparticles. Using Au nanoparticles functionalized with the peptide H
2N-Leu-Aib-Tyr-OMe, pH-sensitive reversible aggregation has been demonstrated result in a visible red to violet color change [
13]. This same peptide Au nanoparticle system was also found to be responsive to dosages of Hg
2+ ions above 4 ppm [
14], while other peptides functionalized to Au nanoparticles have demonstrated detection of other metal ions of interest [
15,
16,
17]. In other manifestations, the coating of the receptive peptides to Au or ZnO nanoparticles enabled their high surface area display, enabling their use as a coating deposited onto quartz crystal microbalances for the sensing of different gases, including volatile alcohols and esters [
18,
19]. Peptides displayed on polymer nanoparticles in the form of polydiacetylene vesicles have also been implemented for detecting the presence of trinitrotoluene at relatively high concentrations [
20], while utilizing the same vesicles for the coating of carbon nanotube-based sensors facilitated orders of magnitude increases in sensitivity with retained selectivity for trinitrotoluene [
8]. Polydiacetylene vesicles provide a class of responsive materials that are particularly fitting for sensing applications due to their ability to provide optical responses in the form of changes in their florescence or absorbance in response to given stimuli depending on their structure [
21]. Functionalizing the surface of polydiacetylene vesicles with receptive moieties thus provides a well-known approach for customizing their chromic and fluorogenic responses to targets [
22]; however, an alternative mode of detection also exists, by which the colloidal stability of polydiacetylene vesicles can be modulated by the presence of target analytes, resulting in aggregation [
23]. Using this aggregation strategy, the pendant side-chains of polydiacetylene vesicles have previously been decorated with chemical groups, including cyanuric acid for the detection of melamine [
24], or oxime modifications to facilitate the detection of organophosphates [
25]. In addition, we have recently shown that the peptides displayed on polydiacetylene vesicles that mimic biotin could facilitate the detection of streptavidin through multi-vesicle binding, which results in aggregation [
26].
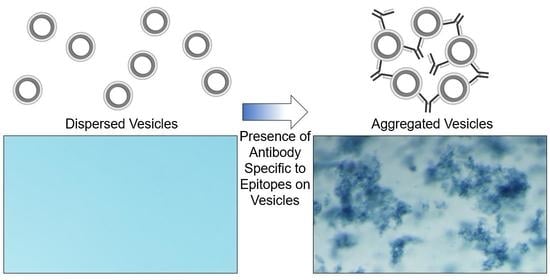
In the following work, we extend this strategy to demonstrate that polydiacetylene-based vesicles displaying peptide epitopes can facilitate a straightforward aggregation test for the detection of specific antibodies, as seen in
Figure 1. Immunoassays are widely used in biology and medicine for identifying antibodies in samples with common assays, including latex agglutination tests, lateral flow assays, and Luminex bead-based assays. Latex agglutination tests, for example, have been used to reveal the presence of antibodies within samples by the clumping or aggregation of antigen-coated latex beads, and depending on the format of the assay, have been used for determining immunity to viruses including SARS-CoV-2 [
27,
28,
29,
30], as well as detection of infection by bacteria or parasites [
31,
32]. Applying this principle, we reveal a strategy for the generation of epitope displaying polydiacetylene vesicles capable of being used in the visual detection of the presence of specific antibodies by target-specific binding to induce aggregation. In this proof-of-concept, we utilized the influenza hemagglutinin (HA) peptide epitope [
33] (Tyr-Pro-Tyr-Asp-Val-Pro-Asp-Tyr-Ala) appended to an amphiphile of 10,12 pentacosadiynoic acid (PCDA) via a Gly-Gly-Ser-Gly spacer for the formation of epitope-decorated vesicles that facilitate the detection of the presence of the anti-HA antibody by aggregation. As there is not yet a single test capable of measuring the presence of each different antigen-specific antibody within a sample, separate antigen-labeled probes must be developed and implemented. The following work describes a streamlined synthetic approach for the rapid development of peptide epitope-labeled vesicles to serve as such antigen-labeled probes, which will facilitate aggregation-based assays in detecting the presence of specific antibodies. We believe this strategy will serve as a foundation for future work by developing a high-throughput approach for simultaneously detecting the presence of multiple distinct antigen-specific antibodies when loading the vesicles with unique reporter components.
