
Ultrasensitive Electrochemical Sensor Based on SnO2 Anchored 3D Porous Reduced Graphene Oxide Nanostructure Produced via Sustainable Green Protocol for Subnanomolar Determination of Anti-Diabetic Drug, Repaglinide

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Apparatus
2.3. Material Characterization
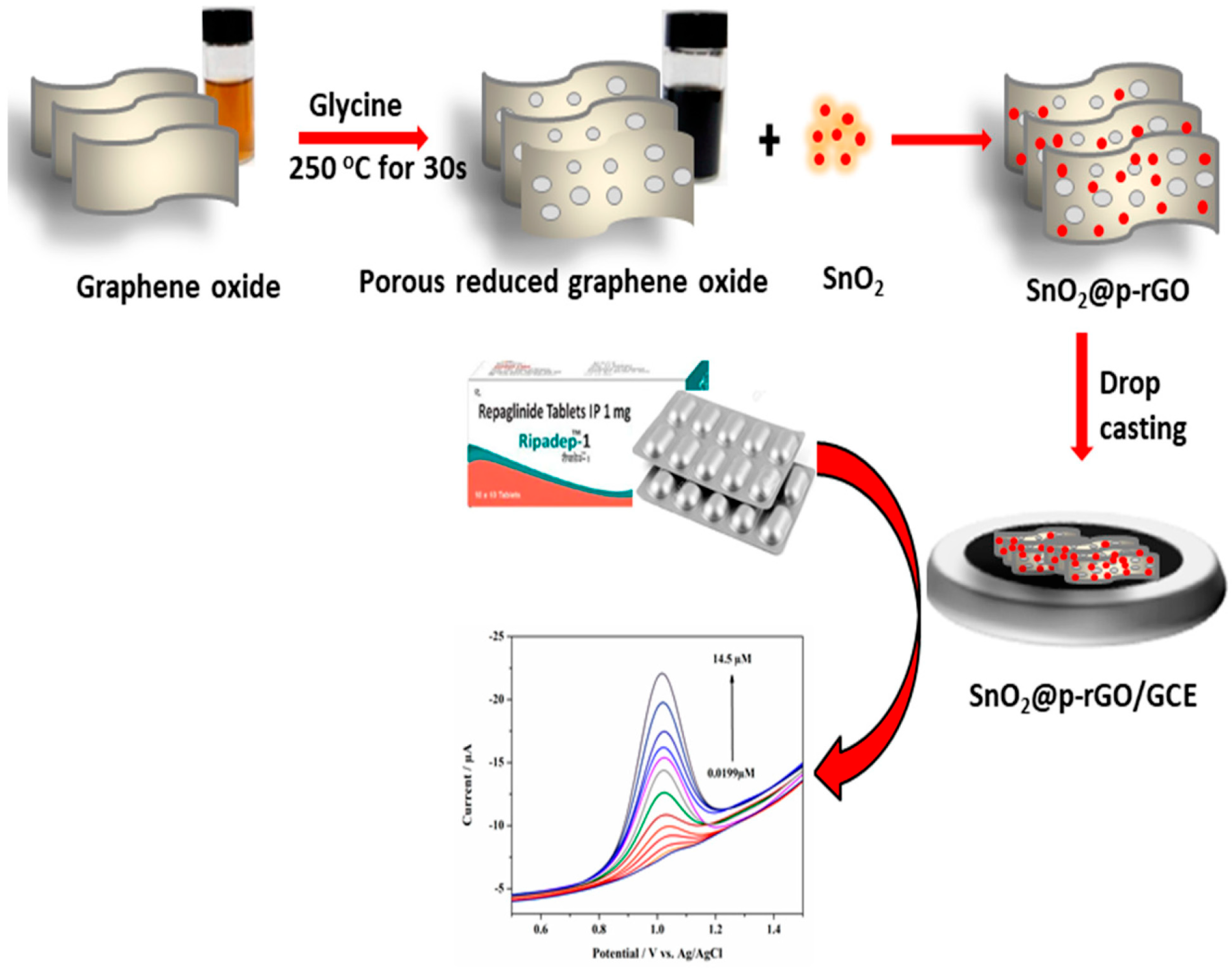
2.4. Preparation of p-rGO
2.5. Preparation of Cotton Peel Extract
2.6. Green Synthesis of Tin Oxide Nanoparticles Utilizing Cotton Boll Peel Extract
2.7. Preparation of SnO2@p-rGO and Fabrication of SnO2@p-rGO/GCE
2.8. Assay of Tablets
2.9. Quantification of RPG in Human Urine Samples
3. Results and Discussion
3.1. Mechanism of Formation of p-rGO and SnO2 Nanoparticles
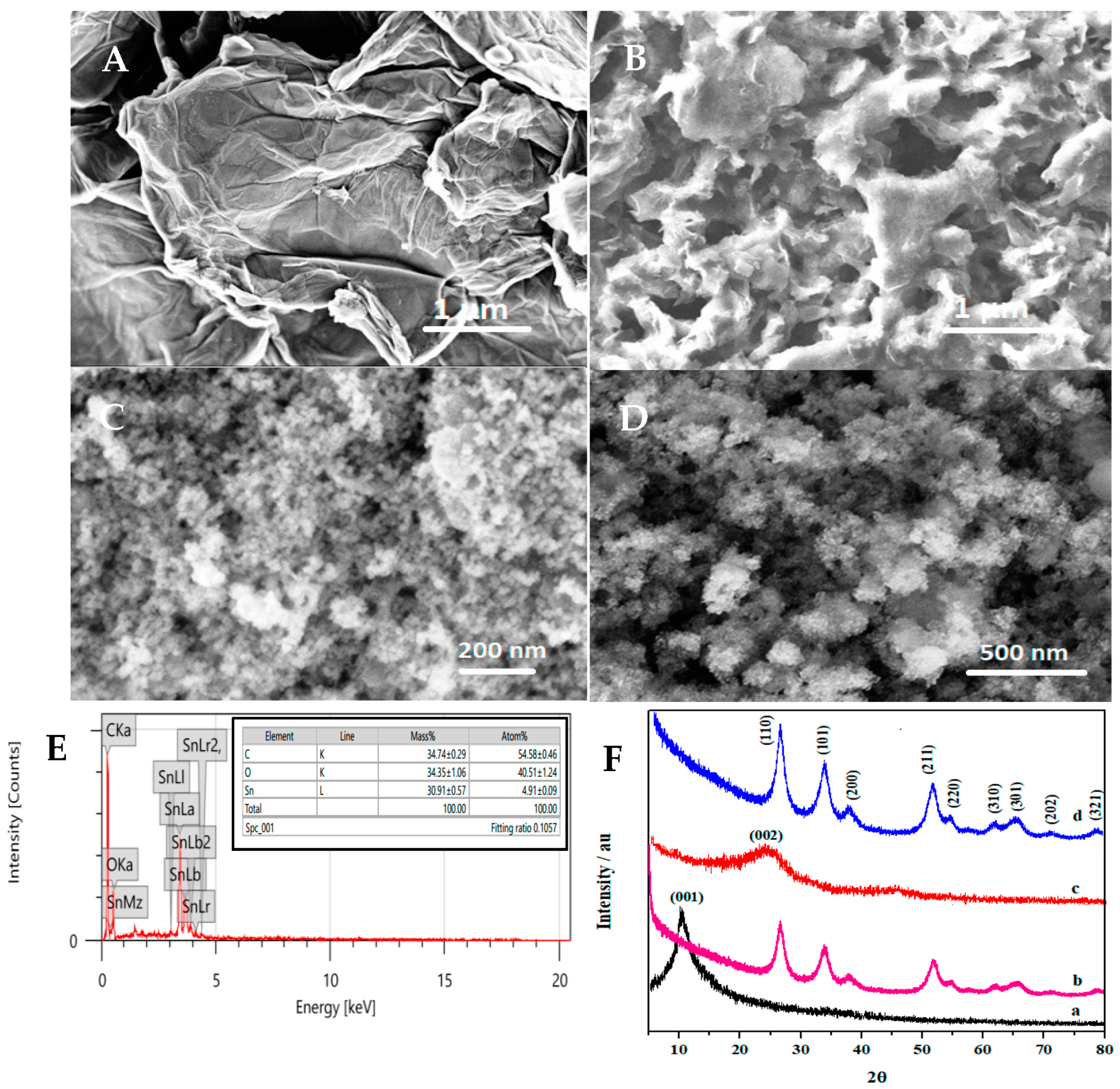
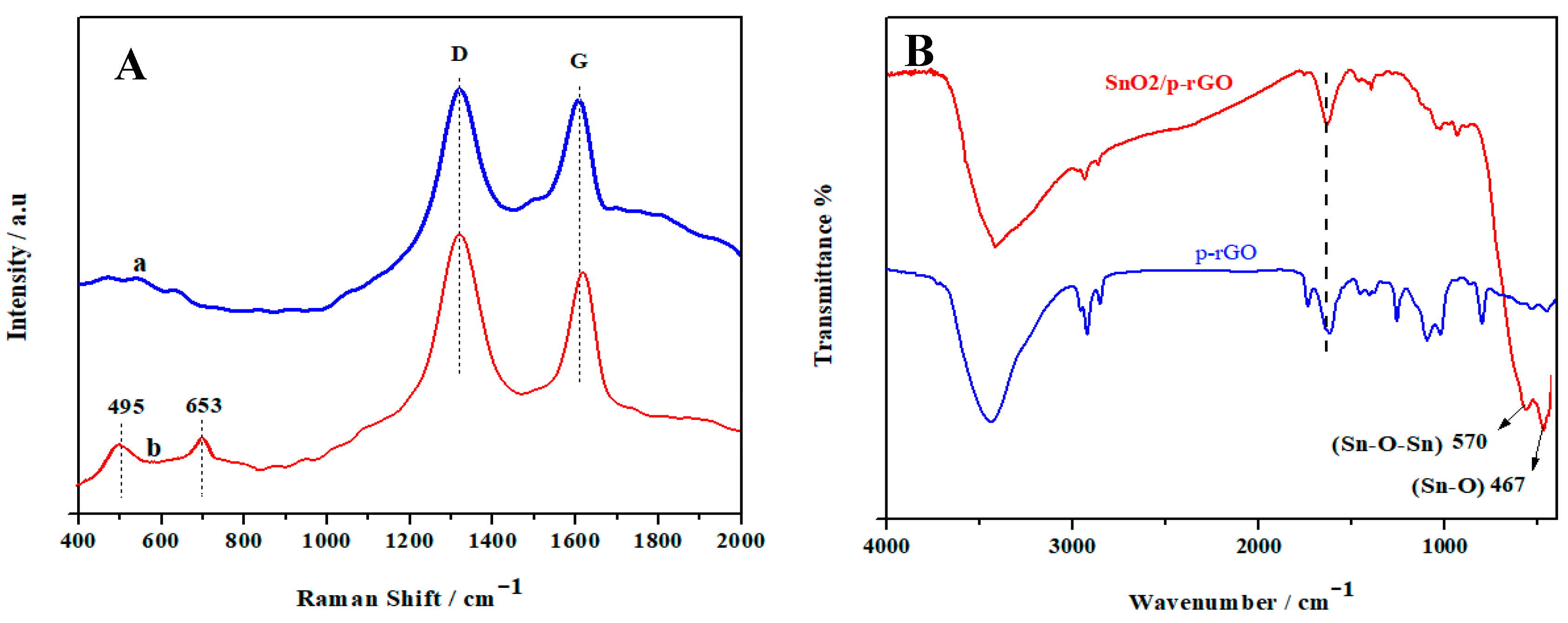
3.2. Structural and Morphological Characteristics
3.3. Electrochemical Characterization of Modified GCEs
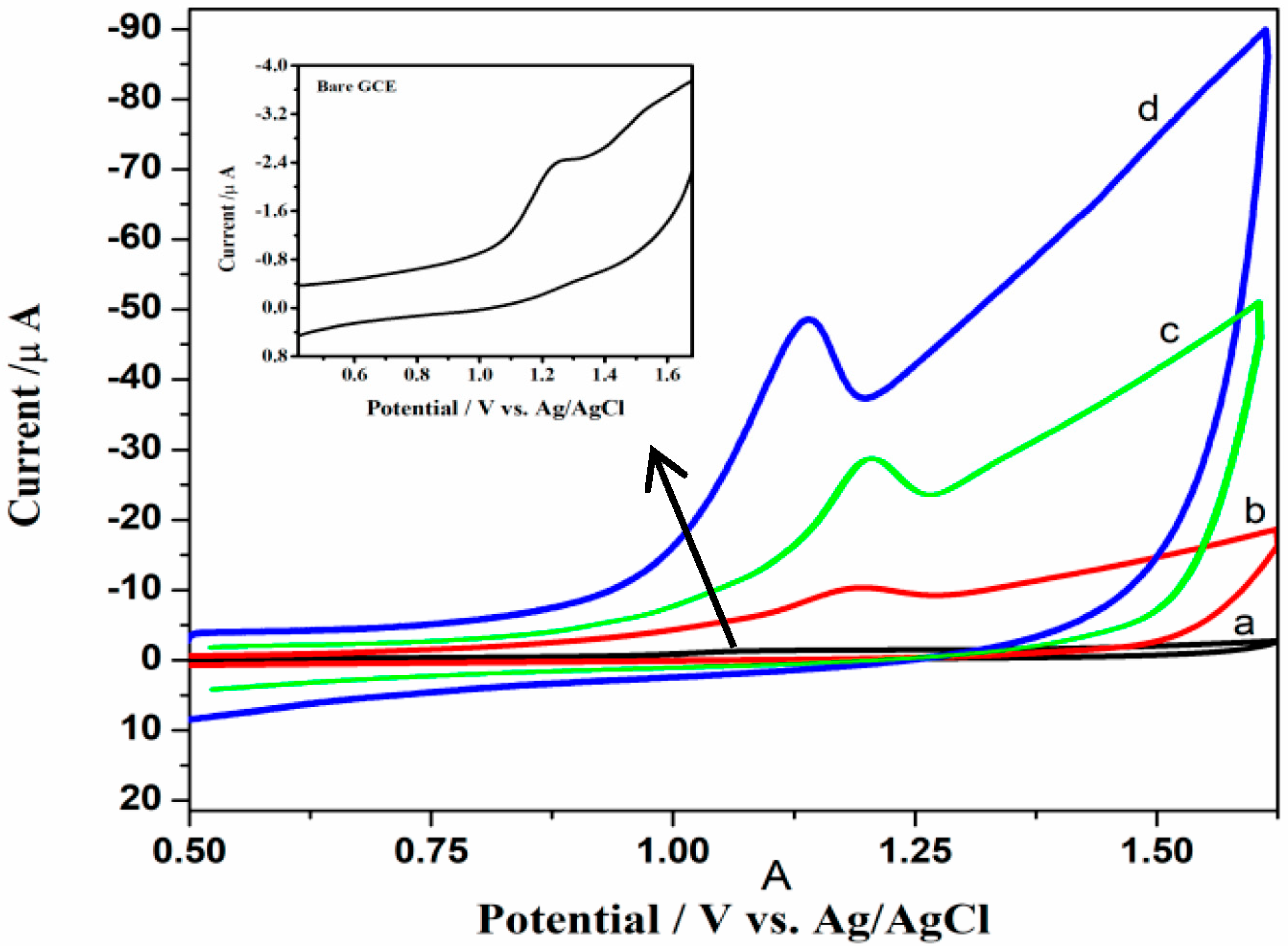
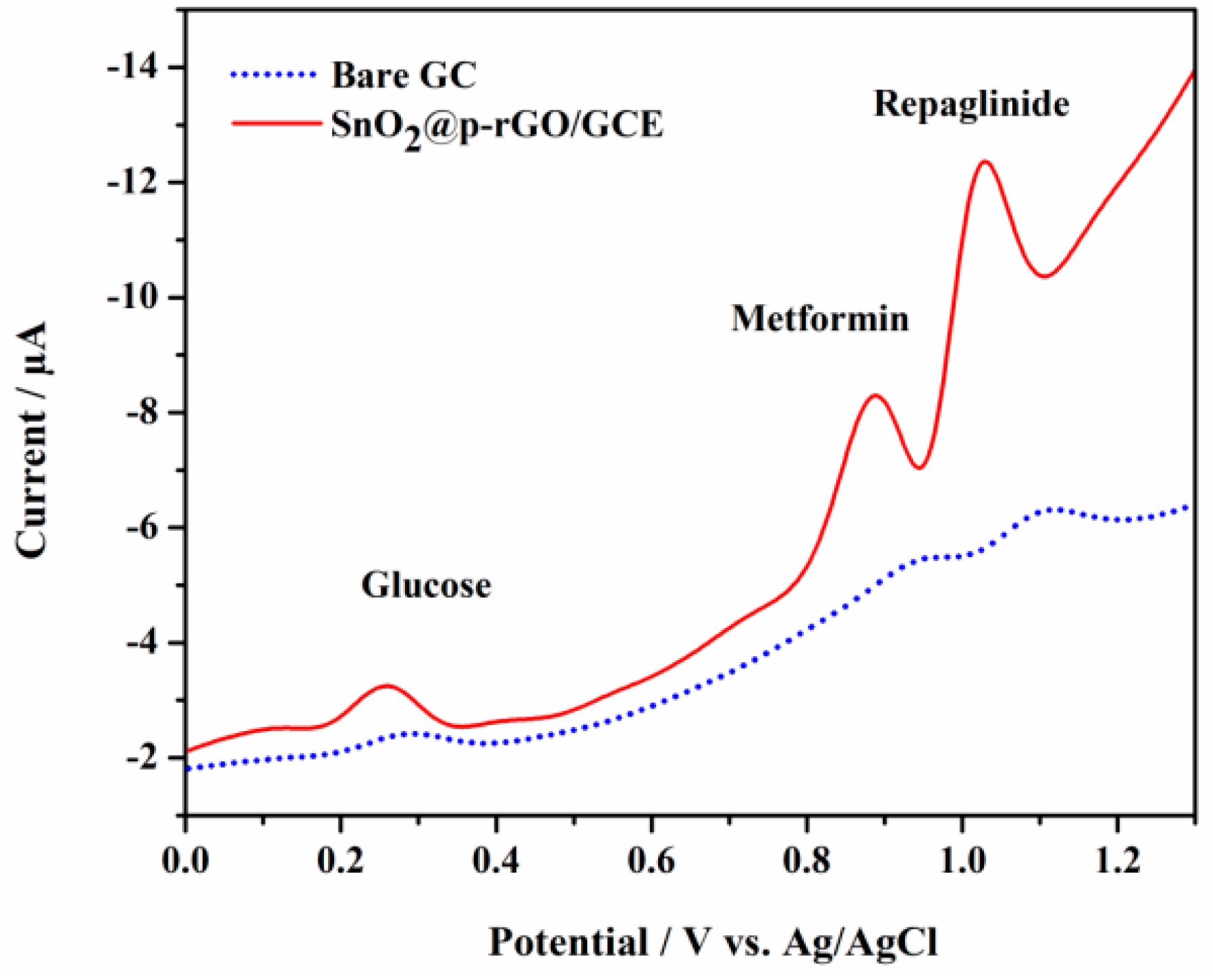
3.4. Electrochemical Behavior of RPG at Bare GCE and Modified GCEs
3.5. Effect of pH and Supporting Electrolyte
3.6. Optimization of the Sensing Response
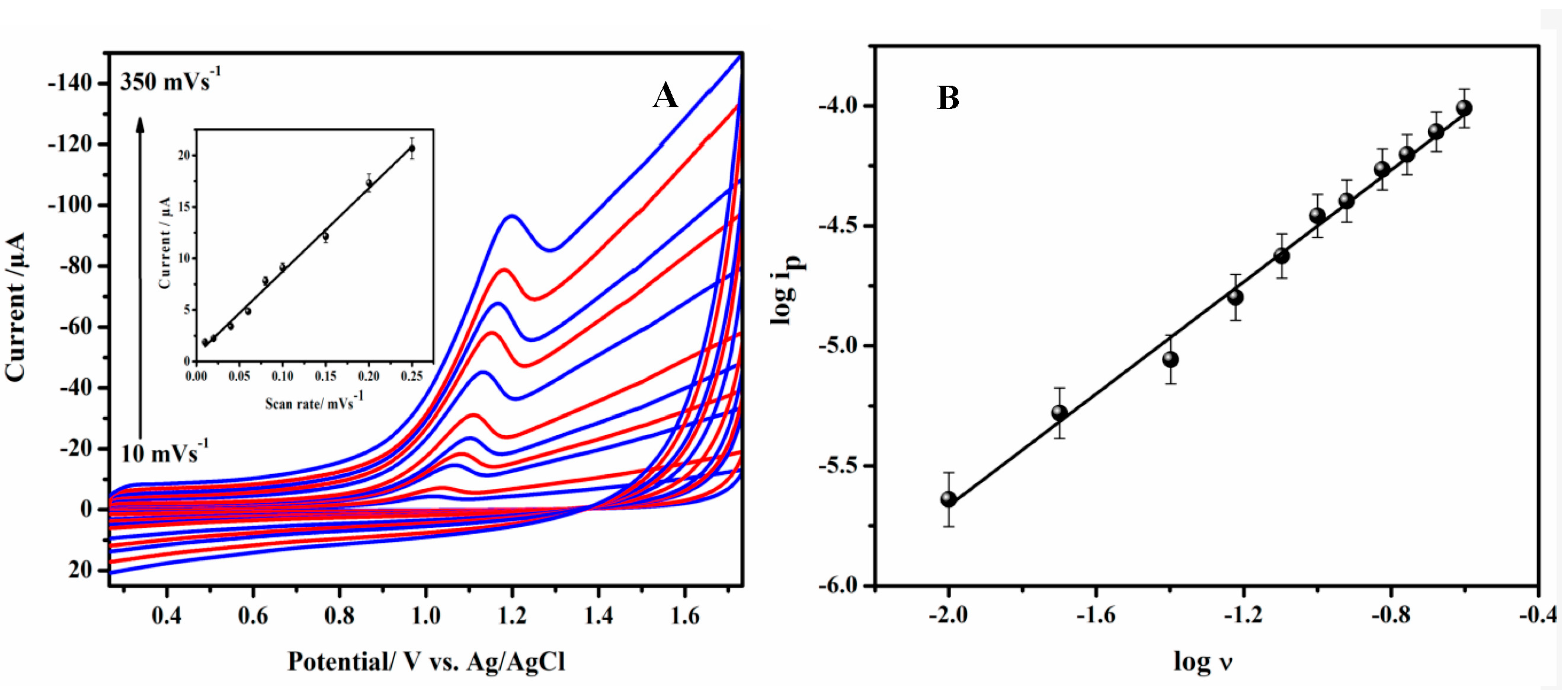
3.7. Scan Rate Studies
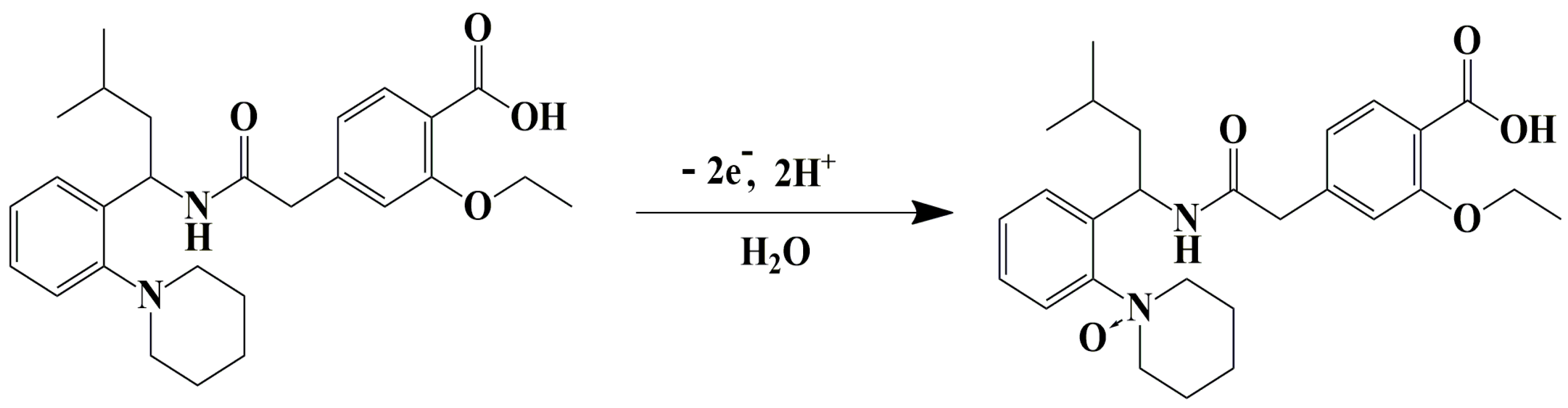
3.8. Electrode Reaction Mechanism
3.9. Construction of Calibration Curve
3.10. Reproducibility and Stability of SnO2@p-rGO/GCE
3.11. Determination of RPG in Urine Samples
3.12. Determination of RPG in Pharmaceutical Formulations
3.13. Selectivity and Interference Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gumieniczek, A.; Berecka, A. Analytical Tools for Determination of New Oral Antidiabetic Drugs, Glitazones, Gliptins, Gliflozins and Glinides, in Bulk Materials, Pharmaceuticals and Biological Samples. Open Chem. 2016, 14, 215–242. Available online: https://doi.org/10.1515/CHEM-2016-0023/ASSET/GRAPHIC/J_CHEM-2016-0023_FIG_007.JPG (accessed on 25 November 2022). [CrossRef]
- Fuhlendorff, J.; Rorsman, P.; Kofod, H.; Brand, C.L.; Rolin, B.; MacKay, P.; Shymko, R.; Carr, R.D. Stimulation of Insulin Release by Repaglinide and Glibenclamide Involves both Common and Distinct Processes. Diabetes 1998, 47, 345–351. [Google Scholar] [CrossRef]
- Kim, J.M.; Seo, S.W.; Han, D.G.; Yun, H.; Yoon, I.S. Assessment of Metabolic Interaction between Repaglinide and Quercetin via Mixed Inhibition in the Liver: In Vitro and In Vivo. Pharmaceutics 2021, 13, 782. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Wang, M.; Wu, J.X.; Kang, Y.; Chen, L. The Structural Basis for the Binding of Repaglinide to the Pancreatic KATP Channel. Cell Rep. 2019, 27, 1848–1857.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jirovský, D.; Bartošová, Z.; Skopalová, J.; Maier, V. Electrochemical Characterization of Repaglinide and Its Determination in Human Plasma Using Liquid Chromatography with Dual-Channel Coulometric Detection. J. Chromatogr. B 2010, 878, 3243–3248. [Google Scholar] [CrossRef]
