
Differential Pulse Voltammetric Detection of Acetaminophen Using Nickel Phthalocyanine/CeO2-Modified ITO Electrodes



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of CeO2 Nanoparticles
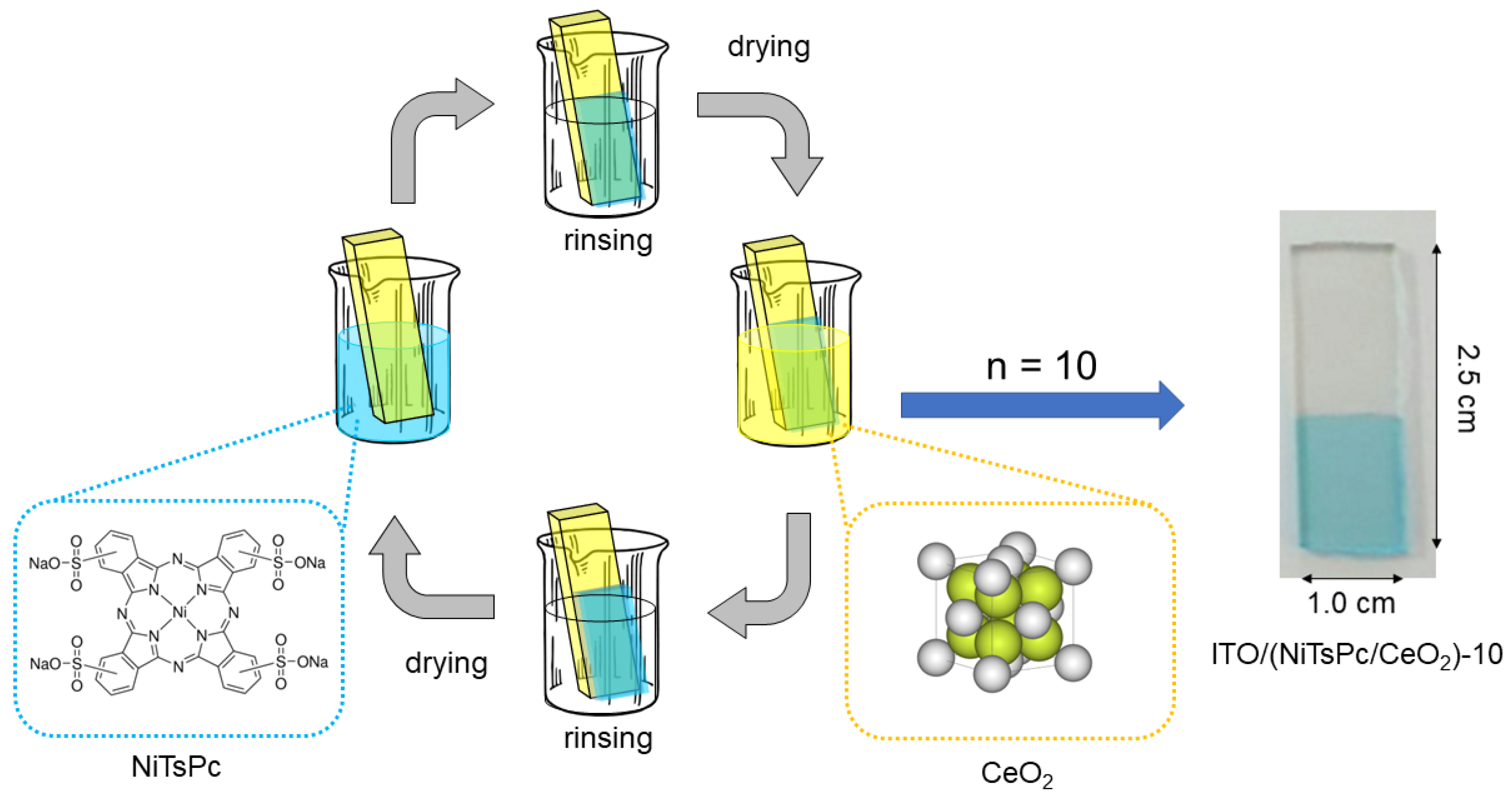
2.3. Film/Electrode Fabrication
2.4. Structural and Morphological Characterizations
2.5. Electrochemical Measurements
3. Results and Discussion
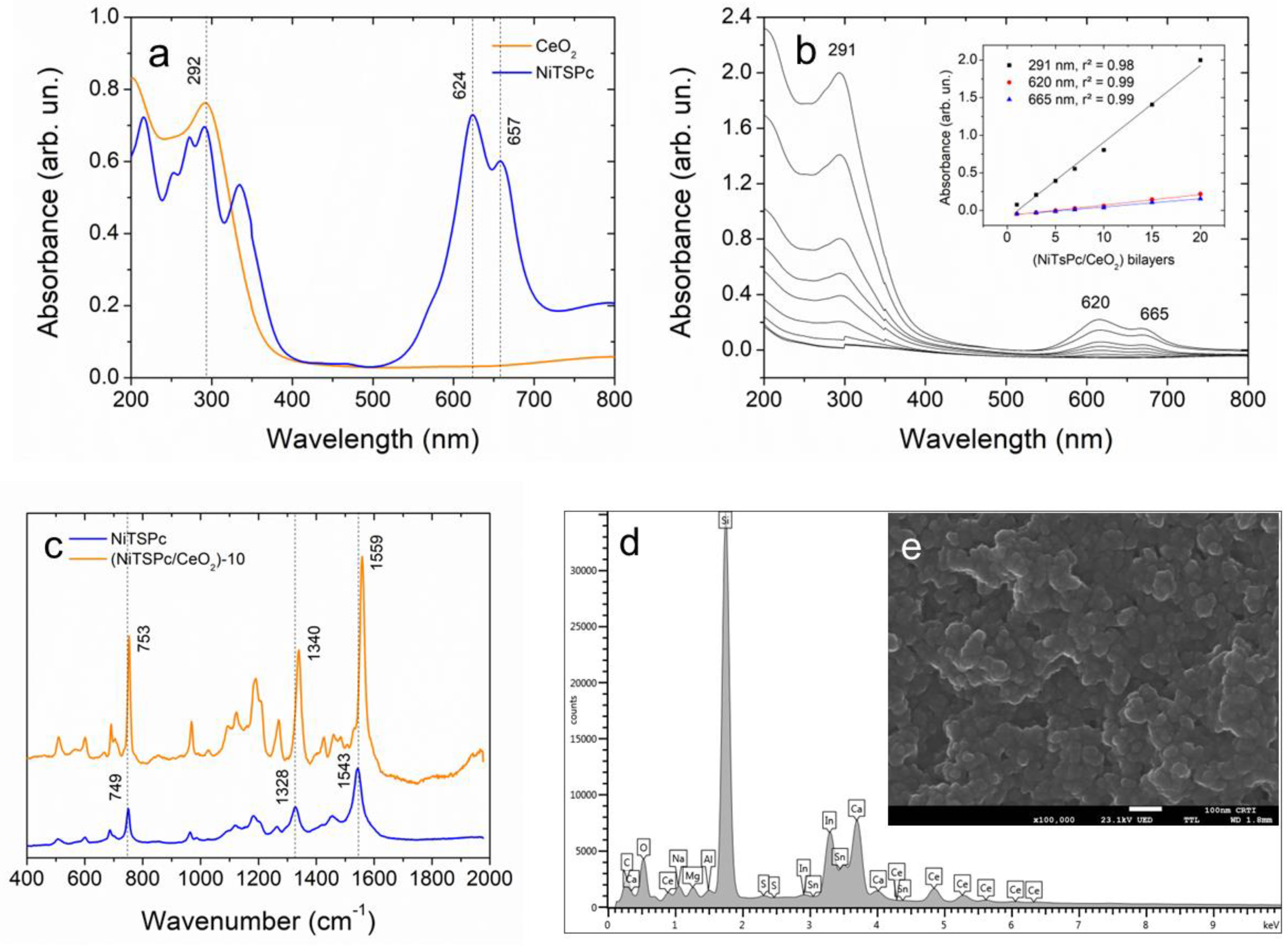
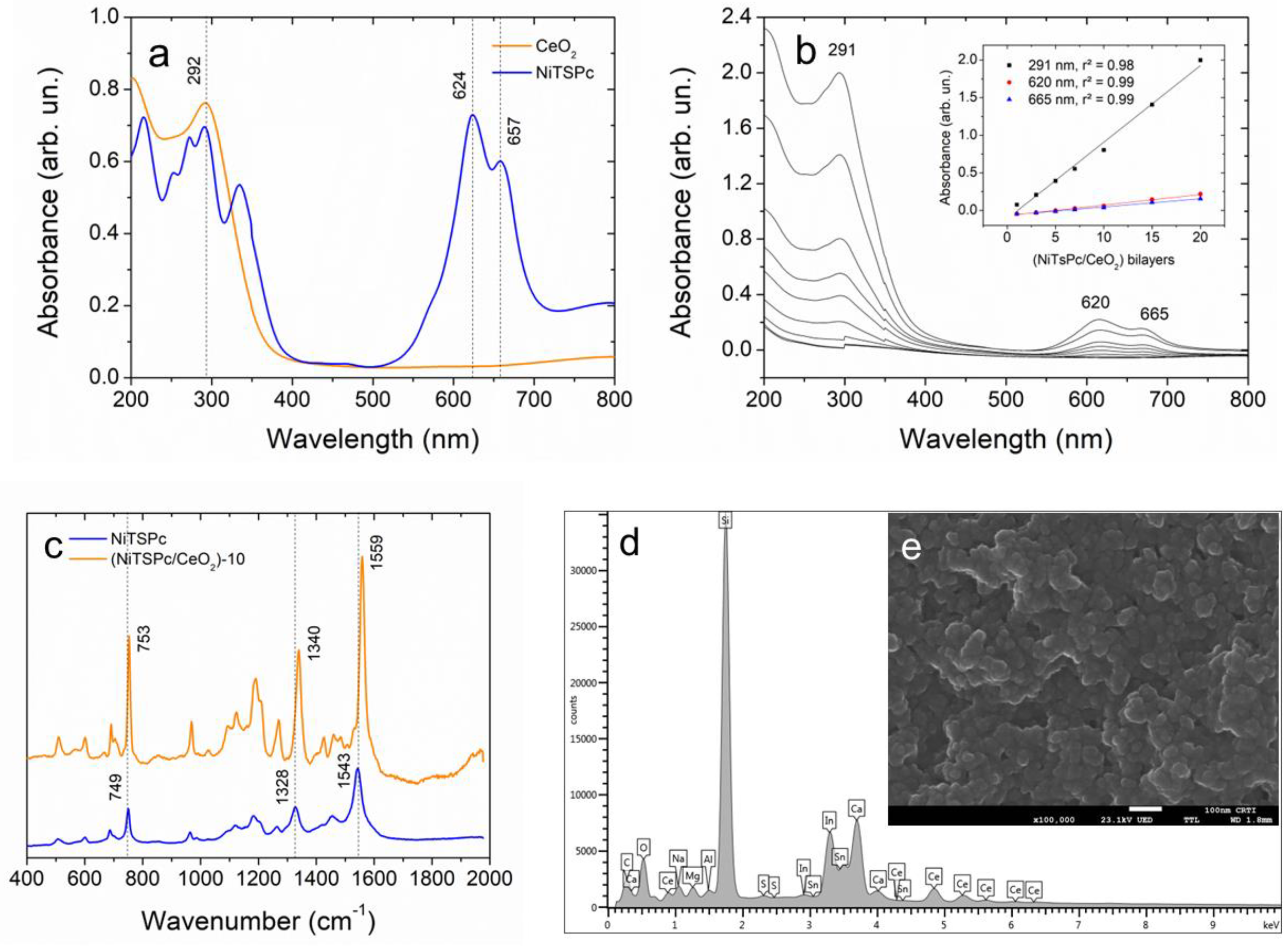
3.1. Structural Characterization of CeO2 Nanoparticles and (NiTsPc/CeO2) Nanofilms
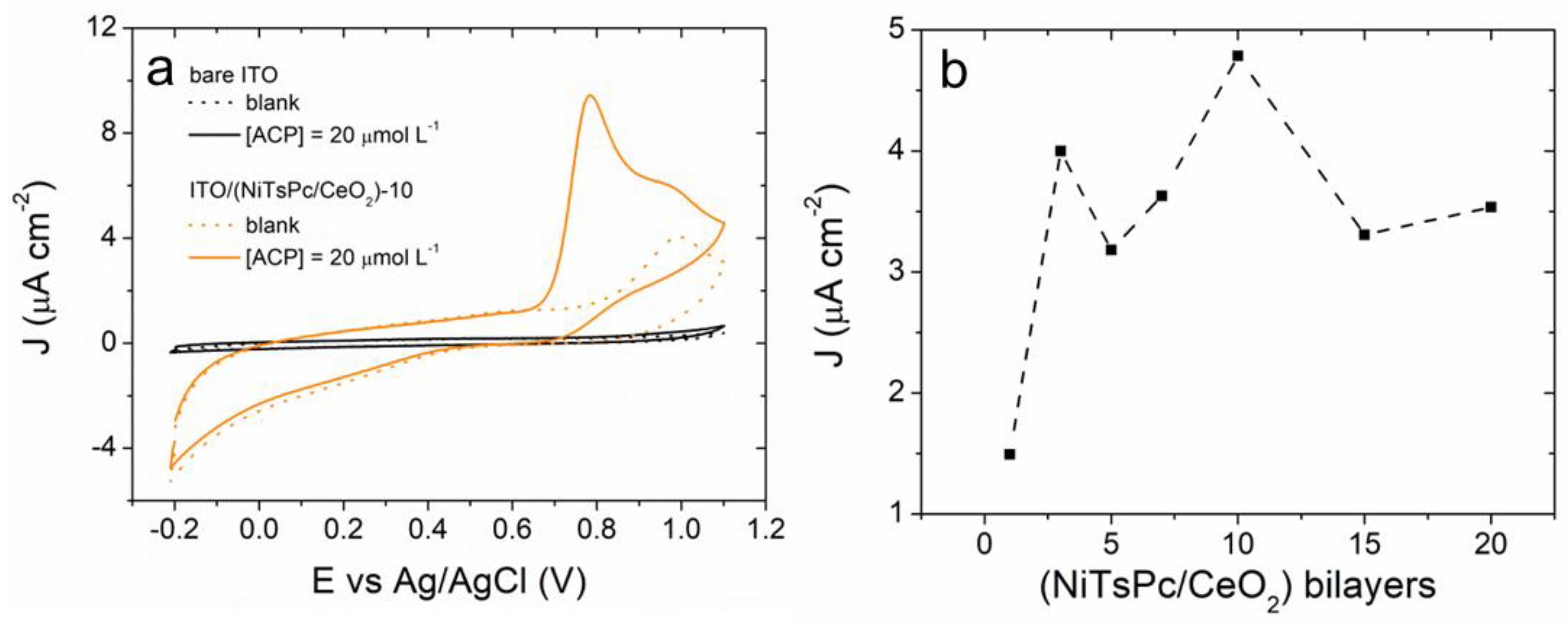
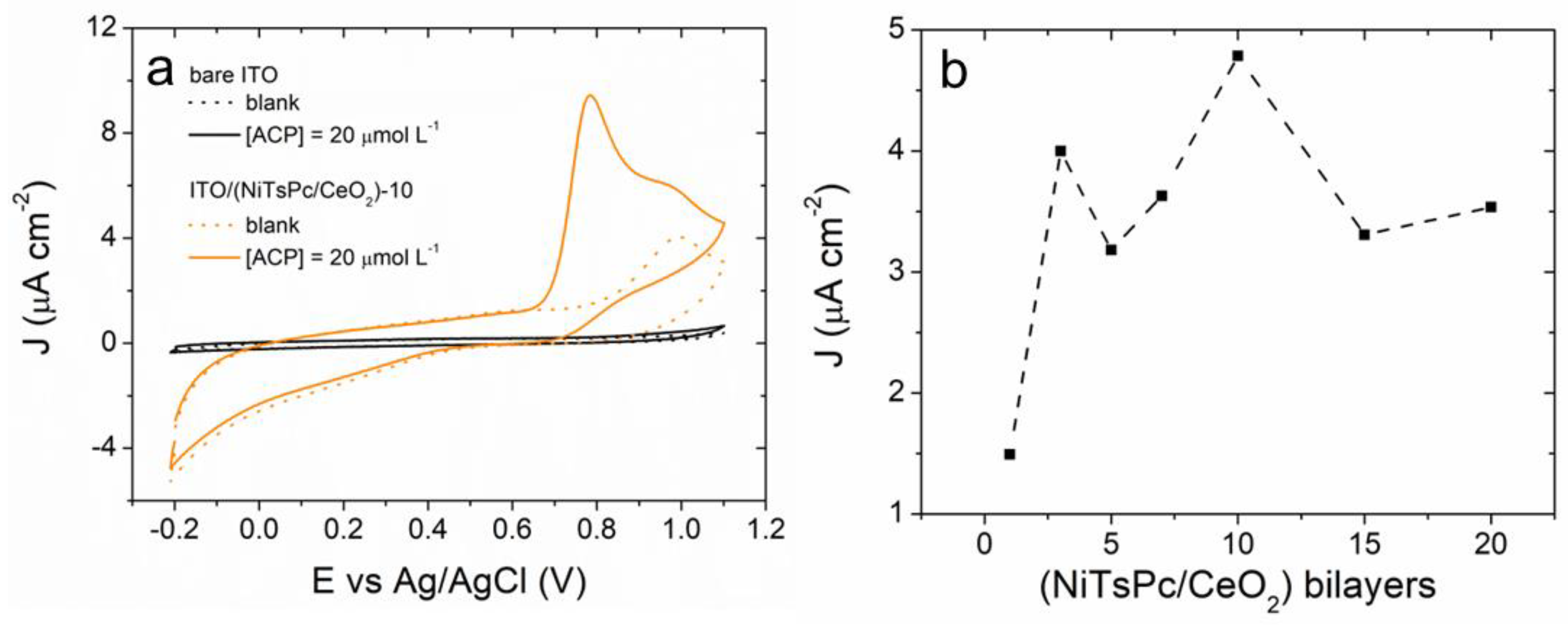
3.2. Electrochemical Behavior of ACP onto ITO/(NiTsPc/CeO2) Electrode
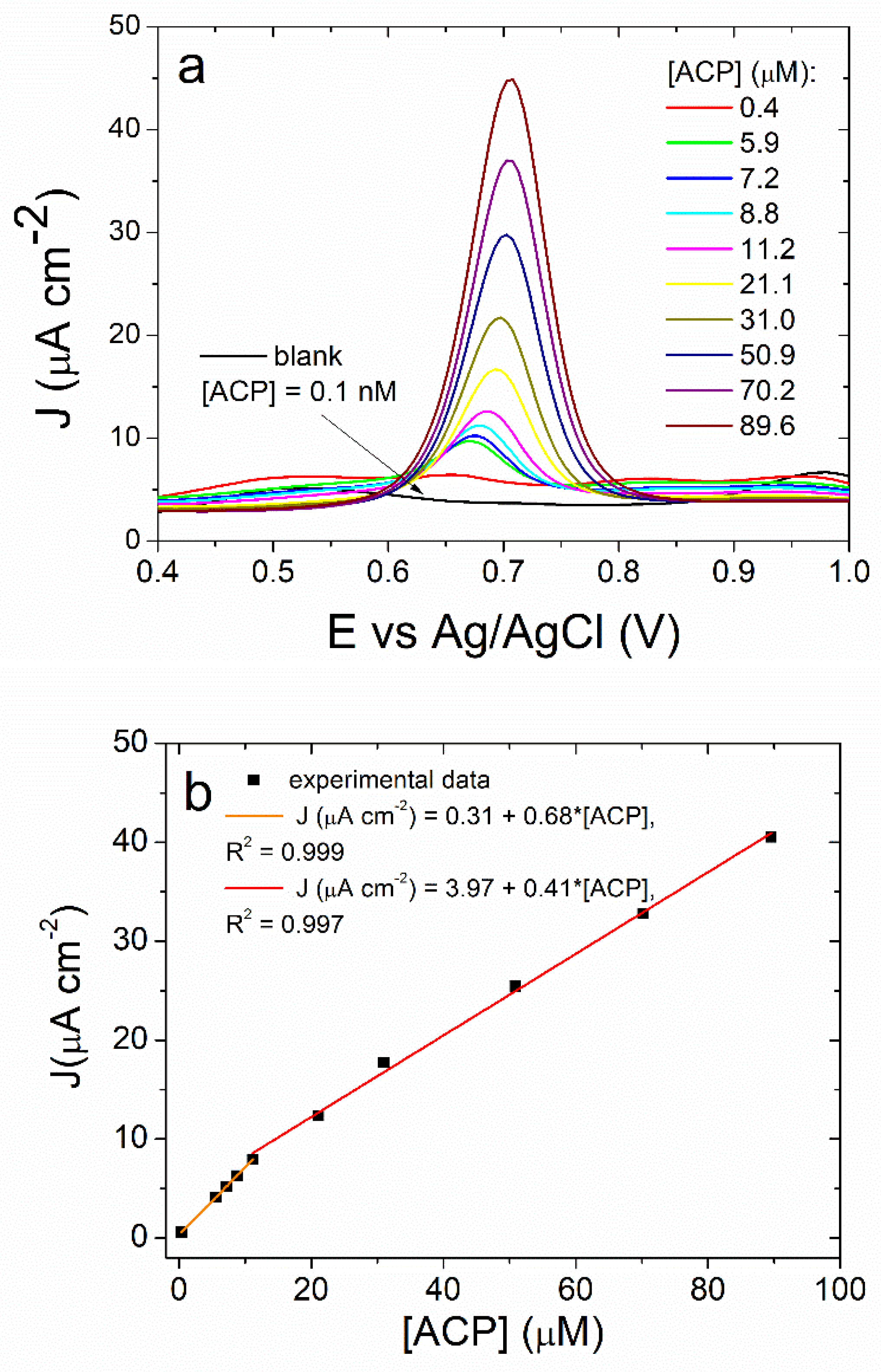
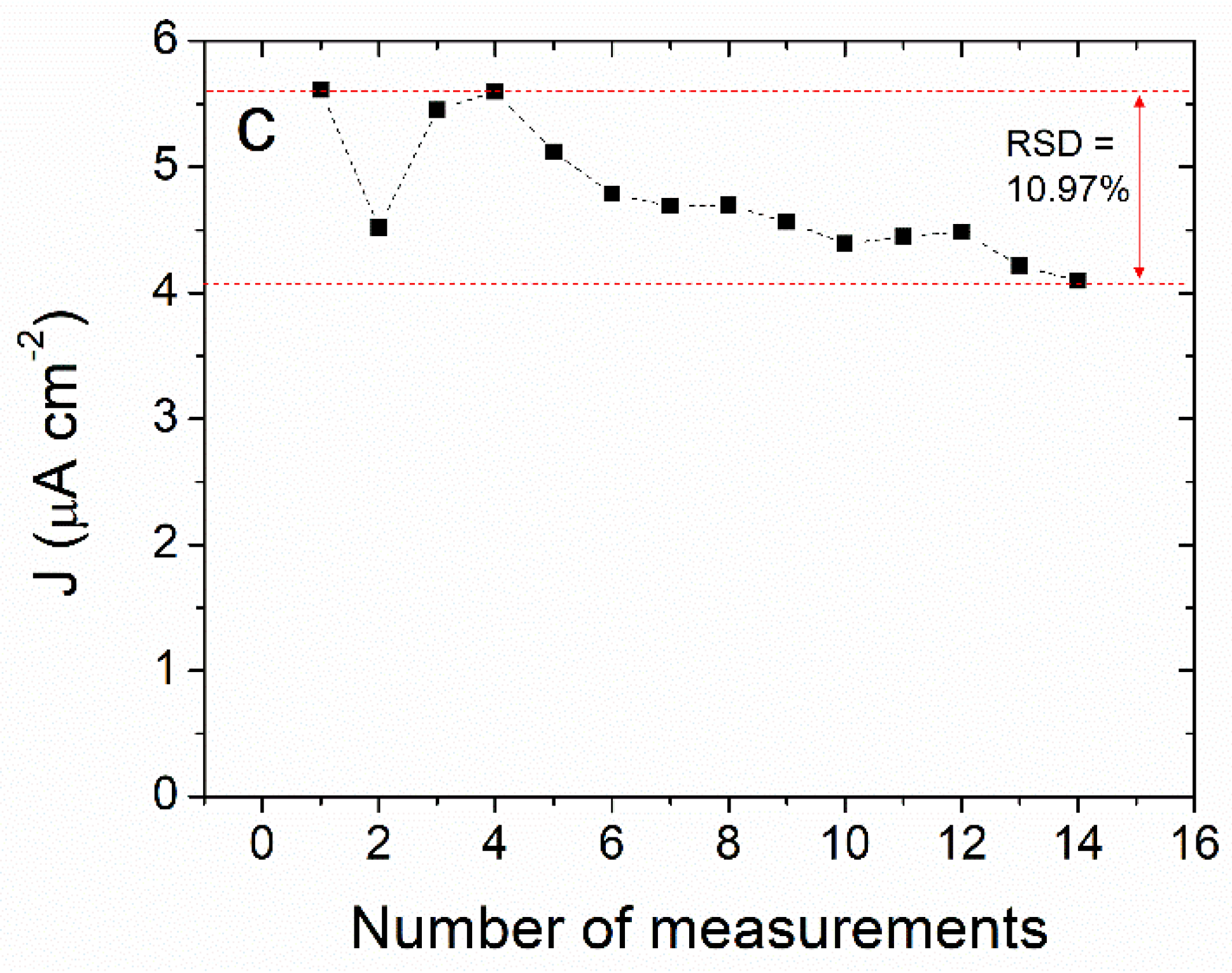
3.3. Differential Pulse Voltammetry Determination of ACP Using the ITO/(NiTsPc/CeO2)-10 Electrode
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Warwick, C. Paracetamol and fever management. J. R. Soc. Promot. Health 2008, 128, 320–323. [Google Scholar] [CrossRef]
- Saragiotto, B.T.; Shaheed, C.A.; Maher, C.G. Paracetamol for pain in adults. BMJ 2019, 367, l6693. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Goh, P.S.; Lau, W.J.; Ismail, A.F. Removal of pharmaceutical contaminants from aqueous medium: A state-of-the-art review based on paracetamol. Arab. J. Sci. Eng. 2020, 45, 7109–7135. [Google Scholar] [CrossRef]