2. Materials and Methods
2.1. Materials and Vesicle Fabrication
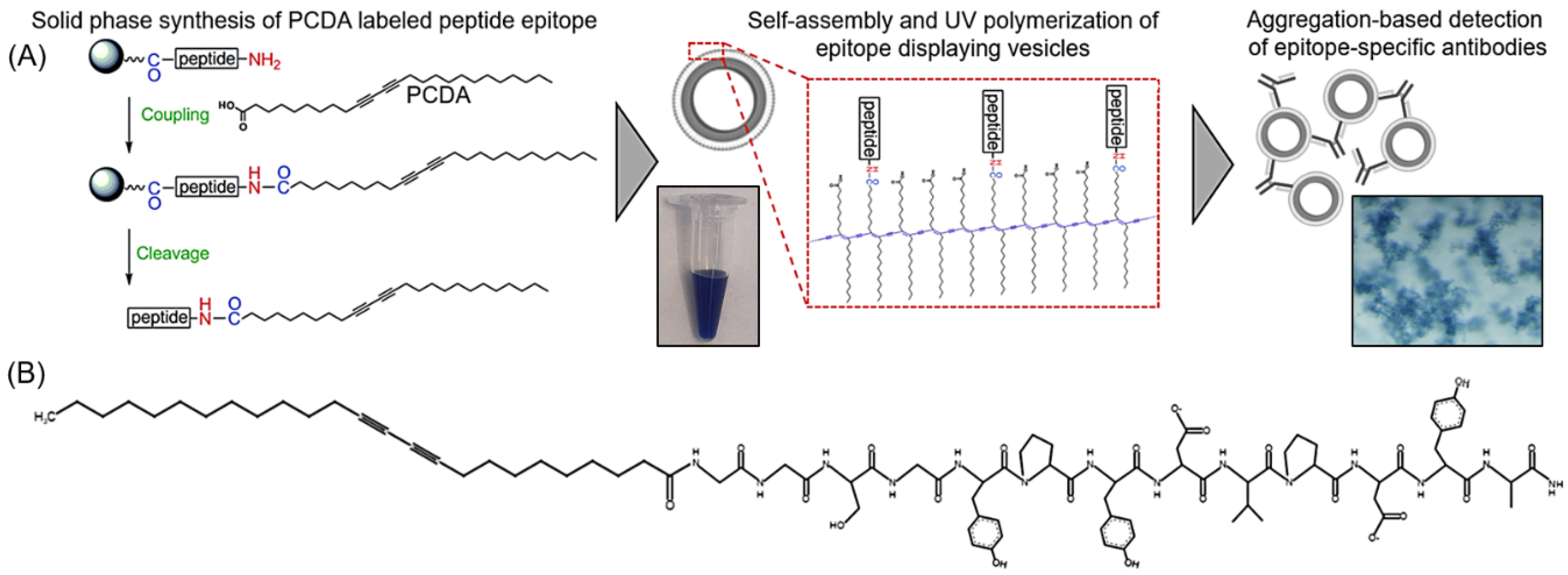
The synthesis of hemagglutinin (HA) epitope-labeled amphiphiles was carried out using the solid-phase peptide synthesis of the HA epitope sequence (Tyr-Pro-Tyr-Asp-Val-Pro-Asp-Tyr-Ala) with an N terminal Gly-Gly-Ser-Gly spacer prior to the final coupling reaction of 10,12 pentacosadiynoic acid (PCDA) to provide the diacetylene-containing amphiphile (
Figure 1). A second control amphiphile in which the HA epitope was scrambled to have the sequence Tyr-Pro-Tyr-Asp-Pro-Ala-Asp-Val-Tyr with a Gly-Gly-Ser-Gly spacer followed by PCDA was also synthesized. Fmoc-protected amino acids were used for the solid phase synthesis of the peptides on a rink amide resin with HOBT/DIC coupling of the amino acids, deprotection of the Fmoc groups with piperidine, and Kaiser tests for confirmation at each step. After the final deprotection step, the HOBT/DIC coupling reaction was again used to cap PCDA to the free N terminus of the peptide. The samples were cleaved with TFA and precipitated in cold ether overnight at −20 °C. The product was then dried and analyzed by liquid chromatography mass spectrometry (LCMS) (
Figure S1).
For vesicle formation, 50 umole of unlabeled PCDA amphiphile, 50 umole of HA epitope-labeled amphiphile, 50 umole of scrambled HA epitope-labeled amphiphile, and 50 umole of PEG3400-bis-PCDA (synthesized as described previously [
34]) were dissolved separately in 200 uL of DMSO. Volumetrically, mixtures of the HA epitope-labeled amphiphile or the scrambled HA epitope-labeled amphiphile were combined with the unlabeled PCDA amphiphile and PEG3400-bis-PCDA to yield a total 50 umole of amphiphile, with different mole ratios of HA epitope-labeled amphiphile to unlabeled PCDA amphiphile and a constant 0.01% of PEG3400-bis-PCDA. Alternatively, for control vesicles, the 0.01% scrambled HA epitope-labeled amphiphile was combined with 0.01% PEG3400-bis-PCDA and 99.98% unlabeled PCDA amphiphile. The percentages of HA epitope-labeled PCDA in the mixtures included 0%, 0.01%, 0.1%, 1%, and 5% HA epitope-labeled PCDA. Vesicles were then formed by injecting the 50 umole mixtures into 7 mL of pre-heated (60 °C) 10 mM HEPES. The samples were continuously heated for 30 min, probe-sonicated for 30 min, and filtered with a 0.2 um syringe filter while hot. Vesicle self-assembly proceeded by incubating the samples at 4 °C overnight, and polymerization of the vesicles was carried out by UV irradiation (3–15 min at 254 nm). Vesicle stocks were stored at 4 °C, and an example of the product is shown in
Figure 2.