- Dhole, S.M.; Amnerkar, N.D.; Khedekar, P.B. Comparison of UV Spectrophotometry and High Performance Liquid Chromatography Methods for the Determination of Repaglinide in Tablets. Pharm. Methods 2012, 3, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.; Patel, L.J.; Patel, M. Development and Validation of Stability Indicating Method for the Determination of Repaglinide in Pharmaceutical Dosage Form Using High Performance Liquid Chromatography. Int. J. ChemTech Res. 2011, 3, 539–546. [Google Scholar]
- Lakshmi, K.S.; Rajesh, T. Separation and Quantification of Eight Antidiabetic Drugs on a High-Performance Liquid Chromatography: Its Application to Human Plasma Assay. ISRN Pharm. 2011, 2011, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Gumustas, M.; Coskun, G.; Ozkan, S.A. Selective and sensitive determination of repaglinide in pharmaceuticals by voltammetric and lc methods. Rev. Roum. Chim. 2015, 60, 477–490. [Google Scholar]
- Gandhimathi, M.; Ravi, T.K.; Renu, S.K. Determination of Repaglinide in Pharmaceutical Formulations by HPLC with UV Detection. Anal. Sci. 2003, 19, 1675–1677. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Miksa, I.R. Multi-Component Plasma Quantitation of Anti-Hyperglycemic Pharmaceutical Compounds Using Liquid Chromatography-Tandem Mass Spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 856, 318–327. [Google Scholar] [CrossRef]
- Ho, E.N.M.; Yiu, K.C.H.; Wan, T.S.M.; Stewart, B.D.; Watkins, K.L. Detection of Anti-Diabetics in Equine Plasma and Urine by Liquid Chromatography-Tandem Mass Spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2004, 811, 65–73. [Google Scholar] [CrossRef]
- Gumieniczek, A.; Berecka, A.; Hopkała, H. Quantitative Analysis of Repaglinide in Tablets by Reversed-Phase Thin-Layer Chromatography with Densitometric UV Detection. JPC—J. Planar Chromatogr.—Mod. TLC 2007, 18, 155–159. [Google Scholar] [CrossRef]
- Goyal, A.; Singhvi, I. Visible Spectrophotometric Methods for Estimation of Repaglinide in Tablet Formulation. Indian J. Pharm. Sci. 2006, 68, 656–657. [Google Scholar] [CrossRef] [Green Version]
- Tatiparthi, R.; Ramanjireddy, T.; Dhachinamoorthi, D.; Chandrasekhar, K.B. Pharmacokinetic Study of Repaglinide Floating Drug Delivery System in Rabbits by RP-HPLC Method Improving the Outpatient Medication Adherence towards the Chronic Diseases by Sending SMS to Remind the Dosing Instructions in Local Languages View Project Herbal Medicine Use: Clinical and Economic Outcomes View Project Pharmacokinetic Study of Repaglinide Floating Drug Delivery System in Rabbits by RP HPLC Method. Artic. J. Chin. Pharm. Sci. 2012, 21, 162–168. [Google Scholar] [CrossRef]