- Sharma, R.K.; Raju, G.; Sarkar, P.; Singh, H.; Singla, E. Comparing the environmental impacts of paracetamol dosage forms using life cycle assessment. Environ. Dev. Sustain. 2022, 24, 12446–12466. [Google Scholar] [CrossRef]
- Agarwal, N. Paracetamol-A contaminant of high concern: Existence in environment and adverse effects. Pharmaceut. Drug Regul. Affair J. 2021, 4, 000128. [Google Scholar] [CrossRef]
- Benson, R.; Conerly, O.D.; Sander, W.; Batt, A.L.; Boone, J.S.; Furlon, E.T.; Glassmeyer, S.T.; Kolpin, D.W.; Mash, H.E.; Schenck, K.M.; et al. Human health screening and public health significance of contaminants of emerging concern detected in public water supplies. Sci. Total Environ. 2017, 579, 1643–1648. [Google Scholar] [CrossRef]
- Montaseri, H.; Forbes, P.B.C. Analytical techniques for the determination of acetaminophen: A review. Trends Analyt. Chem. 2018, 108, 122–134. [Google Scholar] [CrossRef]
- Sivaranjanee, R.; Kumar, P.S.; Saravanan, R.; Govarthanan, M. Electrochemical sensing system for the analysis of emerging contaminants in aquatic environment: A review. Chemosphere 2022, 294, 133779. [Google Scholar] [CrossRef]