2.2. HA Peptide Vesicle Assays with Anti-HA Beads
Anti-HA-coated magnetic beads (1, 5, or 10 uL) were used for the comparison of vesicle clearance by binding between the HA epitope presented on the vesicle and the anti-HA beads. We also examined the abilities of the vesicles with different mole percentages of the HA peptide epitope to be capture by the anti-HA antibody presented by the beads. The given amount of anti-HA bead stock was suspended in 50 uL of 10 mM HEPES containing 1% BSA and incubated at room temperature for 30 min with inversion of the tube every 5 min. The beads were collected by magnet followed by removal of the supernatant and resuspension in a vesicle solution containing 25 uL of 10 mM HEPES, 1%BSA, and 5 uL of PCDA vesicles displaying HA epitope ranging from 0 to 5%, as indicated. The samples were then incubated for 30 min with inversion as described above, and the anti-HA beads were collected by magnet. The supernatant was collected and analyzed by spectrophotometer, and the absorbance of the resulting supernatant was compared to that of the original vesicle suspension to assess the extent of vesicle clearance by the anti-HA beads. This assay was utilized to determine which HA epitope concentration displayed by the vesicles would provide superior anti-HA antibody binding, and that formulation was to be used for subsequent testing.
2.3. Vesicle–Antibody Aggregation Test
For aggregation testing with soluble antibody rather than bead-immobilized antibody, the following vesicle formulations were examined: 0% HA epitope vesicles, 0.01% HA epitope vesicles, and 0.01% scrambled HA epitope vesicles. The use of 0.01% HA epitope was selected based on its superior binding in the anti-HA bead assay described above. A mixture of 5 uL of UV polymerized vesicles was added to 20 uL of 10 mM HEPES, and the suspension was then mixed with 5 uL of unconjugated anti-HA antibody (dilutions of 1 mg/mL anti-HA antibody stock in 10 mM HEPES to different concentrations as indicated in the respective figures) and incubated at room temperature for 30 min. The samples were centrifuged at 5000 rpm for 5 min to expedite the precipitation of aggregates. Supernatant was collected and analyzed using an Epoch2 microplate spectrophotometer (BioTek Inc., Winooski, VT, USA) with Take3 plate attachment, and the absorbance of the resulting supernatant compared to that of the original vesicle suspension was used to determine the extent of antibody-induced aggregation relative to control samples. In addition, vesicles with either 0.01% HA epitope vesicles or 0.01% scrambled HA peptide were examined for anti-HA antibody concentration-dependent aggregation.
2.4. Anti-HA Bead Preparation
Anti-HA beads were prepared by non-covalently labeling protein A/G beads with anti-HA epitope antibody. In brief, 5 uL of protein A/G beads was suspended in 150 uL of 10 mM HEPES and the beads were then collected by magnet. The beads were then resuspended in 150 uL of HEPES containing 2 uL of anti-HA antibody. The sample was inverted once every 5 min for a total of 30 min followed by the collection of the beads and resuspension in HEPES containing 1% BSA. The beads were blocked with BSA for 30 min at room temperature with inversion of the tube every 5 min. The beads were collected and resuspended in fresh 1% BSA for immediate use in exposure to the vesicles for binding studies.
2.5. Microscale Thermophoresis
Additional examination of the 0.01% HA epitope vesicles binding to anti-HA antibody was carried out by microscale thermophoresis (MST). Vesicles with 0.01% HA epitope were heated at 75 °C for one minute to induce the characteristic red fluorescence of polydiacetylene to enable use of the vesicles as the reporter for the MST assay. Anti-HA antibody was diluted in 10 mM HEPES and added to the vesicles in equivalent volumes to provide a final antibody concentration of 250 ng/uL, 120 ng/uL, 60 ng/uL, 20 ng/uL, 6.9 ng/uL, or 0.63 ng/uL. The samples were immediately loaded into standard capillaries and allowed to incubate for an hour to allow adequate time for binding and aggregation. The samples were then loaded into a Nanotemper Monolith (NanoTemper Technologies, Inc., South San Francisco, CA, USA) and the MST traces were recorded using the provided MO.Control software.