- Li, G.; Qi, X.; Wu, J.; Xu, L.; Wan, X.; Liu, Y.; Chen, Y.; Li, Q. Ultrasensitive, Label-Free Voltammetric Determination of Norfloxacin Based on Molecularly Imprinted Polymers and Au Nanoparticle-Functionalized Black Phosphorus Nanosheet Nanocomposite. J. Hazard. Mater. 2022, 436, 129107. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wu, J.; Qi, X.; Wan, X.; Liu, Y.; Chen, Y.; Xu, L. Molecularly Imprinted Polypyrrole Film-Coated Poly(3,4-Ethylenedioxythiophene): Polystyrene Sulfonate-Functionalized Black Phosphorene for the Selective and Robust Detection of Norfloxacin. Mater. Today Chem. 2022, 26, 101043. [Google Scholar] [CrossRef]
- Ozcelikay, G.; Kaya, S.I.; Ozkan, E.; Cetinkaya, A.; Nemutlu, E.; Kır, S.; Ozkan, S.A. Sensor-Based MIP Technologies for Targeted Metabolomics Analysis. TrAC Trends Anal. Chem. 2022, 146, 116487. [Google Scholar] [CrossRef]
- Mathad, A.S.; Seetharamappa, J.; Kalanur, S.S. β-Cyclodextrin Anchored Neem Carbon Dots for Enhanced Electrochemical Sensing Performance of an Anticancer Drug, Lapatinib via Host-Guest Inclusion. J. Mol. Liq. 2022, 350, 118582. [Google Scholar] [CrossRef]
- Bilge, S.; Karadurmus, L.; Atici, E.B.; Sınağ, A.; Ozkan, S.A. A Novel Electrochemical Sensor Based on Magnetic Co3O4 Nanoparticles/Carbon Recycled from Waste Sponges for Sensitive Determination of Anticancer Drug Ruxolitinib. Sens. Actuators B Chem. 2022, 367, 132127. [Google Scholar] [CrossRef]
- Genidy, G.; Kamal, A.; Nabi, M.A. Electrochemical Determination of the Antidiabetic Drug Repaglinide. Yakugaku Zasshi 2008, 128, 171–177. [Google Scholar]
- Roushani, M.; Jalilian, Z. Development of Electrochemical Sensor Based on Glassy Carbon Electrode Modified with a Molecularly Imprinted Copolymer and Its Application for Detection of Repaglinide. Electroanalysis 2018, 30, 2712–2718. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, X.; Chai, Z.; Hu, W. Low-Temperature Plasma Synthesis of Carbon Nanotubes and Graphene Based Materials and Their Fuel Cell Applications. Chem. Soc. Rev. 2013, 42, 8821–8834. [Google Scholar] [CrossRef] [PubMed]
- Kemp, K.C.; Seema, H.; Saleh, M.; Le, N.H.; Mahesh, K.; Chandra, V.; Kim, K.S. Environmental Applications Using Graphene Composites: Water Remediation and Gas Adsorption. Nanoscale 2013, 5, 3149–3171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Qian, K.; Yang, J.; Zhang, J.; Li, L.; Yu, C.; Zhao, D. Functional Nanoporous Graphene Foams with Controlled Pore Sizes. Adv. Mater. 2012, 24, 4419–4423. [Google Scholar] [CrossRef] [PubMed]
- Karaman, C.; Karaman, O.; Atar, N.; Yola, M.L. Tailoring of Cobalt Phosphide Anchored Nitrogen and Sulfur Co-Doped Three Dimensional Graphene Hybrid: Boosted Electrocatalytic Performance towards Hydrogen Evolution Reaction. Electrochim. Acta 2021, 380, 138262. [Google Scholar] [CrossRef]