- Nematollahi, D.; Shayani-Jam, H.; Alimoradi, M.; Niroomand, S. Electrochemical oxidation of acetaminophen in aqueous solutions: Kinetic evaluation of hydrolysis, hydroxylation and dimerization processes. Electrochim. Acta 2009, 54, 7407–7415. [Google Scholar] [CrossRef]
- Li, X.; Kang, J.; Ji, H.; Gong, P.; Li, N. Past, present and future of electroanalytical sensor for aspirin, ibuprofen and paracetamol detection. Curr. Pharm. Anal. 2022, 18, 24–33. [Google Scholar]
- Zhang, F.; Wang, P.; Koberstein, J.; Khalid, S.; Chan, S.W. Cerium oxidation state in ceria nanoparticles studied with X-ray photoelectron spectroscopy and absorption near edge spectroscopy. Surf. Sci. 2004, 563, 74–82. [Google Scholar] [CrossRef]
- Kusuma, K.B.; Manju, M.; Ravikumar, C.R.; Raghavendra, N.; Amulya, M.A.S.; Nagaswarupa, H.P.; Murthy, H.C.A.; Kumar, M.R.A.; Shekhar, T.R.S. Photocatalytic degradation of methylene blue and electrochemical sensing of paracetamol using cerium oxide nanoparticles synthesized via sonochemical route. Appl. Surf. Sci. Adv. 2022, 11, 100304. [Google Scholar] [CrossRef]
- Azeredo, N.F.B.; Gonçalves, J.M.; Lima, I.S.; Araki, K.; Wang, J.; Angnes, L. Screen-printed nickel-cerium hydroxide sensor for acetaminophen determination in body fluids. Chemeletrochem 2021, 8, 2505–2511. [Google Scholar] [CrossRef]