4. Discussion
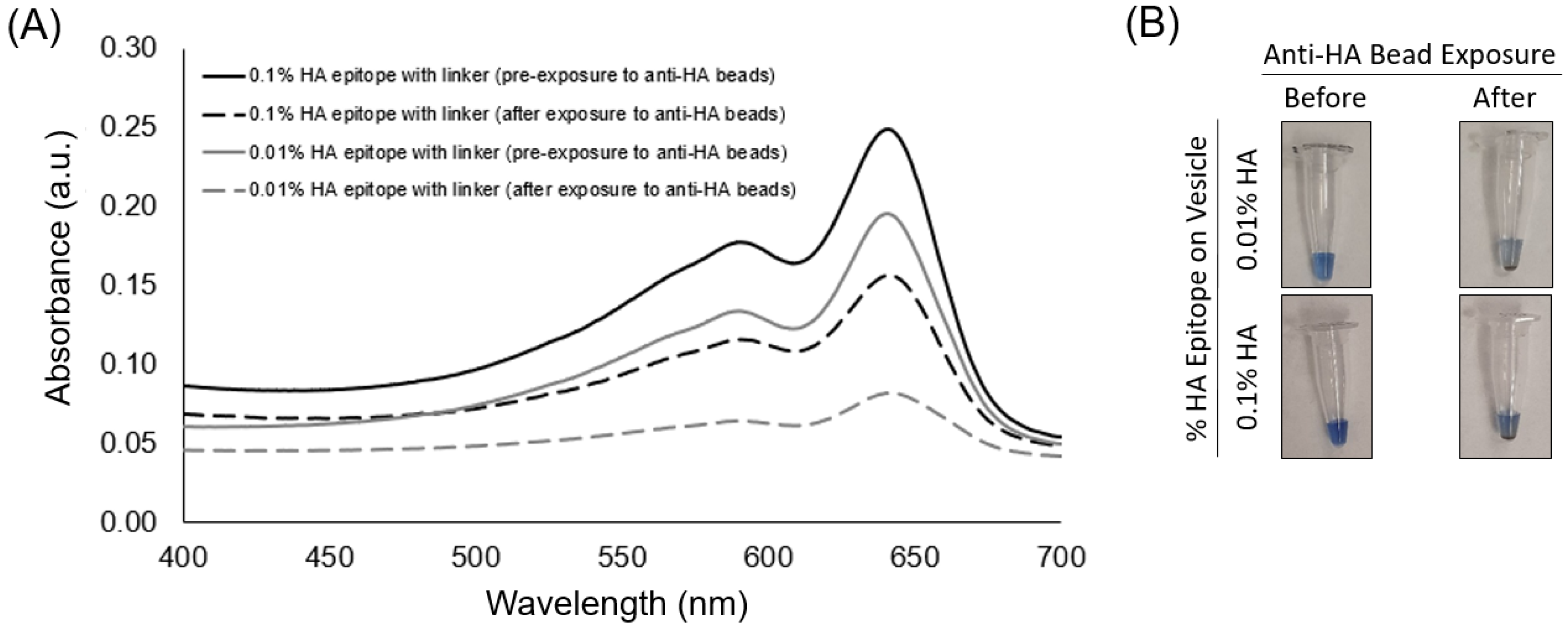
The aim of this study was to examine conditions for using epitope-labeled vesicles for detecting a response to the presence of epitope-specific antibodies in the form of aggregation behavior, which could be observed visually as well as by spectral readout. In this work, for testing purposes, we specifically used the common influenza hemagglutinin (HA) peptide epitope to display on our vesicles via a spacer and with different surface densities in order to examine the aggregation response to anti-HA antibody. As shown in
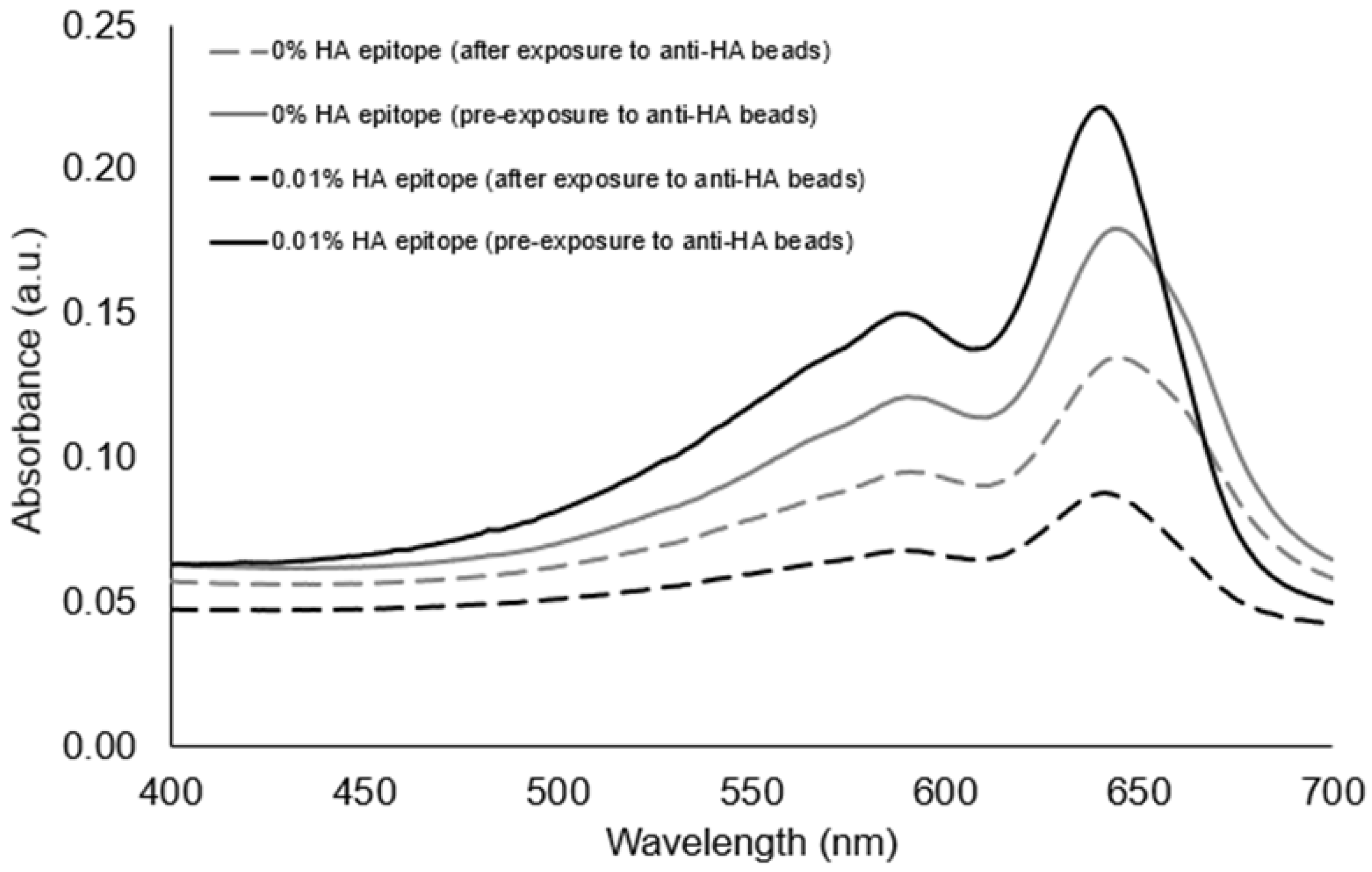
Figure 3, the UV-polymerized vesicles exhibited their characteristic absorption maxima around 645 nm, which is known to result from the overlapping p orbitals within the conjugated diacetylene backbone. Using this absorption peak, we could compare the amount of vesicle aggregation by the loss of color represented as an attenuation in the absorbance spectra. Our first assessment of the HA epitope-displaying vesicles was performed via their ability to bind with anti-HA antibodies by directly examining the spectra intensity of the vesicle solution before and after exposure to anti-HA antibody-coated magnetic beads. The extent of capture of HA epitope-displaying vesicles by anti-HA beads was found to be impacted by the surface density of the HA epitope amphiphile on the vesicles, with a more substantial attenuation in the absorption spectra being observed when using 0.01% HA epitope vesicles as compared to 0.1% HA epitope vesicles. When compared to higher concentrations of 1% and 5% HA epitope (
Figure S4), we observed even less capture by the anti-HA beads, indicating the surface density of epitopes as a critical factor that can be optimized. This trend indicates that the lower percentage of HA epitope displayed on the vesicles provides for better attenuation in signal due to binding and capturing by the anti-HA beads, while the 0% HA epitope vesicles (having no HA epitope) exhibited only marginal attenuation in signal, as seen in
Figure 4. It may be deduced, then, that a high epitope density may cause some form of steric hindrance in which neighboring moieties may block the access of the antibody to the epitope. This is reasonable, given that, in prior works, we have found that as the surface densities of the displayed epitopes on PCDA vesicles increase, there is a point at which vesicle formation becomes hindered, resulting in the inadequate packing of the amphiphiles. Having multiple copies of the epitope site displayed by the vesicles is important in allowing the vesicles to bind with multiple antibodies simultaneously in order to facilitate a mechanism for vesicle aggregation, but displaying too many copies of the epitope can sterically restrict their access to antibodies.