- Yang, Y.; Ma, G.; Huang, J.; Nan, J.; Zhen, S.; Wang, Y.; Li, A. Hollow MnO2 Spheres/Porous Reduced Graphene Oxide as a Cathode Host for High-Performance Lithium-Sulfur Batteries. J. Solid State Chem. 2020, 286, 121297. [Google Scholar] [CrossRef]
- Van Hoa, N.; Dat, P.A.; Van Hieu, N.; Le, T.N.; Minh, N.C.; Van Tang, N.; Nga, D.T.; Ngoc, T.Q. Rapid and Efficient Synthesis of High-Porous Reduced Graphene Oxide/NiCo2S4 Nanocomposites for Supercapacitor Application. Diam. Relat. Mater. 2020, 106, 107850. [Google Scholar] [CrossRef]
- Qiu, H.J.; Guan, Y.; Luo, P.; Wang, Y. Recent Advance in Fabricating Monolithic 3D Porous Graphene and Their Applications in Biosensing and Biofuel Cells. Biosens. Bioelectron. 2017, 89, 85–95. [Google Scholar] [CrossRef]
- Zamani, P.; Higgins, D.C.; Hassan, F.M.; Fu, X.; Choi, J.Y.; Hoque, M.A.; Jiang, G.; Chen, Z. Highly Active and Porous Graphene Encapsulating Carbon Nanotubes as a Non-Precious Oxygen Reduction Electrocatalyst for Hydrogen-Air Fuel Cells. Nano Energy 2016, 26, 267–275. [Google Scholar] [CrossRef]
- Saisahas, K.; Soleh, A.; Somsiri, S.; Senglan, P.; Promsuwan, K.; Saichanapan, J.; Kanatharana, P.; Thavarungkul, P.; Lee, K.; Chang, K.H.; et al. Electrochemical Sensor for Methamphetamine Detection Using Laser-Induced Porous Graphene Electrode. Nanomaterials 2021, 12, 73. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, F.; Sun, Y.; Wan, C. Biomass-Derived Porous Graphene for Electrochemical Sensing of Dopamine. RSC Adv. 2021, 11, 15410–15415. [Google Scholar] [CrossRef] [PubMed]
- Karaman, C.; Aktaş, Z.; Bayram, E.; Karaman, O.; Kizil, C. Correlation between the Molecular Structure of Reducing Agent and PH of Graphene Oxide Dispersion on the Formation of 3D-Graphene Networks. CS J. Solid State Sci. Technol. 2020, 9, 071003. [Google Scholar] [CrossRef]
- Yin, S.; Niu, Z.; Chen, X. Assembly of Graphene Sheets into 3D Macroscopic Structures. Small 2012, 8, 2458–2463. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Sun, B.; Su, D.; Zhao, D.; Wang, G. Soft-Template Synthesis of 3D Porous Graphene Foams with Tunable Architectures for Lithium–O2 Batteries and Oil Adsorption Applications. J. Mater. Chem. A 2014, 2, 7973–7979. [Google Scholar] [CrossRef]
- Hassan Oghli, A.; Soleymanpour, A. Ultrasensitive Electrochemical Sensor for Simultaneous Determination of Sumatriptan and Paroxetine Using Molecular Imprinted Polymer/Sol-Gel/Polyoxometalate/RGO Modified Pencil Graphite Electrode. Sens. Actuators B Chem. 2021, 344, 130215. [Google Scholar] [CrossRef]
- Mehmandoust, M.; Erk, N.; Karaman, O.; Karimi, F.; Bijad, M.; Karaman, C. Three-Dimensional Porous Reduced Graphene Oxide Decorated with Carbon Quantum Dots and Platinum Nanoparticles for Highly Selective Determination of Azo Dye Compound Tartrazine. Food Chem. Toxicol. 2021, 158, 112698. [Google Scholar] [CrossRef]