- Stanković, D.M.; Ognjanović, M.; Jović, M.; Cuplić, V.; Lesch, A.; Girault, H.H.; Jankulović, M.G.; Antić, B. Disposable biosensor based on amidase/CeO2/GNR modified inkjet-printed CNT electrodes-droplet based paracetamol detection in biological fluids for “point-of-care” applications. Electroanalysis 2019, 8, 1517–1525. [Google Scholar] [CrossRef]
- Ponnaiah, S.K.; Prakash, P.; Vellaichamy, B. A new analytical device incorporating a nitrogen doped lanthanum metal oxide with reduced graphene oxide sheets for paracetamol sensing. Ultrason. Sonochem. 2018, 44, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, M.; Sakthivel, M.; Chen, S.-M. One pot synthesis of CeO2 nanoparticles on a carbon surface for the practical determination of paracetamol content in real samples. RSC Adv. 2016, 6, 104227–104234. [Google Scholar] [CrossRef]
- AOAC International. Official Methods of Analysis in Guidelines for Standard Method Performance Requirements (Appendix F); AOAC International: Rockville, MD, USA, 2016. [Google Scholar]
- Goes, E.J.A., Jr.; Roedera, J.S.; Oliveira, K.B.L.; Gois, L.C.; Ferreira, M.P.; Silva, J.G. UV spectrophotometry method validation for quantification of paracetamol in tablet formulations: A proposal of experimental activity for instrumental analysis. Orbital Electron. J. Chem. 2018, 10, 561–567. [Google Scholar]