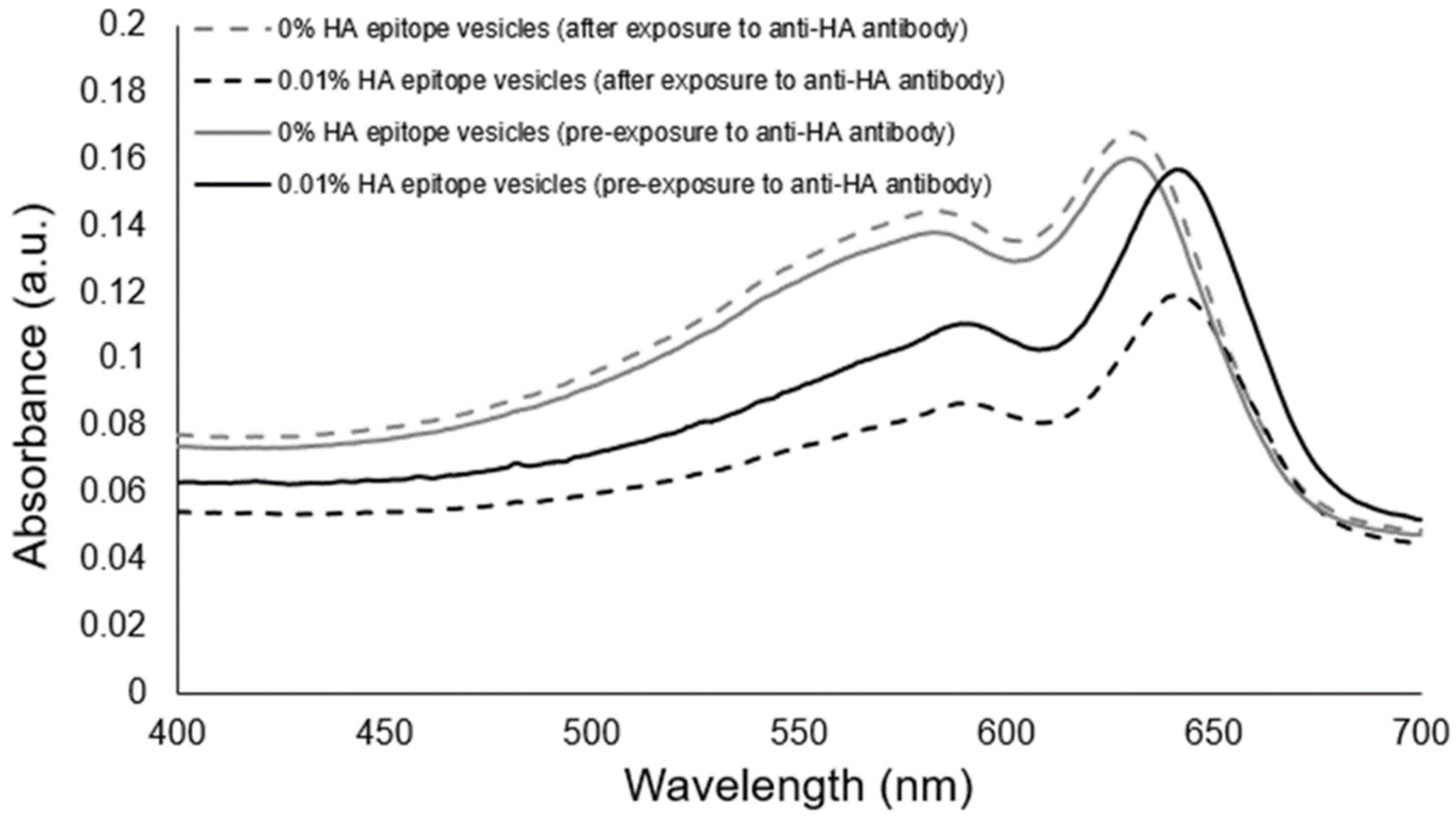
Having confirmed the ability of the HA epitope-displaying vesicles to bind with the anti-HA antibody presented on beads, we then used the 0.01% HA epitope vesicles to test whether they could facilitate the aggregation-based detection of the presence of soluble anti-HA antibody.
Figure 5 reveals that the 0.01% HA-displaying vesicles were able to aggregate within 30 min of exposure to the anti-HA antibody, and that an observable attenuation in the spectra resulted, while no aggregation or attenuation was observed in the spectra of the control PCDA vesicles having no HA epitope.
To examine the specificity of the observed vesicle aggregation, we produced another vesicle formulation displaying a scrambled HA epitope peptide sequence that will not bind specifically to the anti-HA antibody’s complementarity determining regions. As shown in
Figure 6, the formation of aggregates of 0.01% HA epitope vesicles due to specific interactions between the anti-HA antibody and the displayed HA peptide epitope was confirmed, given that the 0.01% scrambled HA peptide did not produce aggregates when the anti-HA antibody was added. The specificity of vesicle aggregation as determined by the peptide epitopes displayed by the vesicles and the presence of corresponding anti-epitope antibodies thereby establishes this approach as a means for detecting the presence of specific antibodies. The antibody-induced aggregation signal could be observed at as low as 33 ng/uL under the current conditions and vesicle configuration.
We have demonstrated that this approach provides for binary sensing in determining if the specific anti-HA antibody is present above a specified concentration. In doing so, this technique may find a use similar to latex agglutination assays to provide semi-quantitative analysis of antibody titers. In binary sensing approaches like latex agglutination tests, the antibody concentration can be determined by setting up a series of dilutions followed by the assessment of aggregation to identify the greatest dilution factor that will still provide a positive aggregation result, which corresponds to the titer. Our vesicles may similarly find use in such a semi-quantitative approach. The use of binary testing alone can have a number of other applications in areas where knowledge of a positive or negative result rather than an exact antibody concentration is worthwhile.