- Xu, C.; Shi, X.; Ji, A.; Shi, L.; Zhou, C.; Cui, Y. Fabrication and Characteristics of Reduced Graphene Oxide Produced with Different Green Reductants. PLoS ONE 2015, 10, e0144842. [Google Scholar] [CrossRef] [Green Version]
- Fan, M.; Zhu, C.; Feng, Z.Q.; Yang, J.; Liu, L.; Sun, D. Preparation of N-Doped Graphene by Reduction of Graphene Oxide with Mixed Microbial System and Its Haemocompatibility. Nanoscale 2014, 6, 4882–4888. [Google Scholar] [CrossRef]
- Wang, J.; Salihi, E.C.; Šiller, L. Green Reduction of Graphene Oxide Using Alanine. Mater. Sci. Eng. C 2017, 72, 1–6. [Google Scholar] [CrossRef]
- Karimi-Maleh, H.; Karimi, F.; Fu, L.; Sanati, A.L.; Alizadeh, M.; Karaman, C.; Orooji, Y. Cyanazine Herbicide Monitoring as a Hazardous Substance by a DNA Nanostructure Biosensor. J. Hazard. Mater. 2022, 423, 127058. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhu, J.; Hu, Y.; Zhu, Y.; Zhu, G.; Hu, L.; Zi, Y.; Huang, W. Tin Oxide (SnO2) Nanoparticles: Facile Fabrication, Characterization, and Application in UV Photodetectors. Nanomaterials 2022, 12, 632. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Ahmed, A.; Singh, A.; Oruganti, S.K.; Khosla, A.; Arya, S. Review—Recent Advances in Tin Oxide Nanomaterials as Electrochemical/Chemiresistive Sensors. J. Electrochem. Soc. 2021, 168, 027505. [Google Scholar] [CrossRef]
- Singh, J.; Dutta, T.; Kim, K.H.; Rawat, M.; Samddar, P.; Kumar, P. ‘Green’ Synthesis of Metals and Their Oxide Nanoparticles: Applications for Environmental Remediation. J. Nanobiotechnol. 2018, 16, 84. [Google Scholar] [CrossRef]
- Narasaiah, B.P.; Banoth, P.; Sohan, A.; Mandal, B.K.; Dominguez, A.G.B.; Valladares, L.D.L.S.; Kollu, P. Green Biosynthesis of Tin Oxide Nanomaterials Mediated by Agro-Waste Cotton Boll Peel Extracts for the Remediation of Environmental Pollutant Dyes. ACS Omega 2022, 7, 15423–15438. [Google Scholar] [CrossRef]
- Saadat Niavol, S.; Milani Moghaddam, H. SnO2 Nanoparticles/Reduced Graphene Oxide Nanocomposite for Fast Ethanol Vapor Sensing at a Low Operating Temperature with an Excellent Long-Term Stability. J. Mater. Sci. Mater. Electron. 2021, 32, 6550–6569. [Google Scholar] [CrossRef]
- Cheng, J.P.; Wang, J.; Li, Q.Q.; Liu, H.G.; Li, Y. A Review of Recent Developments in Tin Dioxide Composites for Gas Sensing Application. J. Ind. Eng. Chem. 2016, 44, 1–22. [Google Scholar] [CrossRef]
- Hummers, W.S.; Offeman, R.E. Preparation of Graphitic Oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, L.; Hu, Z.; Yu, J.C. Synthesis of 3D Structured Graphene as a High Performance Catalyst Support for Methanol Electro-Oxidation. Nanoscale 2015, 7, 10896–10902. [Google Scholar] [CrossRef]