- Ribeiro, C.D.; de Souza, J.R.; Pereira-da-Silva, M.A.; Paterno, L.G. Voltammetric detection of ethinylestradiol in water and synthetic urine samples using a Ni(II) phthalocyanine/iron oxide nanocomposite electrode. Electroanalysis 2020, 33, 609–617. [Google Scholar] [CrossRef]
- Babitha, K.K.; Sreedevi, A.; Priyanka, K.P.; Sabu, B.; Varghese, T. Structural Characterization and optical studies of CeO2 nanoparticles synthesized by chemical precipitation. Indian J. Pure Appl. Phys. 2015, 53, 596–603. [Google Scholar]
- Spanier, J.E.; Robinson, R.D.; Zhang, F.; Chan, S.-W.; Herman, I.P. Size-dependent properties of CeO2 nanoparticles as studied by Raman scattering. Phys. Rev. B 2001, 64, 245407. [Google Scholar] [CrossRef] [Green Version]
- Snow, A.W. Phtalocyanine aggregation. In The Porphyrin Handbook; Elsevier: New York, NY, USA, 2003; Volume 17, pp. 129–176. ISBN 9780123932273. [Google Scholar]
- Schlenoff, J.B.; Ly, H.; Li, M. Charge and mass balance in polyelectrolyte multilayers. J. Am. Chem. Soc. 1998, 120, 7626–7634. [Google Scholar] [CrossRef]
- Ribeiro, C.D.; de Souza, J.R.; Pereira-da-Silva, M.A.; Santos, V.O.; Paterno, L.G. Electrocatalytic oxidation of ethinyl estradiol by an iron oxide nanoparticle/nickel phthalocyanine supramolecular electrode. J. Phys. Chem. C 2020, 124, 19057–19069. [Google Scholar] [CrossRef]