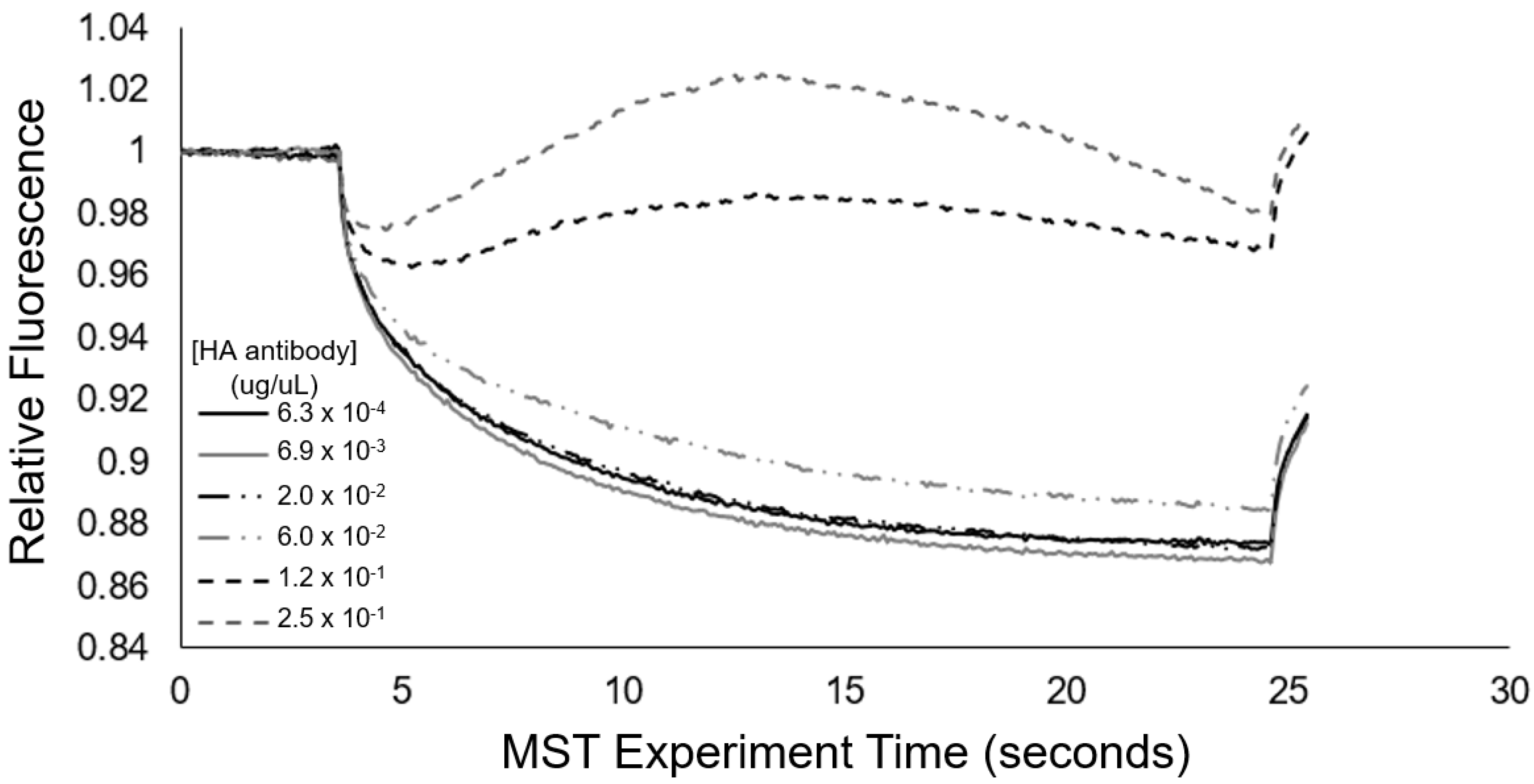
To serve as a mode of cross-validation for the aggregation response behavior and to quantitatively assess the transient aggregation response, we also implemented microscale thermophoresis (MST). From
Figure 7, we see that the MST trace caused a concentration-dependent change in the thermophoretic mobility. Specifically, at high concentrations of antibody (>60 ng/uL), the MST traces revealed substantial increases in the levels of relative fluorescence, which are indicative of positive thermophoresis [
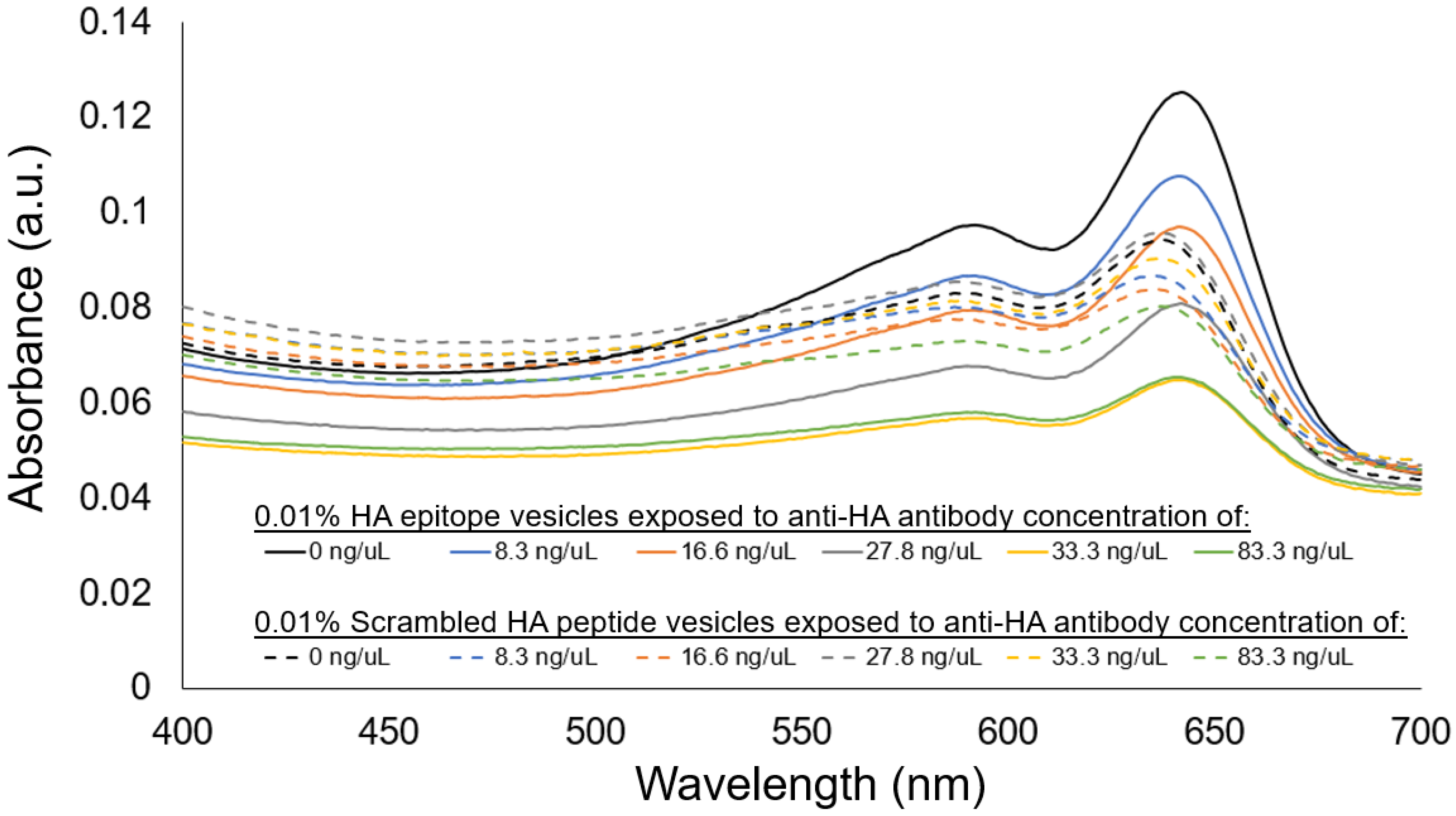
35], and provided indication of 0.01% HA epitope vesicle aggregate formation due to anti-HA antibody binding, resulting in increased aggregate size and less thermophoretic movement. At anti-HA antibody concentrations of 20 ng/uL and below, the aggregate formation of the 0.01% HA epitope vesicles was not observed by MST. This result coincides with our spectral observations (
Figure 8) of the aggregation behavior for the 0.01% HA epitope vesicles, which revealed high levels of aggregation at anti-HA antibody concentrations of 33 ng/uL and above, but less aggregation at lower concentrations.
In looking at the utility of the HA epitope-labeled vesicles for aggregation-based detection, we can see that the use of a soluble anti-HA antibody provides aggregation, but by centrifuging the samples, we can derive a more stark formation of the precipitate. While it is unlikely that centrifugation will be readily available in all contexts, the use of a simple magnetic bead assay may be more widely deployable. Thus, we conducted several optimization experiments to enhance the bead-based assay. Initially we examined the production of our own anti-HA beads by labeling the anti-HA