- Kumar, A.; Khandelwal, M. Amino Acid Mediated Functionalization and Reduction of Graphene Oxide—Synthesis and the Formation Mechanism of Nitrogen-Doped Graphene. New J. Chem. 2014, 38, 3457–3467. [Google Scholar] [CrossRef]
- Suresh, K.C.; Surendhiran, S.; Manoj Kumar, P.; Ranjth Kumar, E.; Khadar, Y.A.S.; Balamurugan, A. Green Synthesis of SnO2 Nanoparticles Using Delonix Elata Leaf Extract: Evaluation of Its Structural, Optical, Morphological and Photocatalytic Properties. SN Appl. Sci. 2020, 2, 1735. [Google Scholar] [CrossRef]
- Narasaiah, B.P.; Mandal, B.K. Remediation of Azo-Dyes Based Toxicity by Agro-Waste Cotton Boll Peels Mediated Palladium Nanoparticles. J. Saudi Chem. Soc. 2020, 24, 267–281. [Google Scholar] [CrossRef]
- Niu, X.; Li, X.; Chen, W.; Li, X.; Weng, W.; Yin, C.; Dong, R.; Sun, W.; Li, G. Three-Dimensional Reduced Graphene Oxide Aerogel Modified Electrode for the Sensitive Quercetin Sensing and Its Application. Mater. Sci. Eng. C 2018, 89, 230–236. [Google Scholar] [CrossRef]
- Feng, H.; Cheng, R.; Zhao, X.; Duan, X.; Li, J. A Low-Temperature Method to Produce Highly Reduced Graphene Oxide. Nat. Commun. 2013, 4, 1539. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Li, H.; Li, Y.; Ma, Y.; Yang, H.; Liu, H.; Ren, X.; Zhao, H. Layered SnO2 Nanorods Arrays Anchored on Reduced Graphene Oxide for Ultra-High and Ppb Level Formaldehyde Sensing. Sens. Actuators B Chem. 2021, 346, 130452. [Google Scholar] [CrossRef]
- Sha, R.; Badhulika, S. Facile Green Synthesis of Reduced Graphene Oxide/Tin Oxide Composite for Highly Selective and Ultra-Sensitive Detection of Ascorbic Acid. J. Electroanal. Chem. 2018, 816, 30–37. [Google Scholar] [CrossRef]
- Fadillah, G.; Wicaksono, W.P.; Fatimah, I.; Saleh, T.A. A Sensitive Electrochemical Sensor Based on Functionalized Graphene Oxide/SnO2 for the Determination of Eugenol. Microchem. J. 2020, 159, 105353. [Google Scholar] [CrossRef]
- Choudhari, A.; Bhanvase, B.A.; Saharan, V.K.; Salame, P.H.; Hunge, Y. Sonochemical Preparation and Characterization of RGO/SnO2 Nanocomposite: Electrochemical and Gas Sensing Performance. Ceram. Int. 2020, 46, 11290–11296. [Google Scholar] [CrossRef]
- Bansode, S.R.; Khare, R.T.; Jagtap, K.K.; More, M.A.; Koinkar, P. One Step Hydrothermal Synthesis of SnO2-RGO Nanocomposite and Its Field Emission Studies. Mater. Sci. Semicond. Process. 2017, 63, 90–96. [Google Scholar] [CrossRef]
- Tao, Y.; Kong, D.; Zhang, C.; Lv, W.; Wang, M.; Li, B.; Huang, Z.H.; Kang, F.; Yang, Q.H. Monolithic Carbons with Spheroidal and Hierarchical Pores Produced by the Linkage of Functionalized Graphene Sheets. Carbon N. Y. 2014, 69, 169–177. [Google Scholar] [CrossRef]
- Li, G.; Shen, Y.; Zhou, P.; Hao, F.; Fang, P.; Wei, D.; Meng, D.; San, X. Design and Application of Highly Responsive and Selective RGO-SnO2 Nanocomposites for NO2 Monitoring. Mater. Charact. 2020, 163, 110284. [Google Scholar] [CrossRef]