- Silva, J.C.O.; Sousa, M.H.; Tourinho, F.A.; Rubim, J.C. Raman spectroscopic investigation of maghemite ferrofluids modified by the adsorption of zinc tetrasulfonated phthalocyanine. Langmuir 2002, 18, 5511–5515. [Google Scholar] [CrossRef]
- Alencar, W.S.; Crespilho, F.N.; Santos, M.R.M.C.; Zucolotto, V.; Oliveira, O.N.; Silva, W.C. Influence of film architecture on the charge-transfer reactions of metallophthalocyanine layer-by-layer films. J. Phys. Chem. C 2007, 111, 12817–12821. [Google Scholar] [CrossRef]
- Mphuthi, N.G.; Adekunle, A.S.; Fayemi, O.E.; Olasunkanmi, L.O.; Ebenso, E.E. Phthalocyanine doped metal oxide nanoparticles on multiwalled carbon nanotubes platform for the detection of dopamine. Sci. Rep. 2017, 7, 43181. [Google Scholar] [CrossRef] [Green Version]
- Coelho, M.K.L.; da Silva, D.N.; Pereira, A.C. Development of electrochemical sensor based on carbonaceal and metal phthalocyanines materials for determination of ethinyl estradiol. Chemosensors 2019, 7, 32. [Google Scholar] [CrossRef] [Green Version]
- Zagal, J.H.; Griveau, S.; Silva, J.F.; Nyokong, T.; Bedioui, F. Metallophthalocyanine-based molecular materials as catalysts for electrochemical reactions. Coord. Chem. Rev. 2010, 254, 2755–2791. [Google Scholar] [CrossRef]