antibody onto magnetic protein A/G beads. To assess whether the protein A/G beads were fully coated with anti-HA antibody, we examined the clearance of HA epitope-containing vesicles when exposed to protein A/G beads produced with increasing amounts of anti-HA antibody. Our data in
Figure S2 suggest that we were able to provide a maximum (saturated) amount of antibody on the protein A/G beads, as only minimal increases in the binding of our 0.01% HA epitope vesicles were observed when using twice the saturating amount of antibody. If this were not the case, we would have expected the capture of a greater amount of the 0.01% HA epitope vesicles when using a higher amount of anti-HA antibody labeling on the beads. To assess whether we were utilizing the optimal amount of anti-HA beads, we also examined the extent of 0.01% HA epitope-labeled vesicle clearance as a function of the amount of anti-HA beads used. From
Figure S3, it can be seen that utilizing a larger amount of anti-HA beads can facilitate a higher degree of clearance of the 0.01% HA epitope-displaying vesicles. This can dramatically improves the clarity of readout, and may be the best way forward in the future use of this vesicle-based sensing approach for the profiling of epitope-specific antibodies. To summarize, we have shown here our proof-of-concept epitope-labeled vesicles for detecting the presence of the epitope-specific antibody; specifically in this case, those antibodies against human influenza agglutinin. We look forward to future work examining the application areas of this form of aggregation-based sensor for antibody detection, and its ability to be customized for other epitopes by virtue of its modularity.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