- Huang, J.; Meng, C.; Wang, H.; Ren, H.; Lu, X.; Joo, S.W. Preparation of Cross-Linked Porous SnO2 Nanosheets Using Three-Dimensional Reduced Graphene Oxide as a Template and Their Gas Sensing Property. J. Alloys Compd. 2022, 910, 164763. [Google Scholar] [CrossRef]
- Yu, X.; Wu, Q.; Zhang, H.; Zeng, G.; Li, W.; Qian, Y.; Li, Y.; Yang, G.; Chen, M. Investigation on Synthesis, Stability, and Thermal Conductivity Properties of Water-Based SnO2/Reduced Graphene Oxide Nanofluids. Materials 2018, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, M.; Mehmandoust, M.; Nodrat, O.; Salmanpour, S.; Erk, N. A Glassy Carbon Electrode Modified Based on Molybdenum Disulfide for Determination of Folic Acid in the Real Samples. J. Food Meas. Charact. 2021, 15, 5622–5629. [Google Scholar] [CrossRef]
- Mehmandoust, M.; Çakar, S.; Özacar, M.; Salmanpour, S.; Erk, N. Electrochemical Sensor for Facile and Highly Selective Determination of Antineoplastic Agent in Real Samples Using Glassy Carbon Electrode Modified by 2D-MoS2 NFs/TiO2 NPs. Top. Catal. 2021, 65, 564–576. [Google Scholar] [CrossRef]
- Yu, L.; Zhang, Q.; Yang, B.; Xu, Q.; Xu, Q.; Hu, X. Electrochemical Sensor Construction Based on Nafion/Calcium Lignosulphonate Functionalized Porous Graphene Nanocomposite and Its Application for Simultaneous Detection of Trace Pb2+ and Cd2+. Sens. Actuators B Chem. 2018, 259, 540–551. [Google Scholar] [CrossRef]
- Yu, X.; Sheng, K.; Shi, G. A Three-Dimensional Interpenetrating Electrode of Reduced Graphene Oxide for Selective Detection of Dopamine. Analyst 2014, 139, 4525–4531. [Google Scholar] [CrossRef] [Green Version]
- Laviron, E.; Roullier, L. General Expression of the Linear Potential Sweep Voltammogram for a Surface Redox Reaction with Interactions between the Adsorbed Molecules: Applications to Modified Electrodes. J. Electroanal. Chem. Interfacial Electrochem. 1980, 115, 65–74. [Google Scholar] [CrossRef]
- Laviron, E. General Expression of the Linear Potential Sweep Voltammogram in the Case of Diffusionless Electrochemical Systems. J. Electroanal. Chem. Interfacial Electrochem. 1979, 101, 19–28. [Google Scholar] [CrossRef]
- Bourlinos, A.B.; Stassinopoulos, A.; Anglos, D.; Zboril, R.; Karakassides, M.; Giannelis, E.P. Surface Functionalized Carbogenic Quantum Dots. Small 2008, 4, 455–458. [Google Scholar] [CrossRef]
- Grimshaw, J. Chapter 6—Oxidation of Aromatic Rings. Electrochem. React. Mech. Org. Chem. 2000, 203–218. Available online: https://books.google.co.in/books?hl=en&lr=&id=kGWj4pLZY6wC&oi=fnd&pg=PP1&dq=+Oxidation+of+Aromatic+Rings.+by++Grimshaw,+&ots=2PcmIf0mjA&sig=RLSwtcuhEBpcuUcheExLQ8vyZzU#v=onepage&q=Oxidation%20of%20Aromatic%20Rings.%20by%20%20Grimshaw%2C&f=false (accessed on 22 November 2022).