- Cheng, Y.; Veder, J.P.; Thomsen, L.; Zhao, S.; Saunders, M.; Demichelis, R.; Liu, C.; De Marco, R.; Jiang, S.P. Electrochemically substituted metal phthalocyanines, e-MPc (M = Co, Ni), as highly active and selective catalysts for CO2 reduction. J. Mater. Chem. A 2018, 6, 1370–1375. [Google Scholar] [CrossRef]
- Thill, A.S.; Figueiredo, W.T.; Lobato, F.O.; Vaz, M.O.; Fernandes, W.P.; Carvalho, V.E.; Soares, E.A.; Poletto, F.; Teixeira, S.R.; Bernardi, F. New horizons in photocatalysis: The importance of mesopores for cerium oxide. J. Mater. Chem. A 2020, 8, 24752–24762. [Google Scholar] [CrossRef]
- Tinh, V.D.C.; Thuc, V.D.; Kim, D. Chemically sustainable fuel cells via layer-by-layer fabrication of sulfonated poly(arylene ether sulfone) membranes containing cerium oxide nanoparticles. J. Membr. Sci. 2021, 634, 119430. [Google Scholar] [CrossRef]
- Azizi, S.; Asadpour-Zeynali, K. Electrochemical synthesis of tungstate bimetallic nanoparticles with application in electrocatalytic determination of paracetamol. ChemistrySelect 2022, 7, e202104548. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode | Method | Linear Range | LOD (nM) | Ref |
|---|---|---|---|---|
| CPE/CeO2 | CV | 1–5 mM | 1 × 106 | [12] |
| SPE/α-Ni0.9Ce0.1(OH)2 | CV | 100–1000 mM | 1700 | [13] |
| CNT/amidase/CeO2/Gr | SWV | 1–100 μM | 180 | [14] |
| GCE/N-CeO2/rGO | DPV | 0.05–0.600 μM | 9.8 | [15] |
| GCE/CeO2-C | DPV | 1–1219 µM | 70 | [16] |
| GCE/CoNiWO4-NS | DPV | 10–1000 µM | 1451 | [33] |
| ITO/(NiTsPc/CeO2) | DPV | 0.4–11.2 µM | 54.7 | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, E.C.; Ribeiro, C.L.; Santos, V.O., Jr.; Paterno, L.G. Differential Pulse Voltammetric Detection of Acetaminophen Using Nickel Phthalocyanine/CeO2-Modified ITO Electrodes. Chemosensors 2023, 11, 154. https://doi.org/10.3390/chemosensors11030154
Gomes EC, Ribeiro CL, Santos VO Jr., Paterno LG. Differential Pulse Voltammetric Detection of Acetaminophen Using Nickel Phthalocyanine/CeO2-Modified ITO Electrodes. Chemosensors. 2023; 11(3):154. https://doi.org/10.3390/chemosensors11030154
Chicago/Turabian StyleGomes, Emiliano C., Camila L. Ribeiro, Vianney O. Santos, Jr., and Leonardo G. Paterno. 2023. "Differential Pulse Voltammetric Detection of Acetaminophen Using Nickel Phthalocyanine/CeO2-Modified ITO Electrodes" Chemosensors 11, no. 3: 154. https://doi.org/10.3390/chemosensors11030154
APA StyleGomes, E. C., Ribeiro, C. L., Santos, V. O., Jr., & Paterno, L. G. (2023). Differential Pulse Voltammetric Detection of Acetaminophen Using Nickel Phthalocyanine/CeO2-Modified ITO Electrodes. Chemosensors, 11(3), 154. https://doi.org/10.3390/chemosensors11030154