- Özkan, S.A.; Özkan, Y.; Şentürk, Z. Electrooxidation of Pimozide and Its Differential Pulse Voltammetric and HPLC–EC Determination. Anal. Chim. Acta 2002, 453, 221–229. [Google Scholar] [CrossRef]
- Zhang, H.; Bo, X.; Guo, L. Electrochemical Preparation of Porous Graphene and Its Electrochemical Application in the Simultaneous Determination of Hydroquinone, Catechol, and Resorcinol. Sens. Actuators B Chem. 2015, 220, 919–926. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Voltammetric Results | Impedance Spectroscopic Results | |||||||
|---|---|---|---|---|---|---|---|---|
| Modified GCE | Area (cm2) | ΔEp (mV) | Ip (µA) | Rct (Ω cm2) | Rs (Ω cm2) | CPE (µF cm−2) | n | W0 (kΩ cm2) |
| SnO2@p-rGO/GCE | 0.594 | 70 | 51.72 | 28.7 | 1.11 | 4.137 | 0.890 | 2.45 × 10−4 |
| p-rGO/GCE | 0.115 | 75 | 41.87 | 162.3 | 1.06 | 3.120 | 0.871 | 1.56 × 10−4 |
| SnO2/GCE | 0.088 | 79 | 30.45 | 486.5 | 1.12 | 3.326 | 0.823 | 1.21 × 10−4 |
| Bare GCE | 0.058 | 85 | 14.20 | 738.6 | 1.18 | 4.372 | 0.798 | 1.26 × 10−4 |
| Parameters | DPV | SWV |
|---|---|---|
| Linearity range, mol L−1 | 4.99 × 10−8–1.83 × 10−5 | 1.99 × 10−8–1.45 × 10−5 |
| LOD, mol L−1 | 9.02 × 10−9 | 8.50 × 10−10 |
| LOQ, mol L−1 | 3.00 × 10−8 | 2.83 × 10−9 |
| Inter-day assay, RSD * (%) | 2.45 | 2.19 |
| Intra-day assay, RSD * (%) | 1.74 | 2.23 |
| Method | Linearity Range | LOD | LOQ | Ref. |
|---|---|---|---|---|
| HPLC a | 5–50 μg/mL | 0.73 μg/mL | 2.21 μg/mL | [6] |
| HPLC | 0.5–3 μg/mL | 0.056 μg/mL | 0.172 μg/mL | [7] |
| HPLC | 50–2000 ng/mL | - | 20 ng/mL | [8] |
| HPLC-EC b | - | 0.017µg/mL | 0.051 µg/mL | [9] |
| HPLC-UV c | 0.1–0.5 μg/mL | - | - | [10] |
| LC-MS/MS d | - | 1.0 ng/mL | - | [12] |
| RPTLC e | 0.6–3.6 µg/10 μL | 0.08 µg/10 µL | 0.27 µg/10 µL | [13] |
| RP-HPLC f | 110–550 ng/mL | - | 110 ng/mL | [15] |
| Differential pulse voltammetry | [19] | |||
| Carbon paste electrode, CPE | 0.36–1.44 µg/mL | 0.0161 µg/mL | 0.203 µg/mL | |
| Glassy carbon electrode, GCE | 0.18–1.81 µg/mL | 0.048 µg/mL | 0.160 µg/mL | |
| MIP-PoDB/PoPD/GCE g | 0.002–0.452 µg/mL | 0.8 ng/mL | - | [20] |
| DPV (SnO2@p-rGO/GCE) | ||||
| Square-wave voltammetry | 0.02–8.14 µg/mL | 4.08 ng/mL | 13.5 ng/mL | |
| SWV (SnO2@p-rGO/GCE) | 0.009–6.56 µg/mL | 0.38 ng/mL | 1.28 ng/mL | Proposed work |
| Proposed Method | Amount of RPG Added, μM | n | Amount Found, μM | Average Recovery (%) | RSD, % |
|---|---|---|---|---|---|
| 0.1 | 5 | 0.098 | 98.0 | 3.02 | |
| DPV | 0.25 | 5 | 0.248 | 99.2 | 1.95 |
| 0.5 | 5 | 0.495 | 99.0 | 2.56 | |
| 0.1 | 5 | 0.099 | 99.0 | 2.50 | |
| SWV | 0.25 | 5 | 0.249 | 99.6 | 1.50 |
| 0.5 | 5 | 0.497 | 99.4 | 2.38 |
| DPV | SWV | |||
|---|---|---|---|---|
| Eurepa MF1 b | Ripadep c | Eurepa MF1 | Ripadep | |
| Labeled amount, mg | 0.5 | 1 | 0.50 | 1.00 |
| Amount found, mg | 0.485 | 0.996 | 0.49 | 0.997 |
| Recovery, % | 97.00 | 99.6 | 98.00 | 99.70 |
| RSD a, % | 2.34 | 1.98 | 2.51 | 2.86 |
| t-Value at 95% confidence level | 1.64 | - | 0.16 | - |
| F-Value at 95% confidence level | 2.37 | - | 4.39 | - |
| Pure RPG added, mg | 0.25 | 0.3 | 0.25 | 0.3 |
| Amount found, mg | 0.244 | 0.294 | 0.248 | 0.299 |
| Recovery, % | 97.60 | 98.00 | 99.20 | 99.60 |
| RSD a, % | 2.46 | 3.06 | 2.60 | 2.76 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathad, A.; Korgaonkar, K.; Jaldappagari, S.; Kalanur, S. Ultrasensitive Electrochemical Sensor Based on SnO2 Anchored 3D Porous Reduced Graphene Oxide Nanostructure Produced via Sustainable Green Protocol for Subnanomolar Determination of Anti-Diabetic Drug, Repaglinide. Chemosensors 2023, 11, 50. https://doi.org/10.3390/chemosensors11010050
Mathad A, Korgaonkar K, Jaldappagari S, Kalanur S. Ultrasensitive Electrochemical Sensor Based on SnO2 Anchored 3D Porous Reduced Graphene Oxide Nanostructure Produced via Sustainable Green Protocol for Subnanomolar Determination of Anti-Diabetic Drug, Repaglinide. Chemosensors. 2023; 11(1):50. https://doi.org/10.3390/chemosensors11010050
Chicago/Turabian StyleMathad, Ayyapayya, Karuna Korgaonkar, Seetharamappa Jaldappagari, and Shankara Kalanur. 2023. "Ultrasensitive Electrochemical Sensor Based on SnO2 Anchored 3D Porous Reduced Graphene Oxide Nanostructure Produced via Sustainable Green Protocol for Subnanomolar Determination of Anti-Diabetic Drug, Repaglinide" Chemosensors 11, no. 1: 50. https://doi.org/10.3390/chemosensors11010050
APA StyleMathad, A., Korgaonkar, K., Jaldappagari, S., & Kalanur, S. (2023). Ultrasensitive Electrochemical Sensor Based on SnO2 Anchored 3D Porous Reduced Graphene Oxide Nanostructure Produced via Sustainable Green Protocol for Subnanomolar Determination of Anti-Diabetic Drug, Repaglinide. Chemosensors, 11(1), 50. https://doi.org/10.3390/chemosensors11010050